
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting protein-loaded CB[8]-mediated supramolecular nanocarriers to cells†
Emanuela
Cavatorta
a,
Jens
Voskuhl
 a,
Dorothee
Wasserberg
b,
Jenny
Brinkmann
a,
Jurriaan
Huskens
*a and
Pascal
Jonkheijm
*a
a,
Dorothee
Wasserberg
b,
Jenny
Brinkmann
a,
Jurriaan
Huskens
*a and
Pascal
Jonkheijm
*a
aMolecular Nanofabrication Group, MESA+ Institute for Nanotechnology, Department of Science and Technology, University of Twente, P. O. Box 217, 7500 AE, Enschede, The Netherlands. E-mail: p.jonkheijm@utwente.nl; j.huskens@utwente.nl
bMedical Cell BioPhysics Group, MIRA Biomedical Technology and Technical Medicine, Department of Science and Technology, University of Twente, Enschede, The Netherlands
First published on 27th November 2017
Abstract
Supramolecular amphiphiles, consisting of ternary complexes of cucurbit[8]uril (CB[8]), an alkylated paraquat derivative and a tetraethylene glycol-functionalized azobenzene, self-assemble into vesicles of about 200 nm in diameter. The outer surface of the vesicles was functionalized with cell-targeting ligands. These vesicles were employed for loading and delivery of proteins into cells. Supramolecular amphiphile-derived vesicles show great promise as nanocarriers of functional molecules to be transferred into cells.
Introduction
The controlled self-assembly of molecules into ordered structures constitutes a powerful approach for the design of novel and functional materials.1 Supramolecular nanocarriers enable the incorporation of multiple characteristics via reversible, yet robust non-covalent interactions into one single system. For example, these features may provide the carriers simultaneously with units for targeting specific places and delivering drugs while imaging these processes.2 In this context cucurbit[8]uril (CB[8]) is a particularly interesting candidate to consider as a component of the non-covalent interaction to assemble the nanocarriers, owing to its biocompatibility and its ability to form ternary complexes by simultaneously including two guest moieties with high overall binding affinities in physiological milieu.3,4 The possibility to form such CB[8]-mediated ternary host–guest complexes provides great synthetic versatility to achieve self-assembled nanocarrier systems, such as vesicles and micelles.5 These supramolecular nanocarriers can readily incorporate cargo by host–guest interactions or simply by encapsulation. Release of the cargo from the nanocarriers can be achieved by reversing the formation of the ternary complex through the use of thermo- and pH-responsive guest moieties, by adding a competing guest or by disassembling the vesicles.5Recently, two types of guests, lipidated trans-azobenzene and methyl viologen, were reported to form vesicles in the presence of CB[8].6 Upon irradiation using UV light, the azobenzene switches to the cis-isomer, which is sterically unable to form the ternary complex and hence the vesicles disassemble and can form new vesicles upon ternary complexation with drug molecules, yet this makes them challenging for use in vitro. These types of CB[8]-mediated vesicles have neither been attempted to be loaded with cargo nor decorated with targeting ligands for cells. Such azobenzene switches have also been attached to mesoporous silica nanoparticles.7 In the trans-form a ternary complex was formed with CB[8] and a 4,4′-dipyridyl-modified peptide ligand. Doxorubicin was included as cargo molecules inside the particles, and the carrier was able to interact with macrophages.7 Photo-irradiation caused the disassembly of the ternary complexes, which in turn caused the release of the cargo from the mesoporous particles.7
In this work, vesicular supramolecular nanocarriers were formed by the formation of amphiphilic hetero-ternary complexes between CB[8], a hydrophobic alkyl-substituted methyl viologen (MV) and a hydrophilic oligo(ethylene glycol)-substituted azobenzene (Azo), which also provides photo-switchability to the nanocarriers (Fig. 1). We introduce fractions of cell targeting ligands onto the nanocarriers by way of supramolecular interactions and load them with fluorescent proteins. We demonstrate that such nanocarriers are capable of cell targeting and can furthermore be used for simultaneous targeted cargo delivery and imaging purposes.
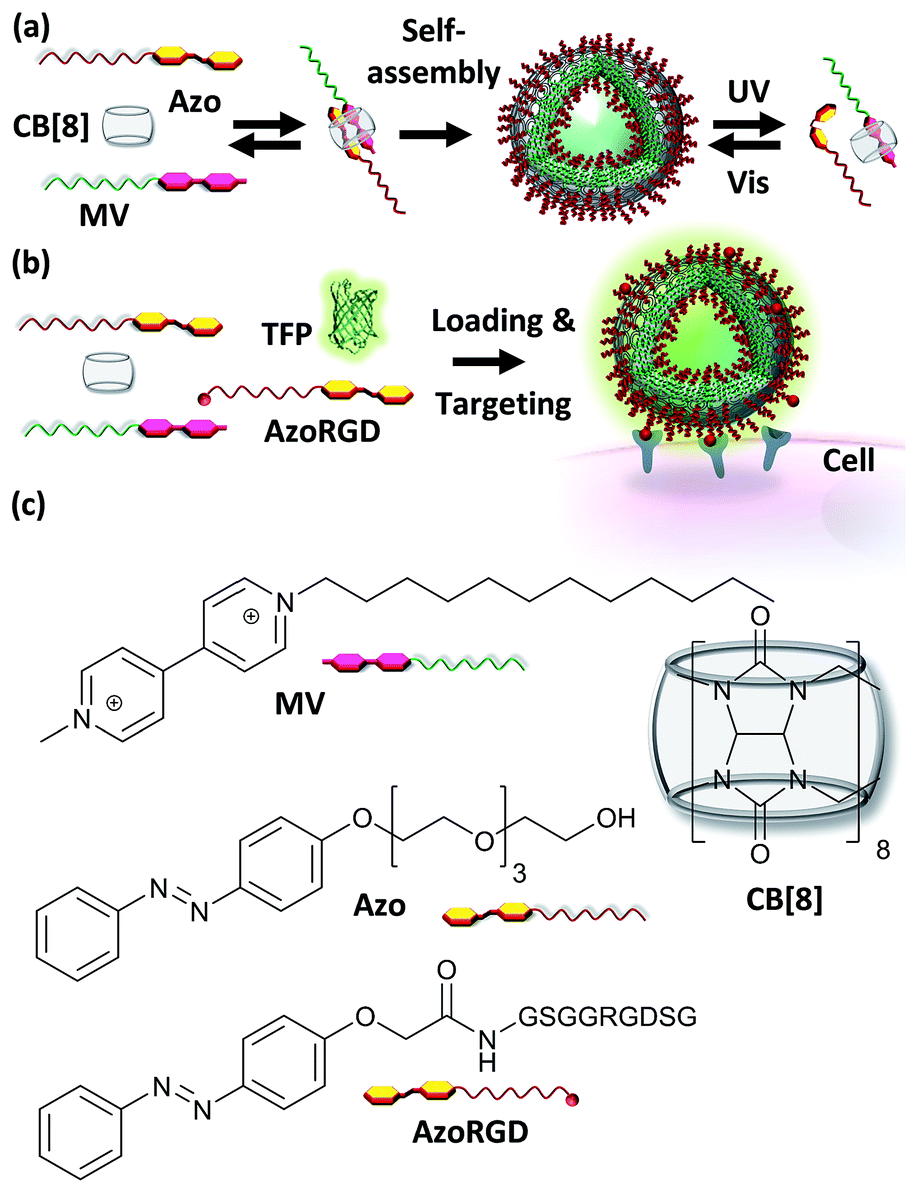 | ||
| Fig. 1 (a) A supramolecular amphiphile is formed as a ternary complex between CB[8], methyl viologen with a hydrophobic alkyl chain (MV) and azobenzene with a hydrophilic oligo(ethylene glycol) chain (Azo). (b) Cell targeting is provided by including AzoRGD peptide ligands, while the vesicles can be loaded with teal, yellow and red fluorescent proteins (TFP, YFP, TagRFP) as cargo (TFP is shown). (c) Molecular structures of MV, Azo, AzoRGD and CB[8]. | ||
Experimental section
Materials, methods and synthetic procedures are reported in the ESI.†Nanoparticle preparation and characterization
To form nanoparticles MV, Azo (or AzoRGD) and CB[8] (or CB[7]) stock solutions were prepared in water at 50 μM and mixed to obtain a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 mixing ratio at final concentrations of 17 μM, each. After a sonication step (1 h, approx. 40 °C) the samples were degassed in vacuo for 15 min and the particles were then characterized by hrSEM, TEM and DLS. Fig. S2† shows the TEM micrographs on negatively stained samples of particles and the theoretical, fully extended, conformations of two supramolecular ternary complexes. For UV switching studies, freshly prepared particles were irradiated for 1 h with UV light (104 mW, with a short pass filter <392 nm) and immediately characterized using DLS or UV-vis spectroscopy. Fluorescent proteins were loaded inside the nanoparticles by adding the proteins to the MV, Azo and CB[8] mixture during vesicle formation. No further purification was performed. A FRET protein pair consisting of yellow and red fluorescent proteins was used as cargo (Fig. S3†). A teal fluorescent protein was used for cell studies.
:1:1 mixing ratio at final concentrations of 17 μM, each. After a sonication step (1 h, approx. 40 °C) the samples were degassed in vacuo for 15 min and the particles were then characterized by hrSEM, TEM and DLS. Fig. S2† shows the TEM micrographs on negatively stained samples of particles and the theoretical, fully extended, conformations of two supramolecular ternary complexes. For UV switching studies, freshly prepared particles were irradiated for 1 h with UV light (104 mW, with a short pass filter <392 nm) and immediately characterized using DLS or UV-vis spectroscopy. Fluorescent proteins were loaded inside the nanoparticles by adding the proteins to the MV, Azo and CB[8] mixture during vesicle formation. No further purification was performed. A FRET protein pair consisting of yellow and red fluorescent proteins was used as cargo (Fig. S3†). A teal fluorescent protein was used for cell studies.
Cell experiments
C2C12 cells (murine myoblast cell line), (Thermo Fisher Scientific) were cultured in DMEM medium with 10% fetal bovine serum, L-glutamine and penicillin-streptomycin. Cells between passages 15 and 25 were used and kept below 90% confluence. For experiments, cells were seeded at densities between 8000–10000 cells per cm2 and left for at least 2 h in the incubator (37 °C with 5% CO2). Cells were then washed once with Live Cell Imaging Solution (LC-buffer, Thermo Fisher Scientific), counterstained with Hoechst (1 μg mL−1, 5 min), then, rinsed twice more with LC-buffer. Cells were then incubated with the nanocarriers loaded with protein (0.5 μM) and functionalized with AzoRGD (0.05–1% of the total content of Azo was replaced by AzoRGD, e.g. resulting in 1.6 μM AzoRGD, 14.5 μM Azo and 16 μM CB[8] and MV for the 1% AzoRGD condition) for 10 min in the incubator. Finally, cells were washed twice with LC-buffer to remove unbound particles. Live cells were then directly imaged using a fluorescence microscope. Fluorescence intensities were quantified after background subtraction using the ImageJ software package.
Results and discussion
Nanocarrier characterization
A supramolecular amphiphile was formed through the formation of a hetero-ternary complex between CB[8], a hydrophobic alkyl-substituted methyl viologen (MV) and a hydrophilic oligo(ethylene glycol)-substituted azobenzene (Azo) (Fig. 1). These three components were mixed in equimolar ratios resulting in a final concentration of the ternary complex of 17 μM, a concentration at which ternary complexes are formed.2g,6–8 The supramolecular amphiphiles self-assemble in aqueous solution into vesicular nanoparticles as was verified using high resolution scanning electron microscopy (hrSEM, Fig. 2a). Inspection of these nanocarriers using transmission electron microscopy (TEM) showed that the particles are hollow, suggesting that the nanocarriers are vesicles (Fig. 2b and S2†). Considering the amphiphilic nature of the building blocks of the nanocarriers, a bilayer structure is hypothesized for the shell of the vesicles in which two leaflets of supramolecular ternary amphiphiles form a bilayer with the hydrophilic oligo(ethylene glycol) chains facing the aqueous medium while shielding the hydrophobic alkyl tails and CB[8] that reside inside this bilayer. From the TEM images, the thickness of this bilayer was estimated to be 8 ± 3 nm and this value agrees well with the calculated width of such a bilayer (Fig. S2†). The average overall diameter of the nanocarriers was determined by hrSEM (128 ± 39 nm averaging 300 particles) and the average hydrodynamic diameter was determined by dynamic light scattering (DLS) (204 ± 38 nm, 3 separate measurements). We note that the samples for hrSEM where dried, which could account for the larger hydrodynamic diameters measured in solution using DLS when compared to those determined using hrSEM. In the case of all negative controls, in absence of CB[8] or using its smaller homologue CB[7], which is unable to complex both guests simultaneously, no particles were observed (Fig. 2c).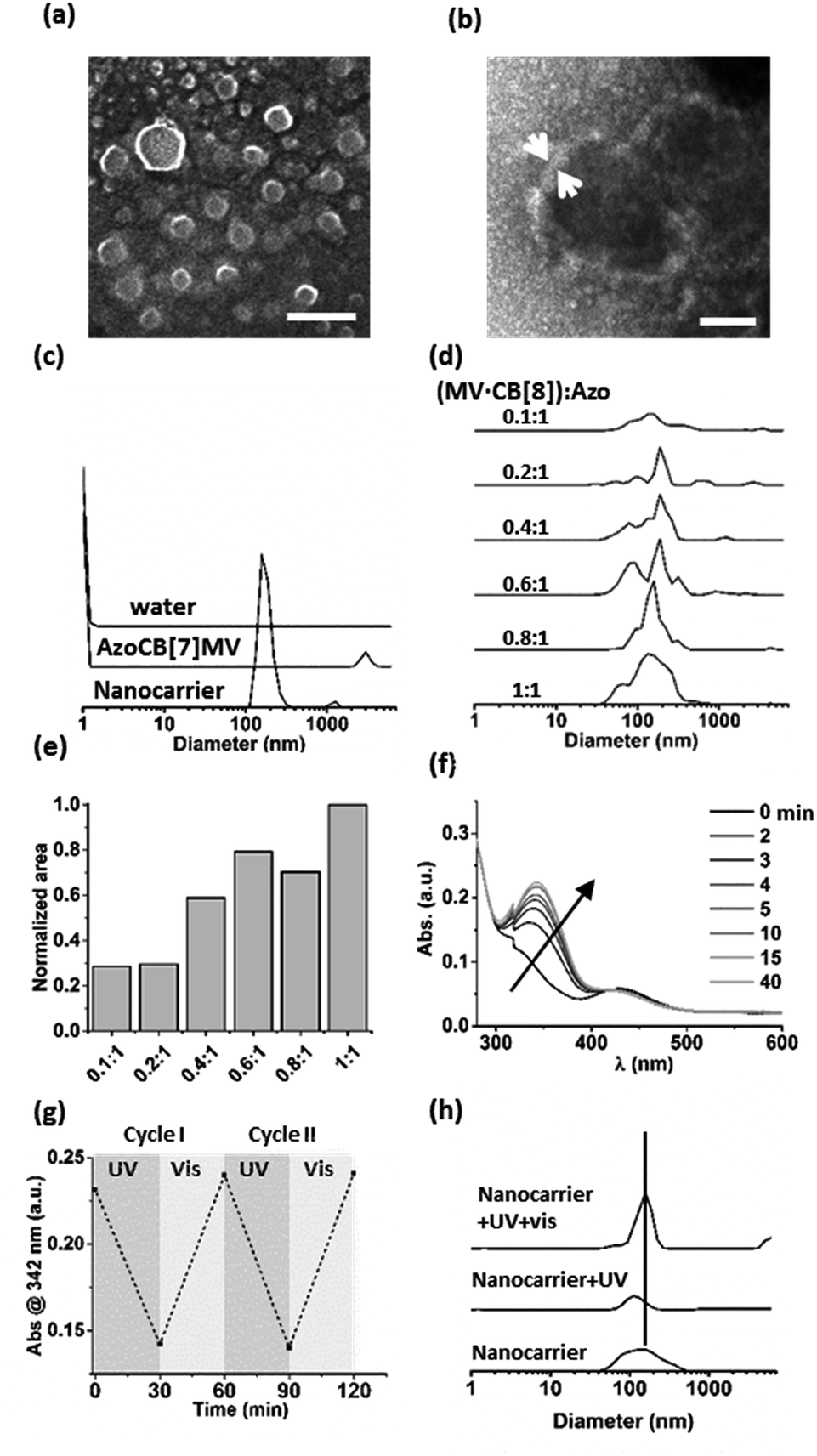 | ||
| Fig. 2 (a) Nanocarriers formed at equimolar ratios of MV, CB[8] and Azo (17 μM) imaged using hrSEM (scale bar 400 nm) and (b) TEM (negative staining with 1% uranyl acetate, scale bar 40 nm). (c) DLS size distributions of the hydrodynamic diameters of nanocarriers formed at equimolar ratios of MV, CB[8] and Azo (17 μM), or with CB[7] (AzoCB[7]MV). The data measured for water is given as a reference. (d) The formation of nanocarriers is monitored using DLS for concentrations of MV·CB[8] between 0 and 17 μM, while keeping the concentration of Azo constant at 17 μM. The mixing ratios between (MV·CB[8]) and Azo are given. (e) Areas under the size distribution curves obtained using DLS for each mixing ratio. (f) UV-vis absorption spectra of nanocarriers after UV irradiation and time-dependent cis–trans interconversion in ambient light (see legend). (g) Absorbance (at λ = 342 nm) of the trans-azobenzene monitored over two cycles of alternating exposure to UV and ambient light (30 min each). (h) DLS size determination of nanocarriers (SNPs) formed from a mixture of MV, CB[8] and Azo at equimolar ratios, after UV irradiation (1 h, 104 mW, with a shortpass filter <392 nm), and after subsequent irradiation with ambient light (15 min). | ||
Importantly, the formation of particles was found to be dependent on the ratio of the three components. DLS was used to monitor samples prepared by mixing a constant concentration of Azo (17 μM) with increasing concentrations of a preformed binary complex of CB[8] and MV ranging from 0 to 17 μM. The closer the concentration of the binary complex MV·CB[8] was to that of Azo, the higher the amount of particles formed (Fig. 2d and e). This indicates that particles were formed by the self-assembly of the amphiphilic hetero-ternary complex.
The light responsiveness of the Azo guest was investigated when included in CB[8] to form the supramolecular amphiphile that forms the nanocarriers. The kinetics of re-equilibration of the photostationary state of azobenzene upon UV irradiation (λ < 392 nm, 1 h, r.t.) and, subsequently, under ambient light were monitored using UV-vis spectroscopy (Fig. 2f and g). Upon UV irradiation the cis-isomer caused dissociation of the amphiphilic ternary complexes thereby triggering partial disappearance of the particles as was observed using DLS measurements. Only about 20% of the particles remained after UV irradiation. The exposure to ambient light for 15 min prompted the reassembly of the particles as can be concluded from the observed increase of the amount of nanocarriers, which indicates that the amphiphilic ternary complexes were formed after back-conversion of the azobenzene unit to the trans-isomer, which is able to form the hetero-ternary complex (Fig. 2h).
Protein loading
Next, loading of the nanocarriers with cargo was explored. As model cargo, fluorescent proteins were chosen for their ease of detection using fluorescence spectroscopy. To this end, a Förster resonance energy transfer (FRET) experiment was performed. Nanocarriers were prepared as described in the experimental section from solutions containing either a yellow fluorescent protein (YFP, donor), or a red fluorescent protein (TagRFP, acceptor) or both (all proteins at 18 μM as final concentration). The same solutions, prepared in the absence of the components that form the nanocarriers, were used as reference. In Fig. 3a the normalized spectra of pure donor and acceptor as well as their mixtures are shown, both in the presence and the absence of the nanocarriers. Although the proteins were used at semi-diluted (μM) concentrations and have short (ns) fluorescence lifetimes, considerable FRET occurred already in the protein mixture of YFP and TagRFP without nanocarriers, which was attributed to protein aggregation.9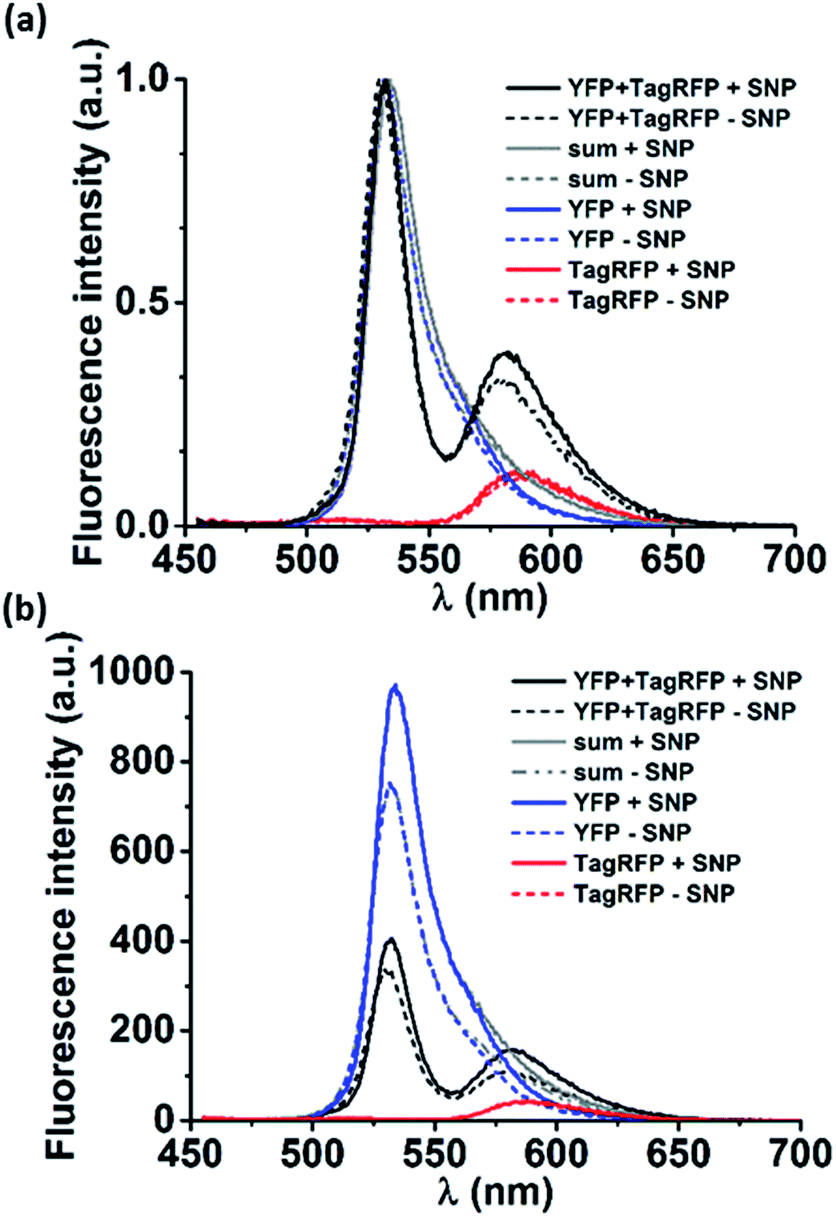 | ||
| Fig. 3 (a) Normalized and (b) non-normalized fluorescence emission spectra (λex = 440 nm) of: YFP/TagRFP mixtures (YFP + TagRFP), pure TagRFP or pure YFP. The sum of the normalized emission spectra of the single proteins is also given (sum). Each condition was measured for proteins (18 μM) mixed with MV, CB[8] and Azo (+supramolecular nanoparticle (SNP), solid lines) and only proteins (−SNP, dashed lines). | ||
To estimate the magnitude of FRET in solutions with nanocarriers, we quantified the change in FRET when particles were formed. The volume fraction actually encapsulated in particles was estimated to be 0.005% of the total volume (see calculation in the ESI†). The solution enclosed in the resulting nanocarriers will have the same concentration as the solution outside the particles as a result of the uniform protein distribution throughout the solution. This means that the fraction of enclosed proteins is equal to the enclosed volume fraction, i.e. 0.005%, which is neither expected to give rise to any observable FRET effect nor any change in FRET. However, we find marked changes in the spectra in solutions where the nanocarriers could form (Fig. 3). To analyze whether the effects can be attributed to FRET, first, the donor and acceptor components of the emission spectra of the mixtures (Fig. 3, YFP and TagRFP with and without nanocarriers, black lines) were deconvoluted using Lorentzian fits (Fig. S4†). Then, the contribution of each acceptor component was corrected by subtracting the emission that originates from direct excitation of the acceptor at the excitation wavelength of the donor (Fig. 3, TagRFP without nanocarrier, red lines). Finally, the intensity ratios (donor/acceptor emission) of the resulting deconvoluted and corrected spectra of the mixtures with and without particles were evaluated to be 0.62 and 0.34, respectively. The fact that the donor/acceptor intensity ratio doubles upon particle formation cannot be caused solely by the encapsulated fraction of 0.005%, even if assuming a 100% FRET efficiency between these donors and acceptors. This indicates that the interaction of the mixed proteins caused by the presence of nanocarriers is much larger than can be explained by the effects expected for purely statistical inclusion. The observed spectral changes can be explained by significant protein interaction (“loading”) of the nanocarriers, which causes an enhanced local protein concentration that in turn causes the observed FRET enhancement.10 However, there are other effects that could cause the FRET enhancement upon formation of the nanocarriers. As shown in Fig. 3b, the (non-normalized) emission spectra of the pure proteins changed in the presence of nanocarriers, in particular that of YFP. This indicates that the nanocarriers have an effect on the individual proteins, which could be due to several factors. Firstly, the refractive index of the surroundings of the proteins (in proximity of the supramolecular amphiphiles) is changed, which would in turn affect the lifetimes of the excited state of the fluorophores.11 Secondly, aggregates of proteins may rupture in the presence of the particles that would reduce self-quenching and increase their emission. Thirdly, a combination of both factors can also occur.
The effect of the nanocarriers on the emission of the pure proteins means that the donor and acceptor emission and their ratios could not be directly used to quantify the energy transfer. In order to compare the FRET in absence and presence of nanocarriers, the data were analyzed in more detail.
The emission spectra of the mixtures of proteins with and without particles were corrected for the effects of the particles on the pure proteins. To this end, the deconvoluted fitted Lorentzian curves to the TagRFP and YFP emission spectra of the mixed proteins (Fig. S4†) and the emission spectra of the pure proteins were integrated. The integrals of the raw spectra of the pure proteins and those of the Lorentzian fits for the proteins in the mixtures should be linearly correlated to the number of photons emitted per second (cps). These integrated emission intensities were hereafter used to quantify the FRET with and without supramolecular nanoparticles.
We determined a factor 1.32 for the increase of YFP emission in the presence of particles. Assuming that the effects of the particles on YFP are the same for the pure protein as for the protein in the mixture, we applied this factor to the YFP component of the emission of the mixture without particles (7130 cps). This yields an expected emission of YFP in the mixture with particles of 9410 (7130 × 1.32) cps. The emission measured for YFP in the mixture with particles is 7540 cps, thus 1870 cps less than the maximum possible (expected) value. This difference of 1870 cps represents the maximum possible increase in FRET that is only due to the increase of donor intensity, i.e. not a direct effect of the particles on the efficiency of the energy transfer.
In addition, the TagRFP acceptor emission was corrected for direct excitation (2300 cps with SNPs, 2140 cps without particles, Fig. 3 and S4† red lines). These values were subtracted from the respective emission values of the TagRFP component spectra, resulting in TagRFP emission solely due to FRET of 4700 cps with particles and 2450 cps without particles. The difference between 4700 and 2450 cps of 2250 cps represents the actual enhancement of FRET in the presence of particles. This value is higher than the maximum possible contribution from the increased donor emission in the presence of particles (1870 cps, see above). This indicates that the observed increase in FRET is indeed related to the presence of particles.
Clearly, the encapsulation of 0.005% (see ESI†) of the proteins in solution via statistical inclusion cannot lead to such a significant increase in FRET. The results of our calculations, therefore, indicate an interaction of proteins with the bilayer structure of the particles. If this interaction indeed exists and would lead to closely packed proteins, it would cause a strong energy transfer between proteins. Considering an average diameter of the β-barrel of the fluorescent proteins of 2.4 nm and a Förster radius of 5.7 nm for YFP with TagRFP (see ESI†), the FRET efficiencies could theoretically become close to 100% if proteins were densely packed. Moreover, the close proximity of the proteins on the surface of the supramolecular particles would cause a change of the refractive index of the medium surrounding the proteins, which in turn would cause changes in lifetimes and consequently quantum yields.11 This in turn could explain the increase in emission intensity of the pure proteins in the presence of the nanocarriers. Taken together, all observed spectral changes can be explained by assuming protein binding to the membranes of the nanocarriers.
Cellular targeting and delivery of TFP
After having verified successful loading of our nanocarriers, we set out to introduce cellular targeting groups on such protein-loaded nanocarriers. For this purpose an Arg-Gly-Asp (RGD) motif was grafted onto an azobenzene guest moiety (AzoRGD, Fig. 1). RGD peptides bind to integrin receptors on the cell membrane.12 Integrin receptors are overexpressed in tissues where angiogenesis is stimulated as is common in tumor tissue, making RGD ligands a promising tool in cancer therapies and imaging.13 Supramolecular particles were prepared by mixing teal fluorescent protein (TFP, 0.5 μM) with the components of our photo-switchable hetero ternary complex: MV, CB[8] and Azo, resulting in particles loaded with TFP. The targeting moiety RGD was introduced by replacing 1% of Azo with AzoRGD during initial mixing (Fig. 1 and Experimental section). Myoblast C2C12 cells were seeded into standard tissue culture plates and allowed to adhere for 2 h. Cells were then stained with a nuclear dye and incubated for 10 min with the TFP-loaded SNPs. After washing, the cells were directly imaged using a fluorescence microscope (Fig. 4a). In the absence of the nanocarriers, i.e. omitting the supramolecular building blocks (MV, CB[8] and Azo), the green fluorescence intensity was significantly reduced, independent of the presence of targeting ligand AzoRGD (Fig. 4b). More notably, when the targeting ligand AzoRGD was missing on the nanocarriers nearly no TFP emission from the cells was observed (Fig. 4b). These controls confirmed the specific roles of the particles and the RGD ligands (Fig. 4b). The TFP green fluorescence intensity of the cells after washing was quantified as averages of triplicate experiments (75 ≤ n ≤ 220 cells), and the results are reported in Fig. 4c. The specificity of the delivery of TFP by the nanocarriers bearing RGD was confirmed by the observed trend in TFP emission recorded when lowering the percentage of AzoRGD in the particles. When the percentage of AzoRGD in the nanocarriers was lowered to 0.05%, the green emission inside the cells was significantly less intense when compared to cells treated with nanocarriers bearing 1% AzoRGD. The emission intensity when using nanocarriers with 0.05% AzoRGD was still significantly larger than that of the negative controls (Fig. S7†). Interestingly, the overlap of the bright field micrographs of the cells showed that the TFP emission is localized in the proximity of the nuclei, rather than dispersed over the whole cell membrane or in the cytosol (Fig. 4a). Multivalent RGD-functionalized particles are known to induce the active endocytosis of the RGD–integrin complex, and with it, the particles, resulting in the observed accumulation in a perinuclear compartment14 as it has previously been employed for imaging and drug delivery.13b After careful consideration of the results from FRET experiments, we propose that a significant fraction of the fluorescent protein cargo, resides in close proximity to, or contact with, the membranes of the nanocarriers (enhanced fluorescence emission). Then, fusion to a targeted cell would lead to initial endocytosis and later localization of the cargo with nanocarrier close to the nucleus, rather than a classic release, precluding standard studies of release kinetics. Potential use of photoswitching to enable release of cargo is challenging as the current wavelengths for azobenzene isomerization are not necessarily biocompatible. Overall, the results presented here confirm the specific interaction between AzoRGD-functionalized and TFP-loaded nanocarriers with the cells. This in turn confirms the interaction between TFP and the nanocarriers as discussed above.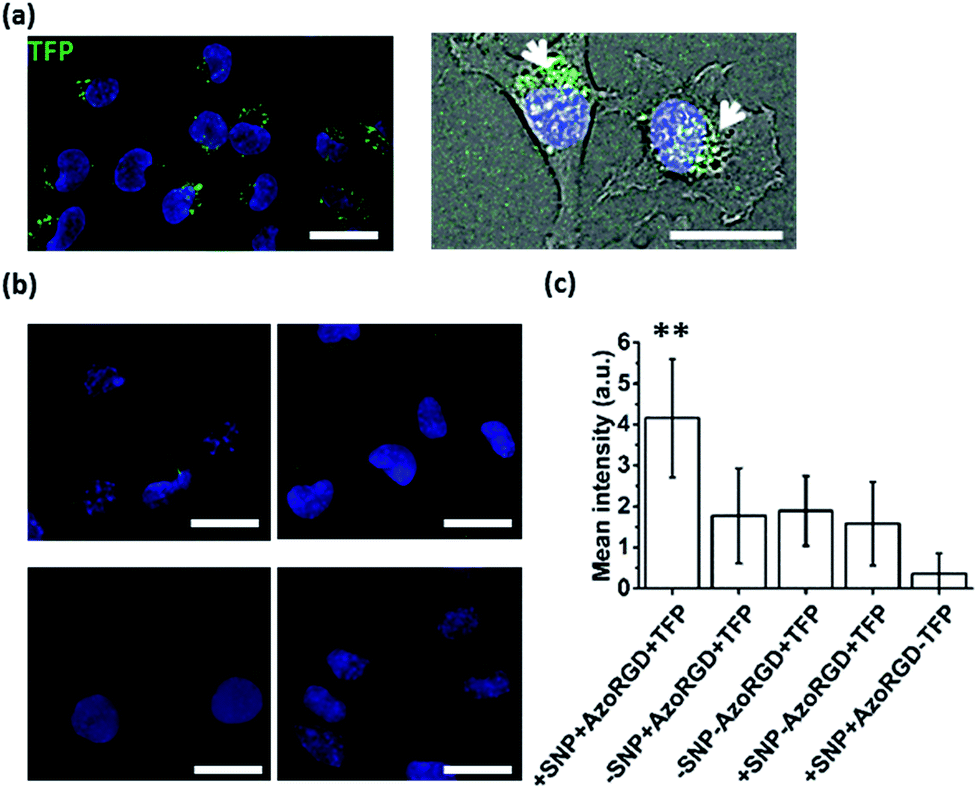 | ||
| Fig. 4 (a) Fluorescence image (left, (nuclear stain, blue)) overlaid with bright field micrograph (right) of cells treated with nanocarriers loaded with TFP (green) and functionalized with AzoRGD. (b) Fluorescence images (DAPI, blue) of cells treated with AzoRGD and TFP, without nanocarriers (top-left), TFP only (top-right), TFP-loaded nanocarriers, without AzoRGD (bottom-left) and nanocarriers with AzoRGD, without TFP (bottom-right). Scale bars 50 μm. (c) Average green fluorescence intensity (background corrected, 75 ≤ n ≤ 220 cells) for all conditions plotted. Three independent experiments in duplicate. Error bars represent standard deviations of the distributions and ** indicates a significant difference with a 95% level of confidence. | ||
Conclusions
We achieved protein-loaded supramolecular nanoparticles for targeted protein delivery into the cell interior. We employed CB[8] hosts, mixed with equimolar amounts of alkylated methyl viologen and oligo(ethylene glycol)-derivatized azobenzene guests to form supramolecular amphiphiles that self-assemble in aqueous media into vesicles of ca. 200 nm in diameter. By loading the particles with fluorescent proteins and by incorporating targeting ligands into the nanoparticles, uptake of the loaded particles was observed using fluorescence microscopy. The strategy to make the supramolecular particles offers a versatile, modular approach for introducing targeting and imaging functionalities that are appealing to be further explored for future biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Shrikrishnan Sankaran and Mark de Ruiter are acknowledged for preparation of the fluorescent proteins and Mark Smithers and Rico Keim for the hrSEM and TEM experiments. This study was supported by a Starting ERC grant (Sumoman 259183).Notes and references
- T. Aida, E. W. Meijer and S. I. Stupp, Science, 2012, 335, 813–817 CrossRef CAS.
- (a) C. E. Ashley, E. C. Carnes, G. K. Phillips, D. Padilla, P. N. Durfee, P. A. Brown, T. N. Hanna, J. Liu, B. Phillips, M. B. Carter, N. J. Carroll, X. Jiang, D. R. Dunphy, C. L. Willman, D. N. Petsev, D. G. Evans, A. N. Parikh, B. Chackerian, W. Wharton, D. S. Peabody and C. J. Brinker, Nat. Mater., 2011, 10, 389–397 CrossRef CAS; (b) F. Tantakitti, J. Boekhoven, X. Wang, R. V. Kazantsev, T. Yu, J. Li, E. Zhuang, R. Zandi, J. H. Ortony, C. J. Newcomb, L. C. Palmer, G. S. Shekhawat, M. O. de la Cruz, G. C. Schatz and S. I. Stupp, Nat. Mater., 2016, 15, 469–476 CrossRef CAS; (c) J. Voskuhl and B. J. Ravoo, Chem. Soc. Rev., 2009, 38, 495–505 RSC; (d) K. M. Park, D. W. Lee, B. Sarkar, H. Jung, J. Kim, Y. H. Ko, K. E. Lee, H. Jeon and K. Kim, Small, 2010, 6, 1430–1441 CrossRef CAS; (e) X. Mei, S. Yang, D. Chen, N. Li, H. Li, Q. Xu, J. Ge and J. Lu, Chem. Commun., 2012, 48, 10010–10012 RSC; (f) J. Li, Z. Yu, H. Jiang, G. Zou and Q. Zhang, Mater. Chem. Phys., 2012, 136, 219–224 CrossRef CAS; (g) C. Stoffelen, J. Voskuhl, P. Jonkheijm and J. Huskens, Angew. Chem., Int. Ed., 2014, 53, 3400–3404 CrossRef CAS; (h) X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS; (i) J. Moratz, A. Samanta, J. Voskuhl, S. K. M. Nalluri and B. J. Ravoo, Chem.–Eur. J., 2015, 21, 3271–3277 CrossRef CAS; (j) J. Brinkmann, E. Cavatorta, S. Sankaran, B. Schmidt, J. van Weerd and P. Jonkheijm, Chem. Soc. Rev., 2014, 43, 4449–4469 RSC; (k) S. K. M. Nalluri, J. Voskuhl, J. B. Bultema, E. J. Boekema and B. J. Ravoo, Angew. Chem., Int. Ed., 2011, 50, 9747–9751 CrossRef CAS; (l) J. Cabanas-Danés, E. Dooms Rodrigues, E. Landman, J. van Weerd, C. A. van Blitterswijk, T. Verrips, J. Huskens, M. Karperien and P. Jonkheijm, J. Am. Chem. Soc., 2014, 136, 12675–12681 CrossRef.
- (a) E. Masson, X. Ling, R. Joseph, L. Kyeremeh-Mensah and X. Lu, RSC Adv., 2012, 2, 1213–1247 RSC; (b) L. Isaacs, Acc. Chem. Res., 2014, 47, 2052–2062 CrossRef CAS; (c) S. J. Barrow, S. Kasera, M. J. Rowland, J. del Bario and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS.
- (a) Q. An, J. Brinkmann, J. Huskens, S. Krabbenborg, J. de Boer and P. Jonkheijm, Angew. Chem., Int. Ed., 2012, 51, 12233–12237 CrossRef CAS; (b) S. Sankaran, M. C. Kiren and P. Jonkheijm, ACS Nano, 2015, 4, 3579–3586 CrossRef; (c) E. Cavatorta, M. L. Verheijden, W. van Roosmalen, J. Voskuhl, J. Huskens and P. Jonkheijm, Chem. Commun., 2016, 52, 7146–7149 RSC; (d) K. M. Park, J. A. Yang, H. Jung, J. Yeom, J. S. Park, K. H. Park, A. S. Hoffman, S. K. Hahn and K. Kim, ACS Nano, 2012, 6, 2960–2968 CrossRef CAS; (e) C. Kim, S. S. Agasti, Z. Zhu, L. Isaacs and V. M. Rotello, Nat. Chem., 2010, 2, 962–966 CrossRef CAS.
- (a) Y. J. Jeon, P. K. Bharadwaj, S. Choi, J. W. Lee and K. Kim, Angew. Chem., Int. Ed., 2002, 41, 4474–4476 CrossRef CAS; (b) D. Jiao, J. Geng, X. J. Loh, D. Das, T. C. Lee and O. A. Scherman, Angew. Chem., Int. Ed., 2012, 51, 9633–9637 CrossRef CAS; (c) X. J. Loh, M. H. Tsai, J. D. Barrio, E. A. Appel, T. C. Lee and O. A. Scherman, Polym. Chem., 2012, 3, 3180–3188 RSC; (d) X. J. Loh, J. Del Barrio, P. P. C. Toh, T. C. Lee, D. Jiao, U. Rauwald, E. A. Appel and O. A. Scherman, Biomacromolecules, 2012, 13, 84–91 CrossRef CAS; (e) X. J. Loh, J. Del Barrio, T. C. Lee and O. A. Scherman, Chem. Commun., 2014, 50, 3033–3035 RSC.
- J. H. Mondal, S. Ahmed, T. Ghosh and D. Das, Soft Matter, 2015, 11, 4912–4920 RSC.
- N. Ma, W. J. Wang, S. Chen, X. S. Wang, X. Q. Wang, S. B. Wang, J. Y. Zhu, S. X. Cheng and X. Z. Zhang, Chem. Commun., 2015, 51, 12970–12973 RSC.
- (a) N. L. Weineisen, C. A. Hommersom, J. Voskuhl, S. Sankaran, A. M. A. Depauw, N. Katsonis, P. Jonkheijm and J. J. L. M. Cornelissen, Chem. Commun., 2017, 53, 1896–1899 RSC; (b) S. Sankaran, J. van Weerd, J. Voskuhl, M. Karperien and P. Jonkheijm, Small, 2015, 11, 6187–6196 CrossRef CAS; (c) F. Tian, D. Jiao, F. Biedermann and O. A. Scherman, Nat. Commun., 2012, 3, 1207 CrossRef; (d) Z. Yu, Y. Lan, R. M. Parker, W. Zhang, X. Deng, O. A. Scherman and C. Abell, Polym. Chem., 2016, 7, 5996–6002 RSC.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer Science+Business Media, LLC, New York, USA, 3rd edn, 2006 Search PubMed.
- E. S. Douglas, R. A. Chandra, C. R. Bertozzi, R. A. Mathies and M. B. Francis, Lab Chip, 2007, 7, 1442–1448 RSC.
- S. J. Strickler and R. A. Berg, J. Chem. Phys., 1962, 37, 814–822 CrossRef CAS.
- N. de Franceschi, H. Hamidi, J. Alanko, P. Sahgal and J. Ivaska, J. Cell Sci., 2015, 128, 839–852 CrossRef CAS.
- (a) R. Pasqualini, E. Koivunen and E. Ruoslahti, Nat. Biotechnol., 1997, 15, 542–546 CrossRef CAS; (b) E. Garanger, D. Boturyn, Z. Jin, P. Dumy, M. C. Favrot and J. L. Coll, Mol. Ther., 2005, 12, 1168–1175 CrossRef CAS; (c) F. Danhier, A. L. Breton and V. Préat, Mol. Pharm., 2012, 9, 2961–2973 CrossRef CAS.
- E. J. Ezratty, C. Bertaux, E. E. Marcantonio and G. G. Gundersen, J. Cell Biol., 2009, 187, 733–747 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures; nanoparticles preparation and characterization; calculations of the encapsulated protein fraction; details of cells experiments. See DOI: 10.1039/c7ra10980f |
| This journal is © The Royal Society of Chemistry 2017 |