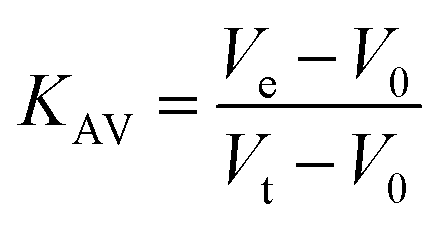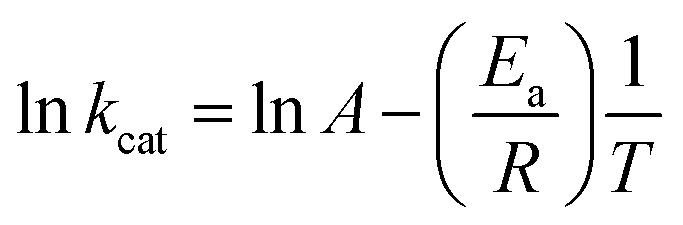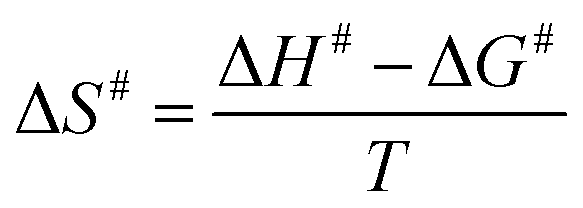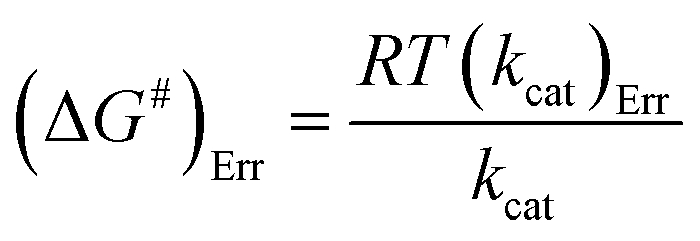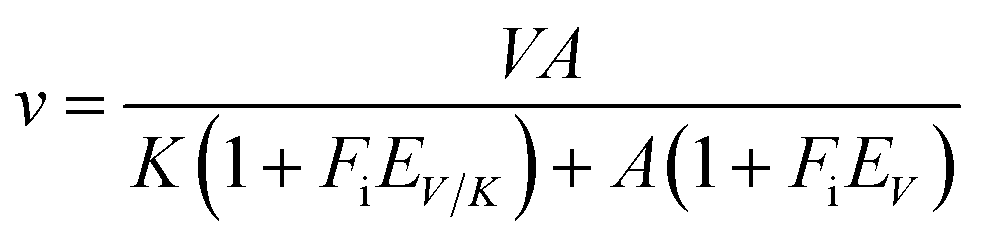DOI:
10.1039/C7RA10526F
(Paper)
RSC Adv., 2017,
7, 54347-54360
Biochemical, thermodynamic and structural studies of recombinant homotetrameric adenylosuccinate lyase from Leishmania braziliensis†
Received
22nd September 2017
, Accepted 22nd November 2017
First published on 27th November 2017
Abstract
Adenylosuccinate lyase (ASL) is involved in both de novo and salvage pathways of purine biosynthesis. ASL belongs to the argininosuccinate lyase/fumarase C superfamily of enzymes which share a general acid–base catalytic mechanism with β-elimination of fumarate as the common product. Cloning, expression, and a method to obtain homogeneous recombinant ASL from Leishmania braziliensis (LbASL) are described. Mass spectrometry analysis of recombinant LbASL, oligomeric state determination and multiple sequence alignment are presented. Steady-state kinetics of LbASL showed a Michaelis–Menten pattern. Isothermal titration calorimetry binding assays suggested that LbASL follows a Uni-Bi ordered kinetic mechanism, in which release of fumarate is followed by AMP to yield free enzyme. Initial velocity data for the reverse reaction and the Haldane relationship allowed calculation of an unfavorable equilibrium constant for the LbASL-catalyzed chemical reaction. The activation energy and thermodynamic activation parameters were estimated. Solvent kinetic isotope effects V/K and V suggest a modest contribution of solvent proton transference during the rate-limiting step of the reaction. Proton inventory data show that the modest normal effect on V arises from a single protonic site, and the transition state fractionation factor value of 0.74 suggests participation of solvent proton transfer in transition-state vibrations perpendicular to the reaction coordinate. pH-rate profiles for kcat and kcat/KM suggested amino acid residues involved in, respectively, catalysis and substrate binding. A model of LbASL was built to provide a structural basis for the experimental data. A better understanding of the mode of action of LbASL is useful for the rational design of antileishmaniasis agents.
Introduction
Leishmaniasis is regarded as one of the most burdensome of the neglected tropical diseases.1 The disease is endemic in 98 countries and three continents, and it is estimated that 350 million people are at risk.2 Approximately 0.2 to 0.4 million cases of visceral leishmaniasis (VL) and 0.7 to 1.2 million cases of cutaneous leishmaniasis (CL) occur each year. CL is more widely distributed, with about one-third of cases occurring in the Americas, the Mediterranean basin, and Western and Central Asia.3 In Brazil, American tegumentary leishmaniasis (ATL) is predominantly caused by Leishmania (Viannia) braziliensis,4 which is responsible for four distinct forms of ATL: localized CL, mucosal leishmaniasis (ML), disseminated leishmaniasis (DL) and diffuse CL (DCL).5,6 Like other Leishmania species, L. braziliensis is a digenetic protozoan parasite that is a flagellated, extracellular promastigote in the phlebotomine sandfly vector, while it is an immotile, intracellular amastigote within phagolysosomes of macrophages of the infected mammalian host.4 The main drug treatments of leishmaniasis include pentavalent antimonials, like sodium stibogluconate (Pentostam) and meglumine antimoniate (Glucantime) (Croft et al. 2011, Croft et al. 2003; McGwire 2014). However, these antimonials have multiple toxicities and are increasingly ineffective due to the development of parasite resistance.7–9 Although second-line drugs, such as amphotericin-B either as deoxycholate or liposomal form, paromomycin and miltefosine show fewer side effects;7,10 these therapies are very expensive and are far from ideal.11 There is thus an urgent need for new treatments to combat this disease.
The development of new effective antiparasitic drugs can be based on exploring the biochemical and physiological differences between the pathogen and its host. One of these metabolic differences lies in the biosynthesis of purine nucleotides.12,13 While mammal cells hold the capacity to synthetize purine nucleotides by the de novo and salvage pathways, Leishmania species are completely dependent on the salvage pathway to supply their purine requirements.14,15 The enzyme adenylosuccinate lyase (ASL; EC 4.3.2.2) belongs to the aspartase/fumarase protein superfamily, all members of which are homotetramers with approximately 200 kDa that share a high level of structural similarity.16–19 ASL is the only enzyme in the purine nucleotide metabolism that catalyzes two distinct reactions, both involving β-elimination of fumarate: (1) conversion of 5-aminoimidazol-4(N-succinylcarboxamide) ribonucleotide (SAICAR) into 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR) and fumarate, and (2) conversion of succinyl-adenosine monophosphate (S-AMP) into AMP and fumarate. The latter reaction is part of the two-reaction pathway that converts inosine monophosphate (IMP) into AMP.16 ASL is the last enzyme in the conversion of IMP to AMP in Leishmania, representing therefore a critical bottleneck in purine salvage (Boitz et al. 2013). Previous studies showed that an L. donovani parasite containing the ASL gene knocked-out exhibited a severely reduced parasite burden in both macrophages and mice, which could be explained by the toxic accumulation of adenylosuccinate.13 These results indicate that ASL could be a promising drug target for anti-leishmaniasis drug development.
Here, we describe cloning, expression and purification to homogeneity of recombinant L. braziliensis ASL (LbASL). Determination of the true steady-state kinetic parameters, thermodynamic constants of substrate and products interaction, pre-steady-state kinetics, energy of activation, solvent kinetic isotope effect (SKIE) and proton inventory studies are also presented. A three-dimensional model has been built to provide a structural basis for interpretation of experimental results. These results contribute to a better understanding of the mode of action of LbASL, which should inform the rational design of chemotherapeutic agents to treat leishmaniasis.
Experimental
Materials
All chemicals were of analytical or reagent grade and were used without further purification, unless stated otherwise. Buffers, S-AMP, AMP, fumarate, ammonium sulfate, streptomycin sulphate, sodium chloride, potassium chloride, EDTA, DTT, glycerol, D2O and lysozyme were purchased from Sigma Aldrich® (Saint Louis, USA). Bacterial growth media and antibiotics were from Gibco. Liquid chromatography columns, low molecular weight (LMW) and high molecular weight (HMW) Gel Filtration Calibration Kits were purchased from GE Healthcare® Life Sciences (Pittsburg, USA). All kinetic data analyses were carried out using SigmaPlot 10.0 (Systac Software, Inc., Melbourne, USA). Data are presented as mean ± standard deviation unless stated otherwise. ITC data analysis was evaluated utilizing the Origin 7 SR4 software (Microcal, Inc.). All enzyme activity measurements were carried out at 25 °C in at least duplicates.
Cloning and recombinant protein expression
The LbASL coding gene LbrM.04.0500 (GenBank ID: 5412684) containing NdeI and HindIII restriction sites on, respectively, the 5′ and 3′ ends was synthetized with signal peptide removed by Biomatik® and ligated into the pET23a(+) expression vector pET23a(+):LbrM.04.0500, previously digested with the same restriction enzymes. The construction of pET23a(+):LbrM.04.0500 was submitted to automatic DNA sequencing to confirm identity, integrity and absence of mutations in the cloned gene.
The recombinant plasmid pET23a(+):LbrM.04.0500 was transformed into E. coli BL21(DE3) cells and plated on lysogeny broth (LB) agar containing 50 μg mL−1 ampicillin. A single colony was inoculated into LB medium (50 mL) containing 50 μg mL−1 ampicillin and grown at 37 °C, 180 rpm, overnight. The culture (8.5 mL) was inoculated in LB medium (500 mL) with the same antibiotic concentration and grown in a shaker-incubator at 37 °C, 180 rpm. When the optical density at 600 nm (OD600) reached 0.4–0.6, as suggested by New England BioLabs Inc20 and previous reports,21,22 the cells were induced with 1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) and harvested at 3 h, 6 h, 9 h, 12 h and 24 h after induction. Cells were harvested by centrifugation at 8000 × g|g for 30 min at 4 °C and stored at −20 °C. Frozen cell paste was disrupted by sonication and soluble and insoluble fractions were analyzed by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE).
Protein purification
Protein purification was performed by FPLC using an ÄKTA System (GE Healthcare® Life Sciences, Pittsburg, USA) at 4 °C. Approximately 1.8 g of frozen cells were suspended in 9 mL of 50 mM Tris HCl pH 7.5 (buffer A), and incubated with 0.2 mg mL−1 lysozyme (Sigma-Aldrich) with stirring for 30 min at 4 °C. Cells were disrupted in a VCX 750 ultrasonic processor (Sonics & Materials Inc., Newtown, USA) by sonication (10 pulses of 10 s each at 60% amplitude) and centrifuged at 48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g|g for 30 min at 4 °C. The supernatant was treated with 1% (v/v) streptomycin sulfate for 30 min with slow stirring to precipitate nucleic acids and centrifuged at 48000 × g|g for 30 min at 4 °C. The resulting supernatant was treated with 1.5 mM ammonium sulfate with stirring for 30 min. The fraction containing precipitated molecules was suspended with 8 mL of buffer A and loaded on a HiLoad Superdex 200 26/60 size exclusion column (GE Healthcare® Life Sciences, Pittsburg, USA), previously equilibrated with buffer A. Proteins were isocratically eluted with 1 column volume (CV) of buffer A at flow rate of 0.5 mL min−1, and fractions containing the target protein were pooled and loaded on a HiLoad Q Sepharose High Performance 16/10 anion exchange column (GE Healthcare® Life Sciences, Pittsburg, USA), pre-equilibrated with buffer A. The column was washed with 7 CVs of buffer A, and the adsorbed proteins were eluted with a linear gradient (0–60%) of 25 CV of buffer A containing 1 M NaCl (buffer B) at flow rate of 1 mL min−1. The fractions containing homogeneous LbASL were pooled and dialyzed against 50 mM potassium phosphate buffer pH 7.0, containing 150 mM KCl, 1 mM EDTA, 1 mM DTT and 10% glycerol (storage buffer), and stored at −20 °C. Protein concentration was determined by the method of BCA using a bovine serum albumin as standard (BCA protein Assay Kit, Thermo Scientific Pierce).
000 × g|g for 30 min at 4 °C. The supernatant was treated with 1% (v/v) streptomycin sulfate for 30 min with slow stirring to precipitate nucleic acids and centrifuged at 48000 × g|g for 30 min at 4 °C. The resulting supernatant was treated with 1.5 mM ammonium sulfate with stirring for 30 min. The fraction containing precipitated molecules was suspended with 8 mL of buffer A and loaded on a HiLoad Superdex 200 26/60 size exclusion column (GE Healthcare® Life Sciences, Pittsburg, USA), previously equilibrated with buffer A. Proteins were isocratically eluted with 1 column volume (CV) of buffer A at flow rate of 0.5 mL min−1, and fractions containing the target protein were pooled and loaded on a HiLoad Q Sepharose High Performance 16/10 anion exchange column (GE Healthcare® Life Sciences, Pittsburg, USA), pre-equilibrated with buffer A. The column was washed with 7 CVs of buffer A, and the adsorbed proteins were eluted with a linear gradient (0–60%) of 25 CV of buffer A containing 1 M NaCl (buffer B) at flow rate of 1 mL min−1. The fractions containing homogeneous LbASL were pooled and dialyzed against 50 mM potassium phosphate buffer pH 7.0, containing 150 mM KCl, 1 mM EDTA, 1 mM DTT and 10% glycerol (storage buffer), and stored at −20 °C. Protein concentration was determined by the method of BCA using a bovine serum albumin as standard (BCA protein Assay Kit, Thermo Scientific Pierce).
LbASL identification by mass spectrometry
The homogeneous protein was submitted to shotgun proteomics to confirm the enzyme's identity. In-gel digestion was performed according to Shevchenko et al.23 Tryptic digest of LbASL was separated on a homemade 20 cm reverse-phase column (5 μm ODSAQ C18, Yamamura Chemical Lab, Japan) using a nanoUPLC (nanoLC Ultra 1D plus, Eksigent, USA) and eluted directly to a nanospray ion source connected to a hybrid mass spectrometer (LTQ Orbitrap Discovery, Thermo, USA). The flow rate was set to 300 mL min−1 in 120 min reverse-phase gradient. The mass spectrometer was operated in a data-dependent mode, with full MS1 scan collected in the Orbitrap, with m/z range of 400–1600 at 30000 resolution. The eight most abundant ions per scan were selected to CID MS2 in the ion trap. Mass spectra were analyzed using PatternLab platform.24 MS2 spectra were searched with COMET25 using a non-redundant database containing forward and reverse E. coli DH10B reference proteome and the sequence of LbASL. The validity of the peptide-spectra matches (PSMs) generated by COMET was assessed using PatternLab's module SEPro with a false discovery rate of 1% based on the number of decoys.
Oligomeric state determination
An estimate for the molecular mass of LbASL in solution was obtained by injecting 100 μL of protein suspension (7 μM homogeneous LbASL in 50 mM Tris HCl pH 7.5 containing 200 mM NaCl) into a HighLoad 10/30 Superdex-200 column (GE Healthcare), and isocratically eluted with 1 CV of 50 mM Tris HCl pH 7.5 containing 200 mM of NaCl at 0.4 mL min−1.
Protein elution was monitored at 215, 254 and 280 nm. The low molecular weight (LMW) and high molecular weight (HMW) Gel Filtration Calibration Kits (GE Healthcare) were used to prepare a calibration curve, measuring the elution volumes (Ve) of several standards (ferritin, aldolase, ovalbumin, conalbumin, ribonuclease and carbonic anhydrase A). These values were used to calculate their partition coefficient (Kav, eqn (1)). Blue dextran 2000 (GE Healthcare) was used to determine the void volume (V0). Vt is the total bead volume of the column. The Kav value for each protein was plotted against their correspondent molecular mass to obtain an estimate for LbASL molecular mass in solution.
| |
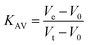 | (1) |
Multiple sequence alignment and homology modeling
Multiple alignment was carried out to compare amino acid sequences of homologous ASL proteins whose residues in the active site were determined by mutagenesis studies or for which three-dimensional structures were solved. The following proteins were included in the alignment: Leishmania braziliensis (LbASL, XP_001561734), Leishmania donovani (LdASL, XP_003858107), Escherichia coli (EcASL, WP_000423742), Plasmodium falciparum (PfASL, XP_001349577), Bacillus subtilis (BsASL, WP_003233955), Homo sapiens (HsASL, NP_000017), and Mycobacterium tuberculosis (MtASL, WP_003898583). The alignment was performed by ClustaW26 using the Blosum62 matrix.
The homology modelling approach, implemented in the MODELLER27 9v19 program, was used to build a 3D model of LbASL. The structure of ASL from E. coli (PDB ID: 2PTQ) associated with the products AMP and fumarate was used as template. This template was selected because the presence of products in the enzyme's active site provides structural information on which to base interpretation of the pH-rate profile data. The protocol used to perform the molecular modelling experiments generated 10 models. All models were submitted to the DOPE energy scoring function28 implemented in the MODELLER 9v19 aiming to select the best structures. The MOLPROBITY webserver29 and PROCHECK30 were used to verify and validate the stereochemical quality of the models.
Steady-state kinetic parameters of LbASL
Recombinant LbASL enzyme activity was monitored by a continuous assay in a UV-2550 UV/visible spectrophotometer (Shimadzu) equipped with a temperature-controlled cuvette holder, using 1.0 cm path length quartz cuvettes. The kinetic data were determined using the difference in absorption between S-AMP and AMP and fumarate, measuring the decrease in absorbance at 282 nm using a difference extinction coefficient value of 10000 M−1 cm−1. One unit of enzyme activity (U) was defined as the amount of enzyme catalyzing the conversion of 1 μmol of substrate into products per second at 25 °C. The enzyme was pre-incubated for 30 min at 25 °C in storage buffer. All enzyme activity assays were performed at 25 °C in 50 mM Tris HCl pH 7.5 containing 200 mM NaCl and 5 mM EDTA (buffer C) (standard conditions), in a total volume of 0.5 mL and reaction course data collected for 60 s. Only for the Superdex 200 eluate step enzyme activity was monitored for 5 min. For the purification steps, S-AMP substrate concentration was fixed at 100 μM (saturation), whereas initial velocity studies employed variable concentrations as described in the next paragraph.
The initial velocity study was carried out to determine the steady-state kinetic parameters for S-AMP conversion into AMP and fumarate (forward reaction). The saturation curve was performed at varying concentrations of S-AMP (5–200 μM) and the reaction was initiated by the addition of 30 nM of recombinant LbASL. Hyperbolic saturation curves were analyzed by non-linear regression of data fitting to the Michaelis–Menten equation (eqn (2)), in which v is the steady-state velocity, V is the maximal velocity, A is the substrate concentration, and KM is the Michaelis–Menten constant.
| |
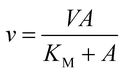 | (2) |
The kcat values were calculated from eqn (3), in which [E]t corresponds to the total concentration of enzyme subunits.
| |
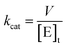 | (3) |
The initial velocities for the reverse reaction were determined varying the concentration of AMP (10–800 μM) at varied-fixed fumarate concentration (100–900 μM). All reactions started with addition of recombinant LbASL, assayed under standards conditions, and all measurements were performed at least in duplicates. Data from initial velocity measurements showing a pattern of lines intersecting to the left of the y-axis in the double-reciprocal plots (or Lineweaver–Burk plots) were fitted to eqn (4), which describes a sequential substrate binding and ternary complex formation (reverse reaction).
| |
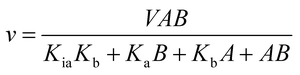 | (4) |
For eqn (4), v is the initial velocity, V is the true maximal initial velocity, A and B are the concentrations of the substrates (AMP and fumarate) for the reverse reaction, Ka(Kq) and Kb(Kp) are their respective Michaelis–Menten constants, and Kia(Kiq) is the dissociation constant for enzyme–substrate A binary complex formation (enzyme–AMP binary complex formation for the reverse reaction).
The initial velocities for the reverse reaction were employed to calculate the equilibrium constant (Keq) using the Haldane equation for an ordered Uni-Bi (or Bi-Uni) mechanism (eqn (5)). Vf is the maximal initial velocity for the forward and Vr for the reverse reaction, Kp represents the Michaelis–Menten constant for the first product to be released from the ternary complex (fumarate), Ka represents the Michaelis–Menten constant for S-AMP (KM of eqn (2)), and Kiq represents the dissociation constant for enzyme–AMP binary complex formation for the reverse reaction.31
| |
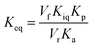 | (5) |
Isothermal titration calorimetry (ITC)
ITC experiments were carried out using an iTC200 Microcalorimeter (Microcal, Inc., Northampton, MA). The reference cell (200 μL) was loaded with water during all the experiments and the sample cell (200 μL) was filled with recombinant LbASL at a concentration of 72 μM in buffer C. The injection syringe (39.7 μL) was filled with either AMP (2 mM) or fumarate (2 mM) in the same buffer, and the ligand binding isotherms were measured by direct titration (ligand into macromolecule). The stirring speed was 500 rpm at 25 °C and constant pressure. Titration first injection (0.5 μL) was not used in data analysis and was followed by 19 injection of 2 μL each at 300 s intervals. Control titrations (ligand into buffer) were performed in order to subtract the heats of dilution prior to data analysis. The Gibbs free energy (ΔG) of binding was calculated using the relationship described in eqn (6), in which R is the gas constant (1.987 cal K−1 mol−1), T is the temperature in Kelvin (T = °C + 273.15), and Ka is the association constant at equilibrium. The entropy of binding (ΔS) can also be determined by this mathematical formula. ΔH represents the enthalpy of binding. The dissociation constant at equilibrium, Kd, was calculated as the inverse of Ka (eqn (7)). All data were evaluated utilizing the Origin 7 SR4 software (Microcal, Inc.)| | |
ΔG0 = −RTlnKa = ΔH0 − TΔS0
| (6) |
| |
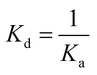 | (7) |
Energy of activation
To determine the energy of activation (Ea) of LbASL for the forward reaction, the dependence of kcat on temperature was measured. Initial velocities were measured in the presence of saturating concentrations of S-AMP (100 μM) at temperatures varying from 15 to 40 °C (from 288.15 to 313.15 K). Prior to data collection, LbASL was incubated for several minutes at all tested temperatures and assayed under standards conditions to ascertain enzyme stability is maintained. All assays were performed in duplicates. Ea was calculated from the slope (Ea/R) of the Arrhenius plot fitting the data to eqn (8), in which R is the gas constant (8.314 J mol−1 K −1), and A is the Arrhenius constant, which represents the product of the collision frequency (Z), and a steric factor (p) based on the collision theory of enzyme kinetics.32,33 A simplistic approach was adopted to explain a complex phenomenon and that A is independent of temperature.| |
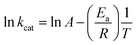 | (8) |
The Ea value allowed to obtain an estimate for the enthalpy of activation (ΔH#) employing eqn (9). The Gibbs free energy (ΔG#) of activation was estimated using eqn (10). These values allowed to obtain an estimate for the entropy of activation (ΔS#) using eqn (11). These equations were derived from the transition state theory of enzymatic reactions.32,33 R, Ea and T are as for eqn (8), kb is the Boltzmann constant (1.380658 × 10−23 J K−1), and h is the Planck's constant (6.626075 × 10−34 J s−1). The error in ΔG# was calculated using eqn (12).
| |
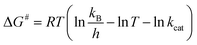 | (10) |
| |
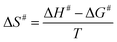 | (11) |
| |
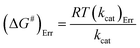 | (12) |
Solvent kinetic isotope effects (SKIE) and proton inventory
All assays were carried out under standard reaction conditions, in duplicate. The solvent kinetic isotope effects on both V/K and V were determined by measuring initial velocities for LbASL reaction using varied concentrations of S-AMP in either H2O or 90% D2O. The SKIE data were fitted to eqn (13),34 in which V is the maximal velocity, A is the substrate concentration, EV/K and EV are, respectively, the isotope effect minus 1 on V/K and V, and Fi is the fraction of deuterium label in the solvent.| |
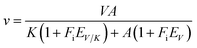 | (13) |
To determine the number of protons contributing to the observed solvent kinetic isotope effect, the proton inventory on the catalytic rate constant (kcat) was measured at saturating concentration of S-AMP at different mole fractions of D2O (0–90%). The data for the relative activity versus mole fraction of D2O plot were fitted to the Gross–Butler equation (eqn (14)),34 in which kn is the rate constant measured at various mole fractions of D2O (e.g., k0 = kcat value in H2O, and k0.9 = kcat value in 90% D2O), n is the isotopic composition of the solvent, and ϕT is the deuterium fractionation factor for transition-state proton exchange relative to bulk water (i.e., exchange equilibrium constant that measures the tendency of a transition-state site to fractionally contain deuterium versus the deuterium fraction of the solvent). It should be pointed out that eqn (14) implies that a single proton contributes to the observed solvent isotope effect and that the reactant-state fractionation factor is equal to unity.
| |
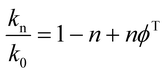 | (14) |
pH-rate profiles
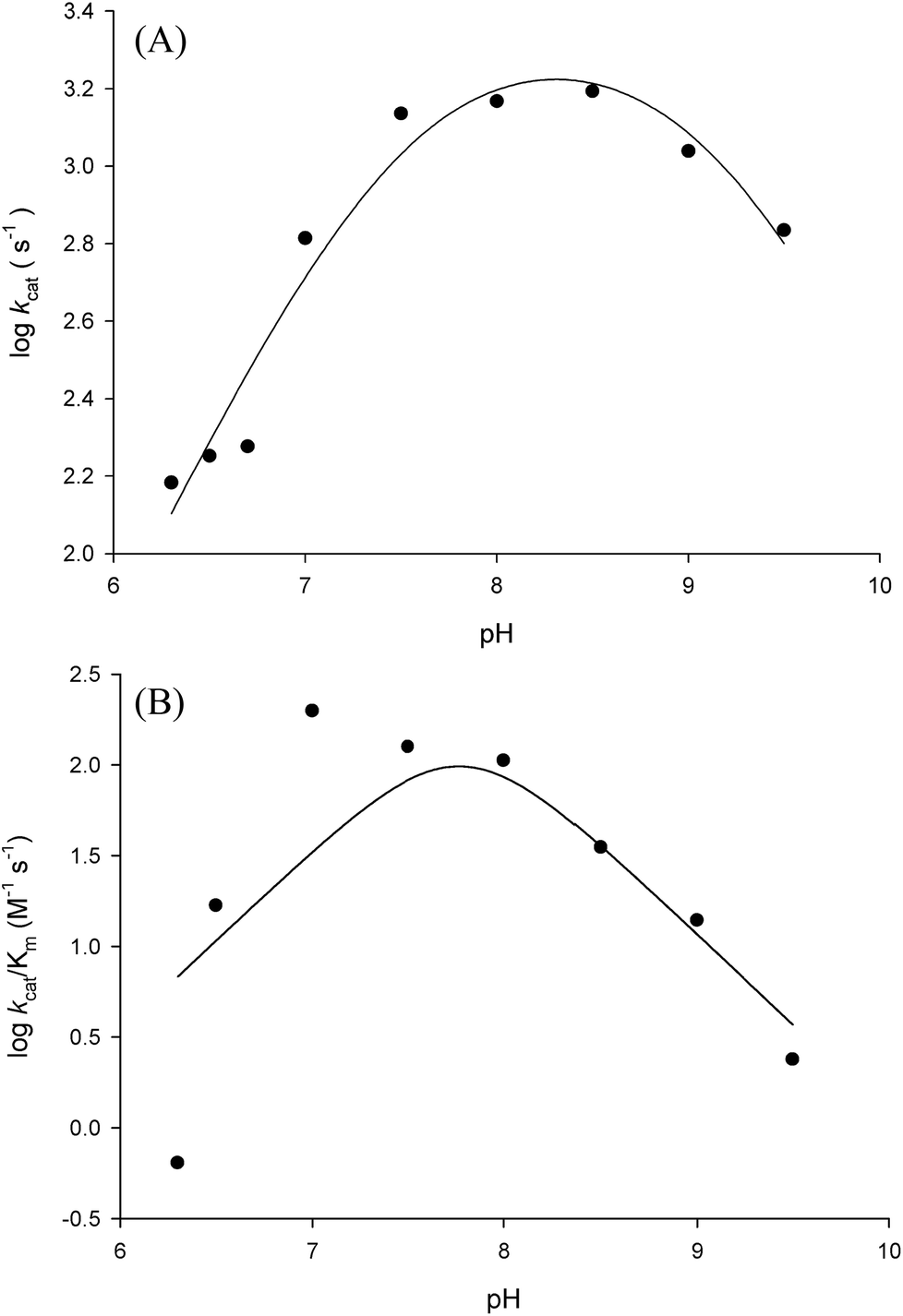
Prior to carrying out pH-rate studies, LbASL was incubated for 2 min at 25 °C in 100 mM 2-(N-morpholino)-ethanesulfonic acid (MES)/N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (Hepes)/2-(N-cyclohexylamino)-ethanesulfonic acid (CHES) buffer mixture over a wide pH range (5.0–10.5),35 and assayed under standard conditions to ensure enzyme stability at the experimental pH values over the course of reaction, thereby showing that changes in enzyme activity were due to changes in proton concentration and not to protein denaturation. Initial velocities measurements were carried out at 25 °C in solutions containing increasing concentrations of S-AMP in 100 mM MES/HEPES/CHES buffer mixture over the following pH values: 6.3 (S-AMP concentration range: 40–150 μM, [LbASL] = 60 nM), 6.5 (S-AMP concentration range: 5–60 μM with 6 or 12 nM of LbASL), 6.7 (S-AMP concentration range: 1–60 μM with 6 or 12 nM of LbASL), 7.0 (S-AMP concentration range: 5–60 μM with 6 or 12 nM of LbASL), 7.5 (S-AMP concentration range: 3–60 μM with 6 or 12 nM of LbASL), 8.0 (S-AMP concentration range: 5–60 μM with 6 or 12 nM of LbASL), 8.5 (S-AMP concentration range: 20–180 μM with 6 or 12 nM of LbASL), 9.0–9.5 (S-AMP concentration range: 20–200 μM, [LbASL] = 12 nM). The pH-rate data for kcat (Fig. 10A) were plotted to eqn (15), in which y represents kcat, C is the pH-independent plateau value of y(kcat), H is the hydrogen ion concentration, and Ka and Kb are, respectively, the apparent acid and base dissociation constant for the ionizing group. Eqn (15) describes a bell-shaped pH profile for a group that must be protonated for catalysis and another group that must be unprotonated for catalysis, and participation of a single ionizing group for the acidic limb (slope value of +1) and participation of a single ionizing group for the basic limb (slope value of −1).35| |
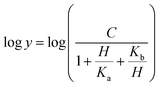 | (15) |
The pH-rate profile for kcat/KM was more complex (Fig. 10B). The data were tentatively either fitted to eqn (15) or (16). The latter equation describes a bell-shaped pH profile that starts with a slope of +2 in the acidic limb which goes to an eventual slope of −1 in the basic limb, suggesting participation of two ionizing groups in the acidic limb.35 K0 is the product of two apparent dissociation constants. Unless the pKs of the groups are at least 3 pH units apart, there will not be both a linear region with a slope of +1 and a flat plateau at intermediate pH values. The intersection of the linear asymptote with slope of 2 and the poorly defined plateau will give the average of the pK values of the two ionizing groups.35
| |
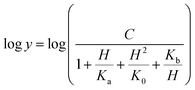 | (16) |
Results and discussion
Cloning and recombinant protein expression
The LbASL-coding DNA sequence LbrM.04.0500 was purchased from Biomatik® and cloned into the pET-23a(+) expression vector. Automated DNA sequencing confirmed the identity and the absence of mutations in the cloned fragment. The best experimental condition for LbASL protein expression was observed in competent E. coli BL21 (DE3) cells, in LB medium after 12 h of growth, without IPTG induction. SDS-PAGE analysis showed that the protein was expressed in the soluble fraction of cellular extracts (∼51 kDa) which is in agreement with the predicted molecular mass value of 51.269 kDa for LbASL subunit. The expressed recombinant LbASL protein was purified to homogeneity (ESI, Fig. S1†). The protein purification protocol (streptomycin sulfate and ammonium sulfate precipitations, and size exclusion and anion exchange columns) yielded approximately 19 mg from 1.8 g of frozen cells indicating a protein yield of 40% (ESI, Table S1†). The recombinant protein was stored at −20 °C in the storage buffer (50 mM potassium phosphate buffer pH 7.0, 150 mM KCl, 1 mM EDTA, 1 mM DTT and 10% glycerol). The storage buffer was identified as the best condition to maintain enzyme stability for up to 3 months. The recombinant enzyme lost more than 50% of initial activity after 3 months when stored at either −20 °C or −80 °C.
LbASL identification by mass spectrometry
The gel band of approximately 51 kDa was excised from SDS-PAGE, submitted to trypsin digestion protocol, and the peptides were analyzed by LC-MS/MS in triplicate. LbASL identity was confirmed, with the identification of 189 unique peptides and sequence coverage of 100% (ESI, Fig. S2†).
Oligomeric state determination
To determine the oligomeric state of recombinant LbASL, 100 μL was loaded on a Superdex 200 HR 10/30 size exclusion column. A single peak was obtained with elution volume corresponding to approximately 223357 kDa, according to data fitting to eqn (1) (ESI, Fig. S3†). This molecular mass value divided by the subunit molecular mass value (51.2699 kDa) indicates that LbASL is a homotetramer in solution. This result is in agreement with the ASL characterized previously from human,36 L. donovani13 and others aspartase/fumarase superfamily members.19
Multiple sequence alignment and homology modeling
The multiple sequence alignment for Leishmania braziliensis (LbASL), Leishmania donovani (LdASL), Escherichia coli (EcASL), Plasmodium falciparum (PfASL), Bacillus subtilis (BsASL),18,37,38 Homo sapiens (HsASL), and Mycobacterium tuberculosis (MtASL),39 allowed to propose the likely amino acid residues involved in LbASL catalysis and substrate binding (Fig. 1). Multiple sequence alignment results showed that LbASL shares 88%, 45%, 35%, 18%, 17% and 12% sequence identity with, respectively, L. donovani, E. coli, Plasmodium falciparum, B. subtillis, H. sapiens and M. tuberculosis.
 |
| | Fig. 1 Multiple sequence alignment of ASL from Leishmania braziliensis (LbASL, XP_001561734), Leishmania donovani (LdASL, XP_003858107), Escherichia coli (EcASL, WP_000423742), Plasmodium falciparum (PfASL, XP_001349577), Bacillus subtilis (BsASL, WP_003233955), Homo sapiens (HsASL, NP_000017), and Mycobacterium tuberculosis (MtASL, WP_003898583). Residues His119, His197, S322, and K328, proposed to be involved in catalysis, are indicated by black solid circles. ASL enzymes present three highly conserved regions, C1, C2, and C3 (residues 147–158, 194–204, and 321–331 in LbASL), which in the structure pack together to form the active site. The blue boxes represent residues conserved in all sequences. The green box represents residues conserved in all sequences, except in Mycobacterium tuberculosis. The alignment was prepared with ClustalW26 and JalView.56 | |
The general mechanism proposed for ASL catalysis is a β-elimination (anti 1,2-addition-elimination reaction), in which a general base of the enzyme abstracts the pro-R hydrogen from the C3 atom (Cβ) of the succinyl moiety of the substrate.40 The resulting carbanion is stabilized as the aci-carboxylate (or enediolate) intermediate with two negative charges on the β-carboxylate group. Cleavage of the C–N bond of the substrate is assisted by leaving group protonation by an enzyme general acid.19 As the reaction occurs via anti elimination, two separate amino acid residues for proton abstraction and donation are required. Conserved His141 and His68 in B. subtilis have been proposed to be, respectively, the base and acid groups.37,41 The sequence comparison showed that residues equivalent to these histidines are conserved (Fig. 1), suggesting that His197 and/or His119 may play a role in LbALS catalysis. Alternatively, the catalytic base residue may be ascribed to Ser322 in LbASL (Fig. 1). Proteins belonging to the aspartase/fumarase superfamily (including ASL) share a characteristic tertiary and quaternary fold as well as similar active site architecture.19 The monomer is comprised of three mainly α-helical domains: N-terminal (D1), central helix (D2) and C-terminal (D3). Three conserved regions are found in the D2 domain: C1 located at the start of D2, and C2 and C3 that are located in the loop regions between the helices of D2 (Fig. 2). Although spatially separated in the monomeric unit, the C1–C3 domains from three different subunits form the active site of the tetrameric protein (Fig. 2). Part of the conserved C3 region is formed by the flexible Ser–Ser loop, which undergoes conformational changes upon substrate binding that is relevant to catalysis in ASL enzymes.42,43 The signature sequence of this Ser–Ser loop in LbASL is 321GSSXXPXKXN330, and is highly conserved among all aligned sequences (Fig. 1). Site-directed mutagenesis studies on B. subtilis indicated that Gln212, Asn270, and Arg301 residues perform critical functions in catalysis by ASL through their contributions to the binding and orientation of the succinyl carboxylate groups of its two substrates SAICAR and S-AMP.34 The corresponding Gln274, Asn330 and Arg362 residues in LbASL are also conserved in the ASLs from other organisms (Fig. 1), except Arg362 that is replaced with a glycine in M. tuberculosis.39
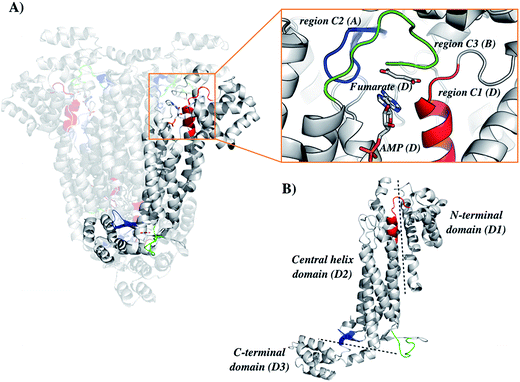 |
| | Fig. 2 Structure organization of the ASL from Leishmania braziliensis (LbASL). LbASL is biologically active as a homotetramer and contains four active sites. Amino acid residues of three subunits contribute to each enzyme active. (A) Quaternary structure of LbASL and a close view of the active site, which is formed by tight packing of regions C1 (red), C2 (blue), and C3 (green), belonging to, respectively, monomers D, A, and B. (B) Representation of the N-terminal (D1), central core (D2) and C-terminal (D3) domains of one enzyme subunit. Protein main polypeptide chain, represented as cartoon, is coloured in grey, and AMP and fumarate are represented as sticks and coloured as CPK. The illustration was generated with PyMOL (the PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC). | |
The homology model of LbASL (Fig. 3) shows a His197 at 4.1 Å of the C–N bond of AMP, suggesting that this residue may act as the catalytic acid. The conserved Ser322 is in close proximity (2.9 Å) to the C(β or α)-H bond of fumarate. This serine is in the highly conserved flexible Ser–Ser loop, which closes the active site upon substrate binding. Accordingly, Ser322 side chain may act as the catalytic base in the LbASL reaction. Although it is tempting to suggest that the corresponding residues may play a role in LbASL mode of action, site-directed mutagenesis efforts will have to be pursued to assign any role to these amino acids.
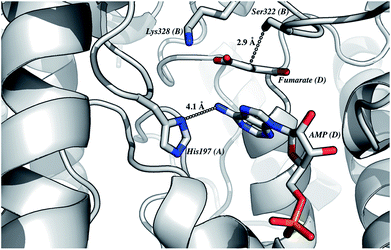 |
| | Fig. 3 The proposed residues involved in the reaction mechanism. This model of LbASL active site shows contacts of the imidazole side chain of His197 with AMP, and the putative catalytic base Ser322 with the C(β or α)-H of the succinyl moiety of the substrate. Protein main-chain, represented as cartoon, is coloured in grey, and AMP, fumarate and amino acid residues are represented as sticks and coloured as CPK. The illustration was generated with PyMOL (the PyMOL Molecular Graphics System, Version 1.8 Schrödinger, LLC). | |
The high conservation of key amino acid residues essential for substrate binding and catalysis for both H. sapiens and L. braziliensis ASL enzymes suggest that the development of selective inhibitors for LbASL might be challenging. Notwithstanding, a better understanding of the mode of action of LbASL may unveil differences in enzyme, chemical and catalytic mechanisms that may contribute to the development of mechanism-based anti-leishmaniasis agents.
Steady-state kinetic parameters
The initial velocity experiments were measured to obtain the true steady-state kinetics parameters and to propose an enzyme mechanism. It has been shown that B. subtilis ASL dissociates into a mixture of monomer–dimer–trimer with decreased enzyme activity at low temperatures (4 and 8 °C), whereas the enzyme is fully active and exists as 100% tetramer at 25 °C.18 Accordingly, recombinant LbASL protein was pre-incubated for 30 min at 25 °C to ascertain maintenance of fully active tetrameric enzyme. Data for liquid chromatography carried out in a cold-room (∼6 °C) using size exclusion column suggested that recombinant LbASL is homotetrameric (Fig. S3†). However, it should be pointed out that whether or not the oligomeric state of LbASL changes as a function of temperature has to await analytical ultracentrifugation data. The specific activity of LbASL was obtained by varying the concentration of S-AMP (5–200 μM) and fixed concentration of enzyme (30 nM), and measuring the decrease in absorbance at 282 nm upon S-AMP conversion into products. Substrate saturation curves were hyperbolic (Fig. 4) and the data were thus fitted to the Michaelis–Menten equation (eqn (2)), and kcat value was calculated using eqn (3). The hyperbolic profile suggests that there is no inhibition for S-AMP at 200 μM (∼10-fold KM). This analysis yielded the following steady-state kinetic parameters: KM = 11.1 (±0.9) μM, Vmax = 6.4 (±0.1) U mg−1 and kcat = 337 (±5.4) s−1. A comparison of the specific activity of ASL from L. donovani,13,43 P. falciparum,44 H. sapiens36 and M. smegmatis39 are summarized in Table 1. LbASL displays higher kcat and specificity constant (kcat/KM) values in comparison to ASL enzymes from different species of Leishmania (Table 1). Interestingly, the larger overall dissociation constant (KM) for S-AMP substrate of LbASL as compared to H. sapiens ASL may suggest differences from substrate binding en route to product formation that may be exploited to increase inhibitor specificity.
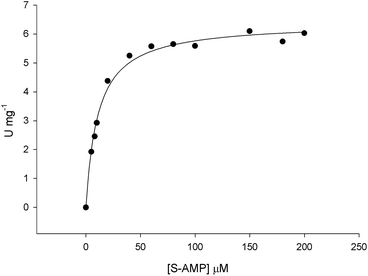 |
| | Fig. 4 Initial velocities for S-AMP conversion into AMP and fumarate. Substrate saturation curve was hyperbolic and data fitted to eqn (2). | |
Table 1 Steady-state kinetic parameters for S-AMP conversion into products catalyzed by ASL homologs
| Specie |
KM (μM) |
Vmax (U mg−1) |
kcat (s−1) |
kcat/KM (M−1 s−1) |
Assay conditions |
| L. braziliensis |
11.1 ± 0.9 |
6.4 ± 0.1 |
337 ± 5.4 |
3.0 (±0.2) × 107 |
25 °C pH 7.5 |
| L. donovani (Spector et al., 1979) |
3.3 ± 0.5 |
100 ± 3 |
87.7 ± 2.6 |
2.7 (±0.4) × 107 |
30 °C pH 7.8 |
| L. donovani (Boitz, et al., 2013) |
24.0 |
2.1 |
28 |
0.12 × 107 |
25 °C pH 7.0 |
| P. falciparum (Bulusu et al., 2009) |
32.0 ± 1.6 |
— |
7.5 ± 0.7 |
0.23 (±0.02) × 106 |
25 °C pH 7.4 |
| H. sapiens (Lee and Colman, 2007) |
1.78 ± 0.05 |
3.88 ± 0.07 |
3.40 ± 0.06 |
1.9 (±0.1) × 106 |
25 °C pH 7.4 |
| M. smegmatis (Banerjee, 2014) |
43.7 ± 2.6 |
— |
0.70 ± 0.01 |
1.6 (±0.1) × 104 |
37 °C pH 7.6 |
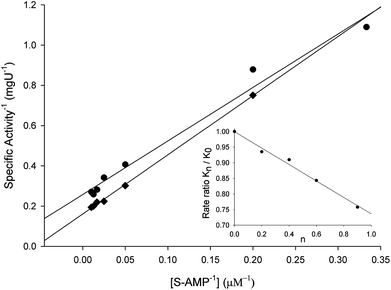
Double-reciprocal plots showed a family of lines intersecting to the left of the y-axis (Fig. 5), suggesting ternary complex formation and a sequential (either random or ordered) mechanism for the reverse reaction. The pattern of straight lines intersecting to the left of the y-axis rules out ping-pong (parallel lines), steady-state random (that gives non-linear reciprocal plots), and rapid-equilibrium ordered (one of the family of lines should cross at a single value on the y-axis) mechanisms. Accordingly, the data were fitted to eqn (4) yielding the following values: KM(AMP) = 13 (±5) μM and KM(fumarate) = 203 (±20) μM, Ki(AMP) = 112 (±20) μM, and kcat = 115 (±3) s−1. The steady-state kinetic parameters for the forward and reverse reactions and the Haldane equation for an ordered Uni-Bi mechanism (eqn (5)) were used to calculate a value of 5720 ± 1041 μM (ca. 5.7 × 10−6 M) for the equilibrium constant (Keq). This result suggests that the forward reaction is not favorable under the in vitro experimental conditions here employed. However, the depletion of products in the physiological context may drive the reaction forward. At any rate, the double-reciprocal plots alone cannot distinguish between rapid-equilibrium random and steady-state compulsory ordered Uni-Bi mechanisms. ITC studies were thus performed to distinguish between these enzyme mechanisms.
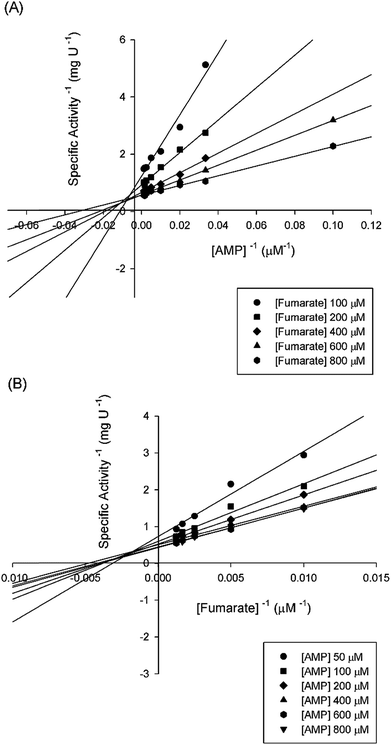 |
| | Fig. 5 Intersecting initial velocity patterns for ASL using either fumarate (a) or AMP (b) as the variable substrate. Each curve represents varied-fixed levels of the substrate. | |
Isothermal titration calorimetry (ITC)
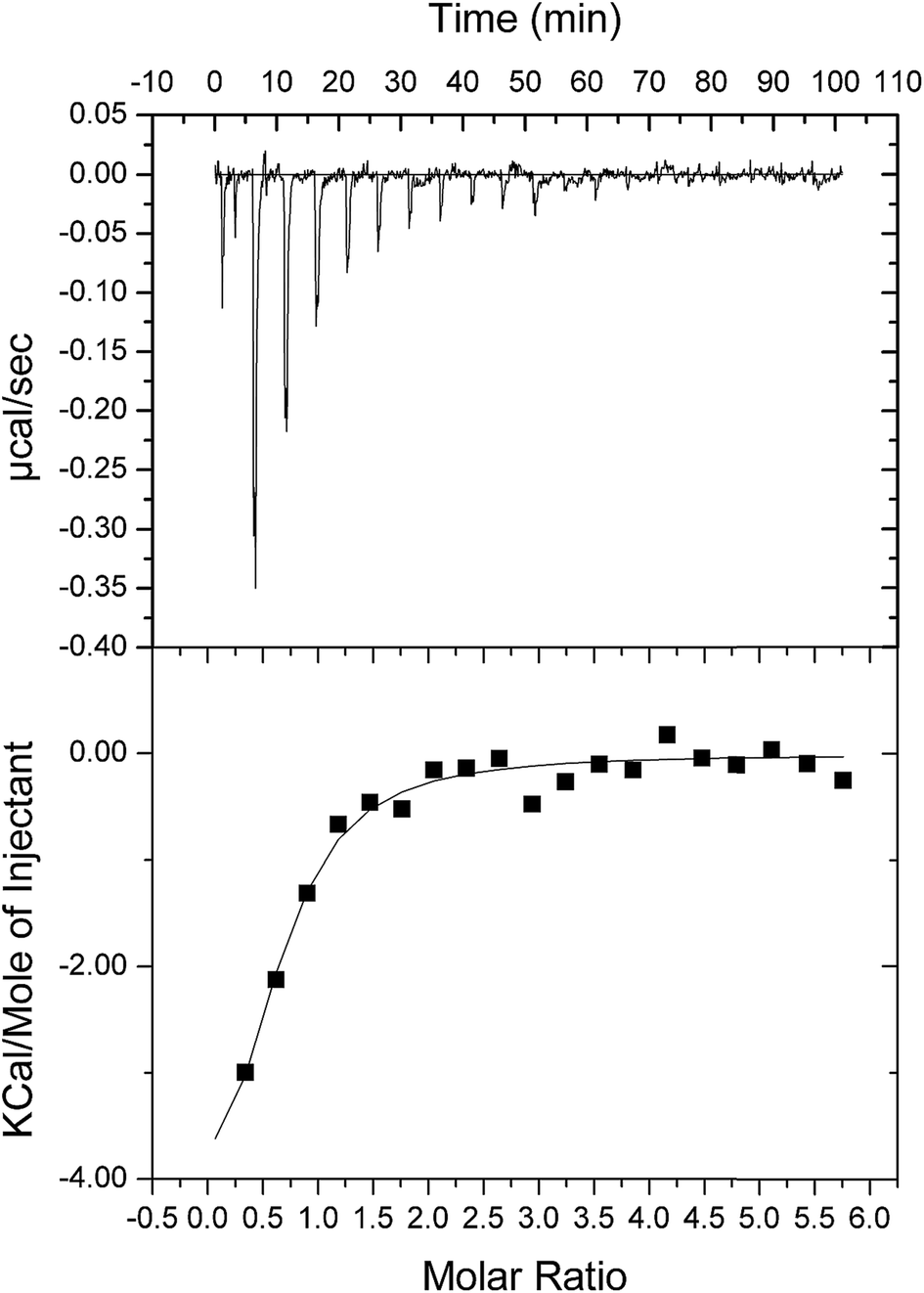
As double reciprocal plots suggested a sequential kinetic mechanism for the reverse reaction, product binding to free enzyme was assessed by ITC to ascertain the order, if any, of chemical compound interaction with free LbASL. Accordingly, binary complex formation of either AMP or fumarate binding to free LbASL enzyme was studied by ITC. No heat change was detected upon addition of fumarate, suggesting that fumarate cannot bind to free LbASL enzyme (ESI, Fig. S4†). An exothermic profile (heat release) was observed for binary complex formation upon AMP binding to free LbASL protein (Fig. 6). The ITC data yielded the following values for LbASL:AMP binary complex formation: ΔH# = −5.5 (±1.5) kcal mol−1 and ΔS# = 3.13 (±0.84) cal mol−1 K−1. The negative enthalpy value indicates a favorable, though small, redistribution of interactomic interactions network (e.g., hydrogen bonds and/or van der Waals interactions) between the reacting species, including solvent. Hydrophobic interactions are related to the relative degrees of disorder in the free and bound systems and thus these interactions are reflected in the entropy change. The release of “bound” water molecules from a surface to the bulk solvent is usually a source of favourable entropy (positive ΔS). A reduction in conformational states in either ligand or protein upon binary complex formation is entropically unfavourable (negative ΔS) because this molecular recognition process limits the external rotational and translational freedom of both partners (for instance, structuring regions of the protein adjacent to the bound ligand and loss of conformational freedom of free ligand).45 The positive entropy value suggests either release of bound water molecules and/or an increase in conformational states in LbASL or AMP upon binary complex formation. The Gibbs free energy ΔG# value of −5.6 (±1.5) kcal mol−1 (Kd ≅ 19 ± 7 μM) suggests a favorable process for LbASL:AMP binary complex formation. ITC data were fitted to one set of site binding model yielding a value of 0.6 (±0.1) for n (stoichiometry, ligands per active site). This result suggests that more than one subunit of LbASL contribute to AMP binding. Structural studies showed that three separate protomers contribute to each binding site of tetrameric M. tuberculosis ASL39 and to other enzymes belonging to the aspartase/fumarase superfamily.19 However, the stoichiometry should be equal to approximately one as there are four active sites per tetrameric LbASL enzyme.
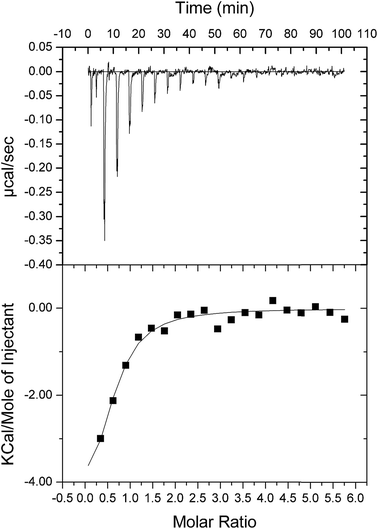 |
| | Fig. 6 ITC analysis of LbASL titration with AMP. The top panels show raw data of the heat pulses resulting from titration of LbASL. The button panels show the integrated heat pulses, normalized per mole of injection as a function of the molar ratio (ligand concentration/LbASL subunit concentration). Data were best fitted to a one binding site model. | |
The steady-state kinetic measurements for the reverse reaction and the ITC data for product binding to LbASL demonstrate that the reaction catalyzed by LbASL follows an ordered Uni-Bi kinetic mechanism, in which fumarate is the first product to dissociate from the ternary complex followed by AMP release to yield free enzyme for the next round of catalysis (Fig. 7). This proposal is in agreement with L. donovani ASL enzyme mechanism.43
 |
| | Fig. 7 Proposed sequential ordered Uni-Bi kinetic mechanism for LbASL, in which fumarate release is followed by AMP dissociation to form free enzyme. | |
Energy of activation
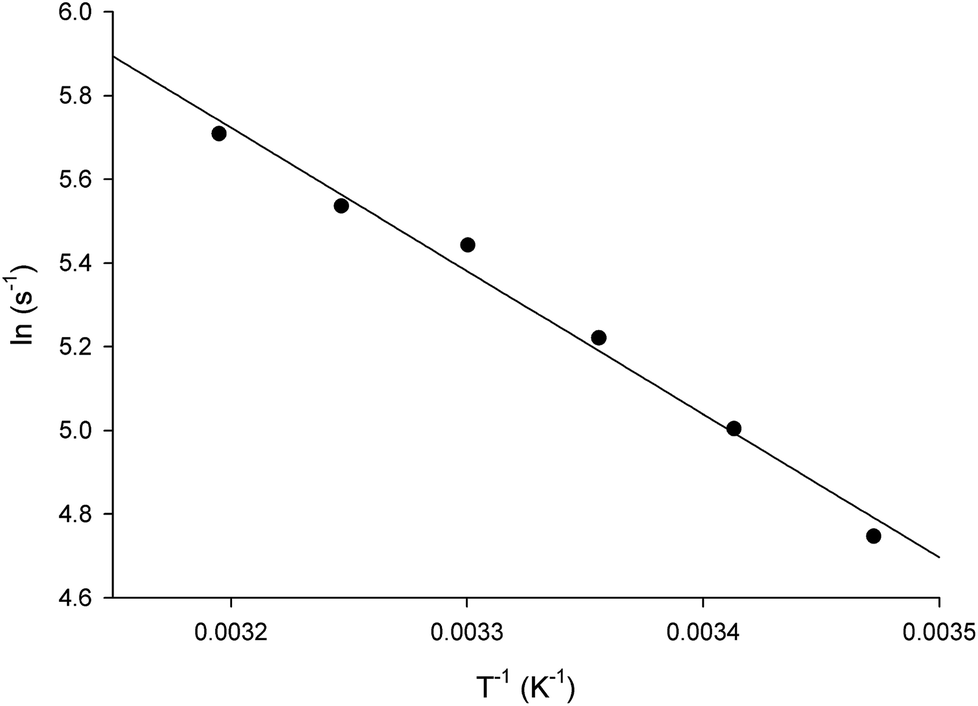
The energy of activation (Ea) for the LbASL-catalyzed chemical reaction was assessed by measuring the dependence of kcat on temperature for S-AMP (Fig. 8). The Ea (6.8 ± 0.3 kcal mol−1) of the reaction was calculated from data fitting to eqn (8) for the slope (−Ea/R) of the Ahrrenius plot (Fig. 8). The transition state enthalpy (ΔH# = 6.2 ± 0.3 kcal mol−1), Gibbs free energy (ΔG# = 16.4 ± 0.5 kcal mol−1) and entropy (ΔS# = −34.4 ± 1.6 cal mol−1 K−1) at 25 °C were calculated using, respectively, eqn (9), (10) and (11). The Ea value of 6.8 kcal mol−1 represents the minimum energy needed to initiate the reaction, and the linearity of the Ahrrenius plot suggests that there is no change in the rate-limiting step over the temperature range employed (15–40 °C). The ΔG# value of 16.4 kcal mol−1 represents the energy barrier required for the reaction to occur and can be regarded as the variation of Gibbs energy between the enzyme–substrate activated complex and enzyme-substrate in the ground state.
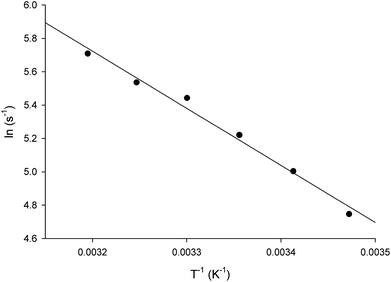 |
| | Fig. 8 Temperature dependence of lnkcat. Saturating concentrations of S-AMP substrate were employed to measure the maximum velocity as a function of temperature ranging from 15 to 40 °C. The linearity of the Arrhenius plot suggests that there is no change in the rate-limiting step over the temperature range utilized in this assay. | |
The negative value for the entropy activation (ΔS#) suggests that the entropy value for the enzyme:substrate activated complex is lower than the one for enzyme:substrate in the ground state, which may be accounted for by a loss of degrees of freedom on going from the ground state to activated state. The constant A (frequency factor that represents the frequency of collisions between reactant molecules) of eqn (8) corresponds to the product of collision frequency (Z) and the probability or steric factor (p) from the collision theory of reaction rates. From the absolute rate theory, A = pZ = (kBT/h)eΔS#/R. This equation enables us to interpret the probability factor (p) in terms of the molar entropy of activation (ΔS#). If reactants are atoms or simple molecules, then relatively little energy is redistributed among the various degrees of freedom in the activated complex (transition-state complex). Consequently, ΔS# will be either a small positive or a small negative number, so that exp(ΔS#/R) or p is close to unity. But if complex molecules are involved in a reaction, ΔS# will be either a large positive or a large negative number. In the former case, the reaction will proceed much faster than predicted by collision theory; in the latter case, a much slower rate will be observed. Note that the probability factor (p) takes into account the fact that in a collision complex molecules must be properly oriented to undergo the reaction (having the proper activation energy is a prerequisite but not a guarantee for a reaction to take place). Thus, the frequency factor (A) of the Arrhenius equation depends also on T and p (that accounts for that fact that colliding molecules must be properly oriented to undergo the reaction). The negative values for the entropy of activation (ΔS#) for S-AMP reaction suggests that this reaction proceeds slower than predicted by the collision theory. Incidentally, a value of 17466582 ± 698663 s−1 was obtained for A which allows to calculate an apparent first-order constant value of approximately 188 ± 8.2 s−1 using the Arrhenius equation (k = Ae−Ea/RT), which is in reasonably good agreement with the kcat value of 337 s−1 from steady-state kinetics data (Table 1).
Solvent kinetic isotope effect (SKIE) and proton inventory
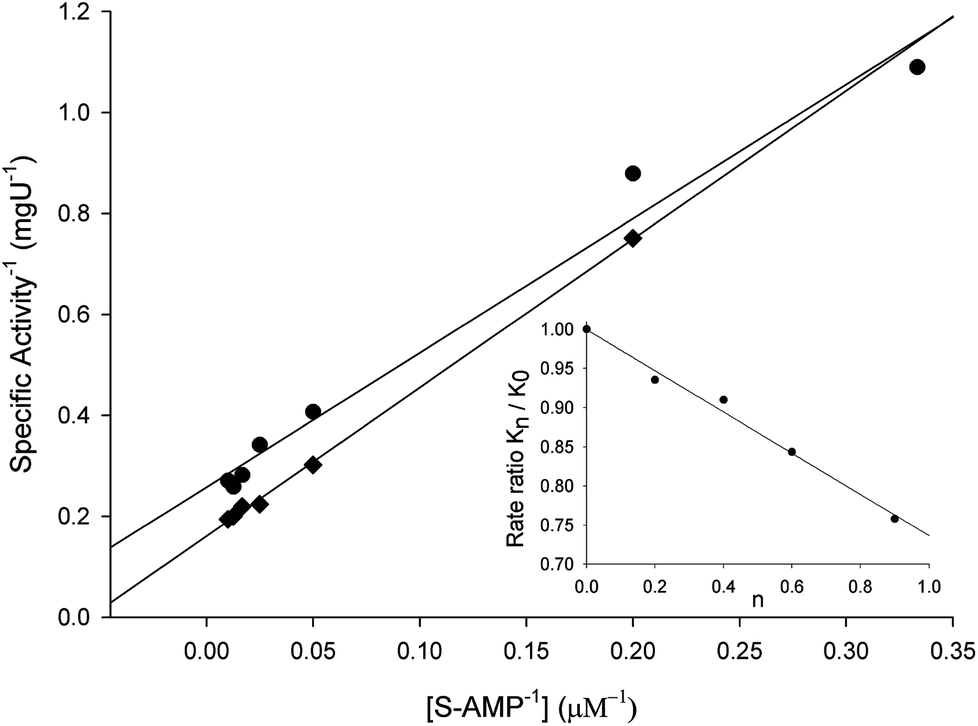
To evaluate the contribution of proton transfer from the solvent to a rate-limiting step of S-AMP conversion into fumarate and AMP for LbASL, SKIE were determined by data fitting to eqn (13) (Fig. 9), yielding a value of 1.40 ± 0.06 for D2OV and of 1.20 ± 0.16 for D2OV/K. Isotope effects on V report on events following the ternary complex formation capable of undergoing catalysis (fully loaded enzyme), which include the chemical steps, possible enzyme conformational changes, and product release (leading to regeneration of free enzyme). Solvent isotope effects on V/K report on the contribution of the proton transfer in steps in the reaction mechanism from binding of the isotopically labeled chemical compound (solvent) to the first irreversible step, usually considered to be the release of the first product (that is, all rate constants from reactant binding until the first irreversible step).34 As rule of thumb, deuterium accumulates where binding is tighter (that is, fractionation factor is larger than one). Transition state proton contributes the reciprocal of its respective fractionation factor to the solvent isotope effect, whereas the contribution of a reactant state proton to the solvent isotope effect is equal to its fractionation factor.34
 |
| | Fig. 9 Solvent kinetic isotope effects for LbASL. S-AMP was used as the variable substrate (5–100 μM). Reaction mixtures contained either 0 (♦) or 90 (●) atom % D2O. The inset represents the proton inventory (0, 20, 40, 60 and 90 atom % D2O) measuring LbASL enzyme activity at fixed-saturating concentration of S-AMP (100 μM), in which the y-axis represents the values for kcat in solutions containing different mole fractions of D2O (kn) divided by kcat in H2O (k0), and the x-axis gives the isotopic composition of the solvent (n). | |
The values of V/K and V suggest that there is a modest contribution of solvent proton transference during the rate-limiting step of the reaction, probably events occurring after formation of the binary complex, as conformational and chemical changes. The proton inventory data show that the modest normal SKIE on V arises from a single protonic site (Fig. 9 inset). The SKIE on V data are similar to the results observed for PfASL.44 On the other hand, there appears to be a modest normal SKIE effect on V/K for LbASL whereas no effect was observed for PfASL.44 The latter suggests that subtle differences of substrate binding and solvent proton participation in this process may play a role in LbASL mode of action. Data fitting to eqn (14) yielded a transition state fractionation factor (φT) value of 0.74 ± 0.05, which is in agreement with the value observed for PfASL enzyme.44 The transition state fractionation factor value suggests that binding of proton solvent to the transition state and/or to LbASL en route to catalysis is looser than the S-AMP substrate in the ground state and/or free enzyme as compared to bulk solvent. Solvation catalytic proton bridges are proton transfers that do not have appreciable proton motional amplitude in the reaction coordinate, but occur in stable, normal modes of the transition state.34 Solvation catalytic proton bridges involve transfers among O, N, and S atoms, for which intrinsic free energy barriers are expected to be small compared to free-energy changes associated with covalent rearrangement of the heavy-atom (nonhydrogenic) framework of the reacting system. Hence, solvation catalytic bridges are strong H bonds with values φT values of 0.3–0.6.34 The φT value thus suggests that solvent proton transfer in transition-state vibrations perpendicular to the reaction coordinate plays a role in LbASL mode of action. As these proton transfers are common for O and N, it is tempting to suggest that protonation of either His197, His119, Lys118, Gln274, Asn330 or Arg362 residues, proton transfer from Ser322 (or Lys118) to N1/N6 of S-AMP may play a role in LbALS-catalyzed chemical reaction. However, it should be pointed out that solvent isotope effects lead to isotope exchanges at hundreds of protic positions of the enzyme, which precludes any assignment to a particular chemical group.
pH-rate profiles
In order to gain information on the chemical mechanism of LbASL and likely residues involved in catalysis and substrate binding, the dependence of kinetic parameters on varying pH values were determined. The pH-rate profile is shown in Fig. 10. The bell-shape pH-rate data for logkcat were fitted to eqn (15) yielding apparent pKa value of 7.5 ± 2.5 and pKb value of 9.1 ± 4.9, which slopes of +1 for the acidic limb and −1 for the basic limb indicate the participation of a single ionizable group in each limb. This ionization could be occurring in either the substrate or enzyme.
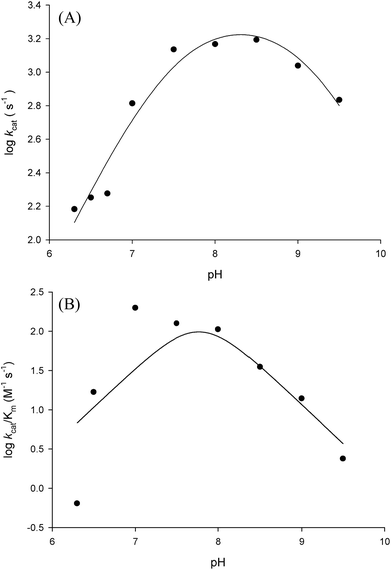 |
| | Fig. 10 Dependence of steady-state kinetic parameters on different pH values for LbASL reaction. (A) Data for pH dependence of logkcat were fitted to eqn (15). (B) Data for the dependence of logkcat/KM on pH values were fitted to eqn (16). | |
It is propose that the cleavage of S-AMP to AMP and fumarate for ASL occurs through a general acid–base mechanism involving a β-elimination of fumarate. The reaction initiates by abstraction of the Cβ-proton from the substrate by the general base, resulting in the formation of a carbanion intermediate, and subsequent proton donation by the catalytic acid to the N1 or N6 atom of the substrate results in cleavage of the Cα–N bond and product release.19,42 The His197 (for LbASL counting) is a highly conserved residue in the conserved region C2 in aspartase/fumarase superfamily. This histidine residue has being proposed to act as the general acid, donating a proton to the leaving group.16,19 In previous site-direct mutagenesis studies, the mutation of correspondent His171 in E. coli ASL,42 and His141 in B. subtilis,41 (His197 in LbASL) showed decrease in enzyme activity, showing that these histidine residues play a role in catalysis. The His197 of LbASL may account for the apparent pKa value of 7.5 in logkcat analysis, which needs to be deprotonated for catalysis, whereas the pK value from the theoretical histidine imidazole group (pK = 6.0) could be shifted due interactions with others residues in the active site.
Another conserved region in the superfamily is flexible a Ser–Ser loop which plays an important role in catalysis, performing a conformational change from open to close conformation upon substrate binding in the active site. A lysine residue (Lys328 LbASL numbering) situated in the Ser–Ser loop, has been propose to interact with the α-carboxylate group of the substrate.19 In addition, this lysine was suggested to be involved in the stabilization of the negative charge of the carbanion intermediate in E. coli ASL,42 E. coli L-aspartase46 and E. coli C-fumarase.47 The apparent pKb value of 9.1 may correspond to Ser322 (LbASL numbering), which could act as the catalytic base. Site-directed mutagenesis studies of the corresponding serines in B. subtilis and H. sapiens ASL proteins have shown that they are essential for catalysis.48,49 Nevertheless, site-directed mutagenesis efforts will have to be pursued to ascertain the amino acid residues that play any role in catalysis.
Fitting the data of the rather complex pH-rate profile for kcat/KM (Fig. 10B) to either eqn (15) or (16) yielded poor estimates. For instance, data fitting to eqn (16) yielded pK values of 6.4 and 9.2 with large errors. The value of 9.2 likely corresponds to the same catalytic group implicated in catalysis by the pH-rate profile for kcat (Fig. 10A). As pH-rate profile studies cannot differentiate between ionizable groups of enzyme and substrate(s), the two ionizing groups in the acidic limb (slope of +2) with a pKa value of approximately 6.4 may correspond to either a protein amino acid residue or to the ionization of carboxyl groups of the succinyl moiety of S-AMP substrate whose dissociation constants may have been perturbed by the amino acid side chains of LbASL. However, caution should be exercised as the standard errors were rather large.
Conclusion
ASLs have been proposed as potential drug targets due to their important role in purine metabolism.13,44,50,51 ASL is the last enzyme in the conversion of IMP to AMP. As Leishmania species lack the de novo pathway and are dependent on the salvage pathway to supply their purine requirements, LbASL could thus represent a drug target for the development of chemical agents to treat leishmaniasis. As point mutations in the human purB ASL-encoding gene causes autosomal recessive disorders such as autism, mental retardation, epilepsy and degeneration of muscles,52–54 it is of paramount importance that species-specific enzyme inhibitor compounds be designed. Accordingly, exploiting differences in the mode of action between human ASL and enzyme from leishmania represents a promising approach to the design of anti-leishmiasis agents with limited host toxicity. The results presented here may contribute to a better understanding of the biology of L. braziliensis, and may aid to the development of LbASL enzyme inhibitors. Incidentally, it has recently been reported a promising vaccine candidate using a polyvalent α-Gal conjugate on Qβ virus-like nanoparticles that showed elimination of Leishmania infection and proliferation of parasites in a C57BL/6 α-galactosyltransferase knockout mouse model.55 Although these preliminary results bode well, there have to be continuing efforts towards the development of alternatives strategies to combat Leishmania infection.
Author contributions
LG, PFD, CDR, ADV and LKBM designed, performed and analyzed all biochemical experiments, and drafted the paper. LFSMT and ONS built the molecular model and interpreted the structure of LbASL. CVB and ADV designed vectors, and performed cloning and expression. AFPM and CVB designed, performed and analyzed the mass spectrometry experiments. PB, EMCF, DSS and LAB designed experiments and revised critically the manuscript.
Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Acknowledgements
This work was supported by funds awarded by Decit/SCTIE/MSMCT-CNPq-FNDCT-CAPES to National Institute of Science and Technology on Tuberculosis (INCT-TB) to D. S. S. and L. A. B. L. A. B. (CNPq, 520182/99-5), D. S. S. (CNPq, 304051/1975-06), O. N. S. (CNPq, 305984/2012-8), and E. M. C. F. (CNPq, 306706/2014-8) are Research Career Awardees of the National Research Council of Brazil (CNPq). L. G. acknowledges a scholarship awarded by CAPES.
References
- P. Kaye and P. Scott, Leishmaniasis: complexity at the host–pathogen Interface, Nat. Rev. Microbiol., 2011, 9, 604–615 CrossRef CAS PubMed.
- WHO, Control of the leishmaniases: report of a meeting of the WHO Expert Committee on the control of Leishmaniases, World Health Organization, Geneva, 2010 Search PubMed.
- J. Alvar, et al., Leishmaniasis worldwide and global estimates of its incidence, PLoS One, 2012, 7(5), e35671 CAS.
- D. E. Teixeira, et al., Atlas didático Ciclo de vida da Leishmania, Fundação CECIERJ, Consórcio CEDERJ, Rio de Janeiro, 2013 Search PubMed.
- L. H. Guimarães, et al., Atypical manifestations of cutaneous Leishmaniasis in a region endemic for Leishmania braziliensis: clinical, immunological and parasitological aspects, PLoS Neglected Trop. Dis., 2016, 10(12), e0005100 Search PubMed.
- R. D. Azulay and D. R. Azulay Jr, Immune-clinical-pathologic spectrum of Leishmaniasis, Int. J. Dermatol., 1995, 34, 303–307 CrossRef CAS PubMed.
- S. L. Croft and G. H. Coombs, Leishmaniasis – current chemotherapy and recent advances in the search for novel drugs, Trends Parasitol., 2003, 19, 502–508 CrossRef CAS PubMed.
- S. L. Croft and P. Olliaro, Leishmaniasis chemotherapy - challenges and opportunities, Clin. Microbiol. Infect., 2011, 17, 1478–1483 CrossRef CAS PubMed.
- B. S. McGwire, et al., Leishmaniasis: clinical syndromes and treatment, Q. J. Med., 2014, 107, 7–14 CrossRef CAS PubMed.
- A. Lemke, et al., Amphotericin B, Appl. Microbiol. Biotechnol., 2005, 68, 151–162 CrossRef CAS PubMed.
- B. Chawla and R. Maghubala, Drug targets in Leishmania, J. Parasit. Dis., 2010, 34(1), 1–13 CrossRef PubMed.
- J. J. Marr, et al., Purine metabolism in Leishmania donovani and Leishmania braziliensis, Biochim. Biophys. Acta, 1978, 544, 360–371 CrossRef CAS.
- J. M. Boitz, et al., Adenylosuccinate synthetase and adenylosuccinate lyase deficiencies trigger growth and infectivity deficits in Leishmania donovani, J. Biol. Chem., 2013, 288(13), 8977–8990 CrossRef CAS PubMed.
- J. M. Boitz, et al., Purine salvage in Leishmania: complex or simple by design?, Trends Parasitol., 2012, 28, 345–352 CrossRef CAS PubMed.
- R. L. Berens, et al., Purine and Pyrimidine Metabolism, Biochemistry and Molecular Biology of Parasites, Academic Press Ltd, 1995, pp. 89–117 Search PubMed.
- E. A. Toth, et al., The structure of adenylosuccinate lyase, an enzyme with dual activity in the de novo purine biosynthetic pathway, Structure, 2000, 8, 163–174 CrossRef CAS PubMed.
- J. B. Palenchar, J. M. Crocco and R. F. Colman, The characterization of mutant Bacillus subtilis adenylosuccinate lyases corresponding to severe human adenylosuccinate lyase deficiencies, Protein Sci., 2003, 12, 1694–1705 CrossRef CAS PubMed.
- L. Z. Ariyananda and R. F. Colman, Evaluation of types of interactions in subunit association in Bacillus subtilis adenylosuccinate lyase, Biochemistry, 2008, 47, 2923–2934 CrossRef PubMed.
- V. V. Puthan, G. Fibriansah, H. Raj, A. W. H. Thunnissen and G. J. Poelarends, Aspartase/Fumarase superfamily: A common catalytic strategy involving general base-catalyzed formation of a highly stabilized aci-carboxylate intermediate, Biochemistry, 2012, 15, 15–22 Search PubMed.
- New England BioLabs Inc, https://www.neb.com/protocols/0001/01/01/protein-expression-using-bl21de3-c2527, accessed November 2017.
- R. C. Schwankea, et al., Molecular cloning, expression in Escherichia coli and production of bioactive homogeneous recombinant human granulocyte and macrophage colony stimulating factor, Int. J. Biol. Macromol., 2009, 45, 97–102 CrossRef PubMed.
- A. D. Villela, et al., Biochemical Characterization of Uracil Phosphoribosyltransferase from Mycobacterium tuberculosis, PLoS One, 2013, 8(2), e56445 CAS.
- A. Shevchenko, H. Tomas, J. Havlis, J. V. Olsen and M. Mann, In-gel digestion for mass spectrometric characterization of proteins and proteomes, Nat. Protoc., 2006, 1(6), 2856–2860 CrossRef CAS PubMed.
- P. C. Carvalho, D. B. Lima, F. V. Leprevost, M. D. Santos, J. S. Fischer and P. F. Aquino, et al., Integrated analysis of shotgun proteomic data with PatternLab for proteomics 4.0, Nat. Protoc., 2016, 11, 102–117 CrossRef CAS PubMed.
- J. K. Eng, T. A. Jahan and M. R. Hoopmann, Comet: an open source tandem mass spectrometry sequence database search tool, Proteomics, 2012, 13, 22–24 CrossRef PubMed.
- M. A. Larkin, G. Blackshields, N. P. Brown, R. Chenna, P. A. McGettigan, H. McWilliam, F. Valentin, I. M. Wallace, A. Wilm, R. Lopez, J. D. Thompson, T. J. Gibson and D. G. Higgins, Clustal W and Clustal X version 2.0, Bioinformatics, 2007, 23, 2947–2948 CrossRef CAS PubMed.
- M.-Y. Shen and A. Sali, Statistical potential for assessment and prediction of protein structures, Protein Sci., 2006, 15, 2507–2524 CrossRef CAS PubMed.
- A. Sali and T. L. Blundell, Comparative protein modelling by satisfaction of spatial restraints, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS PubMed.
- V. B. Chen, W. B. Arendall, J. J. Headd, D. A. Keedy, R. M. Immormino, G. J. Kapral, L. W. Murray, J. S. Richardson and D. C. Richardson, MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 12–21 CrossRef CAS PubMed.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, PROCHECK: a program to check the stereochemical quality of protein structures, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef CAS.
- I. Segel, Enzyme Kinetics, Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, John Wiley and Sons, New York, 1975 Search PubMed.
- T. Lonhienne, E. Baise, G. Feller, V. Bouriotis and C. Gerday, Enzyme activity determination on macromolecular substrates by isothermal titration calorimetry: application to mesophilic and psychrophilic chitinases, Biochim. Biophys. Acta, 2001, 1545, 349–356 CrossRef CAS.
- T. Lonhienne, E. Baise, G. Feller, V. Bouriotis and C. Gerday, Psychrophilic enzymes: revisiting the thermodynamic parameters of activation may explain local flexibility, Biochim. Biophys. Acta, 2000, 1543, 1–10 CrossRef CAS.
- P. F. Cook, Enzyme Mechanism from Isotope Effects, CRC Press, Boca Raton, Florida, 1991 Search PubMed.
- P. F. Cook and W. W. Cleland, Enzyme Kinetics and Mechanism, Garland Science, London and New York, 2007 Search PubMed.
- P. Lee and R. F. Colman, Expression, purification, and characterization of stable, recombinant human adenylosuccinate lyase, Protein Expression Purif., 2007, 51, 227–234 CrossRef CAS PubMed.
- T. T. Lee, C. Worby, J. E. Dixon and R. F. Coman, Identification of His141 in the active site of Bacillus subtilis adenylosuccinate lyase by affinity labeling with 6-(4-Bromo-2,3-dioxobutyl)thioadenisine 5′-Monophosphate, J. Biol. Chem., 1997, 272, 458–465 CrossRef CAS PubMed.
- M. L. Segall and R. F. Colman, Gln212, Asn270, and Arg301 are critical for catalysis by adenylosuccinate lyase from Bacillus subtilis, Biochemistry, 2004, 43, 7391–7402 CrossRef CAS PubMed.
- S. Banerjee, M. J. Agrawal, D. Mishral, S. Sharan, H. Balaram, H. S. Savithri and M. R. N. Murthy, Structural and kinetic studies on adenylosuccinate lyase from Mycobacterium smegmatis and Mycobacterium tuberculosis provide new insights on the catalytic residues of the enzyme, FEBS J., 2014, 281, 1642–1658 CrossRef CAS PubMed.
- W. A. Bridger and L. H. Cohen, The kinetics of adenylosuccinate lyase, J. Biol. Chem., 1968, 243, 644–650 CAS.
- T. T. Lee, C. Worby, J. E. Dixon, Z. Bao and R. F. Colman, His68 and His141 are critical contributors to the intersubunit catalytic site of adenylosuccinate lyase of Bacillus subtilis, Biochemistry, 1999, 38, 22–32 CrossRef CAS PubMed.
- M. Tsai, J. Koo, P. Yip, R. F. Colman, M. L. Segall and P. L. Howell, Substrate and product complexes of Escherichia coli adenylosuccinate lyase provide new insights into the enzymatic mechanism, J. Mol. Biol., 2007, 370, 541–554 CrossRef CAS PubMed.
- T. Spector, et al., Specificity of adenylosuccinate synthetase and adenylosuccinate lyase from Leishmania donovani, J. Biol. Chem., 1979, 254, 8422–8426 CAS.
- V. Bulusu, et al., Elucidation of the substrate specificity, kinetic and catalytic mechanism of adenylosuccinate lyase from Plasmodium falciparum, Biochim. Biophys. Acta, 2009, 1794, 642–654 CrossRef CAS PubMed.
- J. E. Ladbury and M. L. Doyle. Biocalorimetry II, Wiley, London, 2004 Search PubMed.
- S. Saribas, J. F. Schindler and R. E. Viola, Mutagenic Investigation of Conserved Functional Amino Acids in Escherichia coli L-Aspartase, J. Biol. Chem., 1994, 269(9), 6313–6319 Search PubMed.
- T. Weaver and L. Banaszak, Crystallographic Studies of the Catalytic and a Second Site in Fumarase C from Escherichia coli, Biochemistry, 1996, 35, 13955–13965 CrossRef CAS PubMed.
- S. Sivendran and R. F. Colman, Effect of a new non-cleavable substrate analog on wild-type and serine mutants in the signature sequence of adenylosuccinate lyase of Bacillus subtilis and Homo sapiens, Protein Sci., 2008, 17, 1162–1174 CrossRef CAS PubMed.
- M. L. Segall, M. A. Cashman and R. F. Colman, Important roles of hydroxylic amino acid residues in the function of Bacillus subtilis adenylosuccinate lyase, Protein Sci., 2007, 16, 441–448 CrossRef CAS PubMed.
- T. Spector, R. L. Berens and J. J. Marr, Adenylosuccinate synthetase and adenylosuccinate lyase from Trypanosoma cruzi, Specificity studies with potential chemotherapeutic agents, Biochem. Pharmacol., 1982, 31, 225–229 CrossRef CAS PubMed.
- P. K. Fyfe, et al., Structure of Staphylococcus aureus adenylosuccinate lyase (PurB) and assessment of its potential as a target for structure-based inhibitor discovery, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 881–888 CrossRef CAS PubMed.
- J. Jaeken, S. Wadman, M. Duran, F. Van Sprang, F. Beemer, R. Holl, P. Theunissen, P. De Cock, F. Van den Bergh and M. Vincent, Adenylosuccinase deficiency: an inborn error of purine nucleotide synthesis, Eur. J. Pediatr., 1988, 148, 126–131 CrossRef CAS PubMed.
- P. Maaswinkel-Mooij, L. Laan, W. Onkenhout, O. Brouwer, J. Jaeken and B. Poorthuis, Adenylosuccinase deficiency presenting with epilepsy in early infancy, J. Inherited Metab. Dis., 1997, 20, 606–607 CrossRef CAS PubMed.
- D. Verginelli, B. Luckow, C. Crifo, C. Salerno and M. Gross, Identification of new mutations in the adenylosuccinate lyase gene associated with impaired enzyme activity in lymphocytes and red blood cells, Biochim. Biophys. Acta, 1998, 1406, 81–84 CrossRef CAS.
- A. P. V. Moura, L. C. B. Santos, C. R. N. Brito, E. Valencia, C. Junqueira, A. A. P. Filho, M. R. V. Sant'Anna, N. F. Gontijo, D. C. Bartholomeu, R. T. Fujiwara, R. T. Gazzinelli, C. S. McKay, C. A. Sanhueza, M. G. Finn and A. F. Marques, Virus-like particle display of the α-Gal carbohydrate for vaccination against Leishmania infection, ACS Cent. Sci., 2017, 3, 1026–1031 CrossRef CAS PubMed.
- A. M. Waterhouse, J. B. Procter, D. M. Martin, M. Clamp and G. J. Barton, Jalview Version 2–a multiple sequence alignment editor and analysis workbench, Bioinformatics, 2009, 25, 1189–1191 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra10526f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  abc,
Edgar Marcelino de Carvalho Filhod,
Luiz Augusto Basso
abc,
Edgar Marcelino de Carvalho Filhod,
Luiz Augusto Basso