DOI:
10.1039/C7RA09974F
(Paper)
RSC Adv., 2017,
7, 54332-54340
A bifunctional two dimensional TM3(HHTP)2 monolayer and its variations for oxygen electrode reactions†
Received
7th September 2017
, Accepted 13th November 2017
First published on 27th November 2017
Abstract
To achieve renewable energy technologies, low-cost electrocatalysts for the oxygen reduction reaction (ORR) and the oxygen evolution reaction (OER) are required to replace Pt and IrO2/RuO2 catalysts. Based on density functional theory, the catalytic activity of TM3(HHTP)2 (2,3,6,7,10,11-hexahydroxytriphenylene) monolayer and its variations (TMX4, where TM = Fe, Co, Ni, X = O, S, Se) for bifunctional ORR/OER have been investigated. The adsorption ability is dominated by the metal center, in the order of Fe > Co > Ni while the ligand shows the minor contribution. Due to the presence of linear relations between the intermediates, the activity of TMX4 for the ORR/OER follows a dual volcano curve as a function of the OH adsorption strength. Considering the overpotential, CoO4 and CoS4 possess superior bifunctional activity, implying their promise as candidates for the oxygen electrode reaction. This systematical work may open new avenues for the development of high-performance non-PGM catalysts for practical applications of ORR and OER.
1. Introduction
There is growing interest in oxygen electrochemistry as conversions between O2 and H2O play important roles in renewable energy technologies, such as the rechargeable air based battery and devices that require two key electrochemical reactions, oxygen reduction (ORR) and oxygen evolution (OER).1,2 The electrocatalytic oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) play key roles in such renewable energy devices.3 The current spectrum of catalysts utilized for these fundamental electrochemical reactions are Pt for the ORR and IrO2 for applications in the OER.4–6 Their “rare earth” status and associated high cost renders them less than ideal materials for incorporation into commercialization. In addition, the use of two different single function catalysts for the ORR and OER, respectively, makes the air cell significantly more complex as it requires the combination of three electrodes. In this regard, the development of active and affordable bifunctional electrocatalysts remains a challenging task.1
As alternatives, great efforts have been devoted to the development of the functional carbon based materials with specific atomic configuration where the heteroatom-doped, such as the nonmetallic as well as the nonprecious transition metal elements, would activate the inert C material and further boost the ORR/OER activity.7–9 Typical example is shown by J. D. Baran et al. that phthalocyanines, porphyrins and their variations with the active sites composed of TMN4 motif could be acted as the bifunctional catalysts.10 Furthermore, S. Z. Qiao et al. demonstrate that the TMN2 embedded in g-C3N4 promotes the oxygen electrode reactions.2 Based on the mentioned results, the performance is obviously tuned by the selection of TM atom. However, limited investigations have been focused on the influence of the different TM/ligand combination on the activity.
The metal–organic framework (MOF) provide the abundant active sites due to the structural flexibility where the high level of individually coordinated metal and the wide selection of building block.11,12 Besides the TM coordinated with N ligand, other combinations within MOF have been experimentally synthesized,13–16 such as the uniform distribution of TMS4 and TMO4 motifs,14,16 which could offer the prototype for the investigation of the TM/ligand effect. On the other hand, inspired by the attractive bifunctional electrocatalysis exhibited by the TM dichalcogenides,17–20 the active centers consisted by the TM coordinated to the O/S/Se atoms have raised our attention as the ORR/OER electrode.
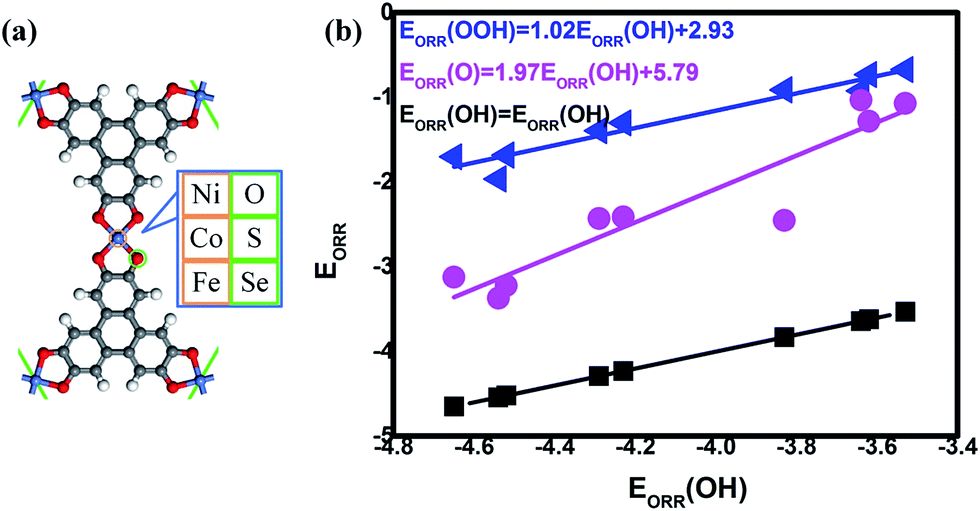
To classify the effect of the mentioned combinations, the primary consideration is the theoretical model where the TM3(HHTP)2 monolayer is selected as the prototype in our investigation.16 In the regard, DFT calculations are used within the electrochemical framework to analyze the ORR/OER reaction. The TM3(HHTP)2 prototype and its variations have been systemically studied to illustrate the critical role of the metal/ligand combination, where the schematic monolayer structures are shown in Fig. 1(a). The corresponding stability of the reaction intermediates is considered, which allows for the evaluation of the free energy and overpotentials. Based on the information, the bifunctional candidates are screened out by a thorough comparison.
 |
| | Fig. 1 The schematic structure (a) and the corresponding adsorption energy of ORR/OER intermediates (b). | |
2. Computational method
All calculations are performed within the DFT framework as implemented in DMol3 code.21,22 The generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) functional is employed to describe exchange and correlation effects.23 The DFT semi-core pseudopots (DSPP) core treat method is implemented for relativistic effects, which replace core electrons by a single effective potential and introduce some degree of relativistic correction into the core.24 The double numerical atomic orbital augmented by a polarization function (DNP) is chosen as the basis set.21 A smearing of 0.005 Ha (1 Ha = 27.21 eV) to the orbital occupation is applied to achieve accurate electronic convergence. In order to ensure high-quality results, the real-space global orbital cutoff radius is set as high as 5.2 Å. In the geometry structural optimization, the convergence tolerances of energy, maximum force and displacement are 1.0 × 10−5 Ha, 0.002 Ha Å−1 and 0.005 Å, respectively. The spin-unrestricted method is used for all calculations. A conductor-like screening model (COSMO) was used to simulate a H2O solvent environment for all calculations,25 which is a continuum model where the solute molecule forms a cavity within the dielectric continuum. The DMol3/COSMO method has been generalized to periodic boundary cases. The dielectric constant is set as 78.54 for H2O. Some previous results have shown that this implicit solvation model is an effective method to describe solvation.15,26 The 15 Å-thick vacuum is added to avoid the artificial interactions between the nanosheet and its images.
In the reaction energy landscape, all ORR/OER intermediates are described as proton/electron (H+ + e−) transfers.4,5 The adsorption energy of the corresponding intermediates are calculated by the following,10
| | |
EORR(OOH) = EOOH − Esubstrate − EO2 − (H+ + e−)
| (1) |
| | |
EORR(O) = EO − Esubstrate − 1/2EO2 + Hf(H2O)
| (2) |
| | |
EORR(OH) = EOH − Esubstrate − 1/2EO2 − (H+ + e−) + Hf(H2O)
| (3) |
where
Esys,
Esubstrate,
EH2O,
EO2 and
EH2 are the total energy of the adsorption systems, the TM
3(HITP)
2 monolayer, H
2O molecule, O
2 molecule and H
2 molecule, respectively.
EORR < 0 corresponds to an exothermic adsorption process.
To study the ORR/OER activity, the Gibbs free energy changes (ΔG) of the ORR elemental steps have been calculated according to the computational hydrogen electrode (CHE) model developed by Nørskov et al. where the chemical potential of proton/electron (H+ + e−) in solution is equal to the half of the chemical potential of a gas-phase H2.4 The ΔG for every elemental step can be determined as following:
| | |
ΔG = ΔE + ΔZPE − TΔS + ΔGpH + ΔGU
| (4) |
where Δ
E is the electronic energy difference based on DFT calculations, ΔZPE is the change in zero point energy,
T is the temperature (equal to 298.15 K here), Δ
S is the change in the entropy, and Δ
GpH and Δ
GU are the free energy contributions due to variation in pH value (pH is set as 0 in acid medium) and electrode potential
U, respectively. In order to decrease the calculation consumption, the approximate correction ΔZPE −
TΔ
S to Δ
E (0.05/0.30/0.35 eV of O*/OH*/OOH*) are used for constructed the Δ
G. As OER are reverse process of the ORR, the corresponding Δ
G of OER intermediates are calculated in the following equation:
| | |
ΔGOER(M) = ΔGORR(M) + 4.92
| (5) |
The thermodynamic CHE model has been applied to interpret the experimental data and design the novel electrocatalysts for metal, oxides as well as carbon-based materials.10,15,27–32 Besides, the present computational method has been applied to illustrate the ORR mechanism of the TM3(HITP)2 (HITP = hexaiminotriphenylene) monolayer. Therein, the 2e− mechanism is predicted to be prevalent for Ni3(HITP)2 system owing to the insufficient O2 activation, which is in accordance with the experimental data established by E. M. Miner.33 Therefore, considered the structural similarity of the TM3(HITP)2 and TM3(HHTP)2 systems, the reliability of our calculation could be confirmed. However, it should be realized that the material stability under the harsh electrochemical environment has been neglected herein. Despite the bifunctional ORR/OER electrocatalytic candidates have been screened out based on our theoretical trend, further performance needs the experimental confirmation.
3. Results and discussion
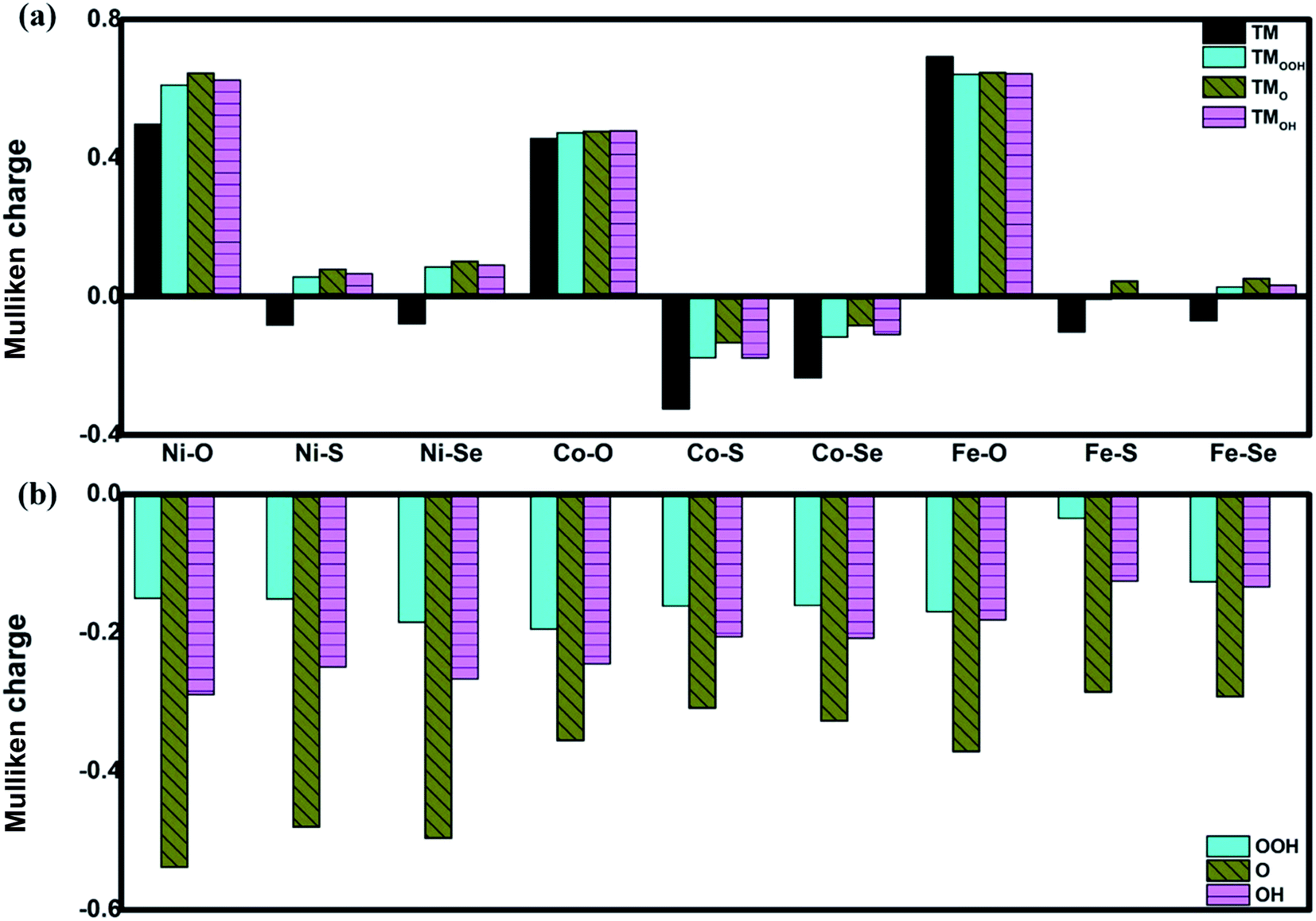
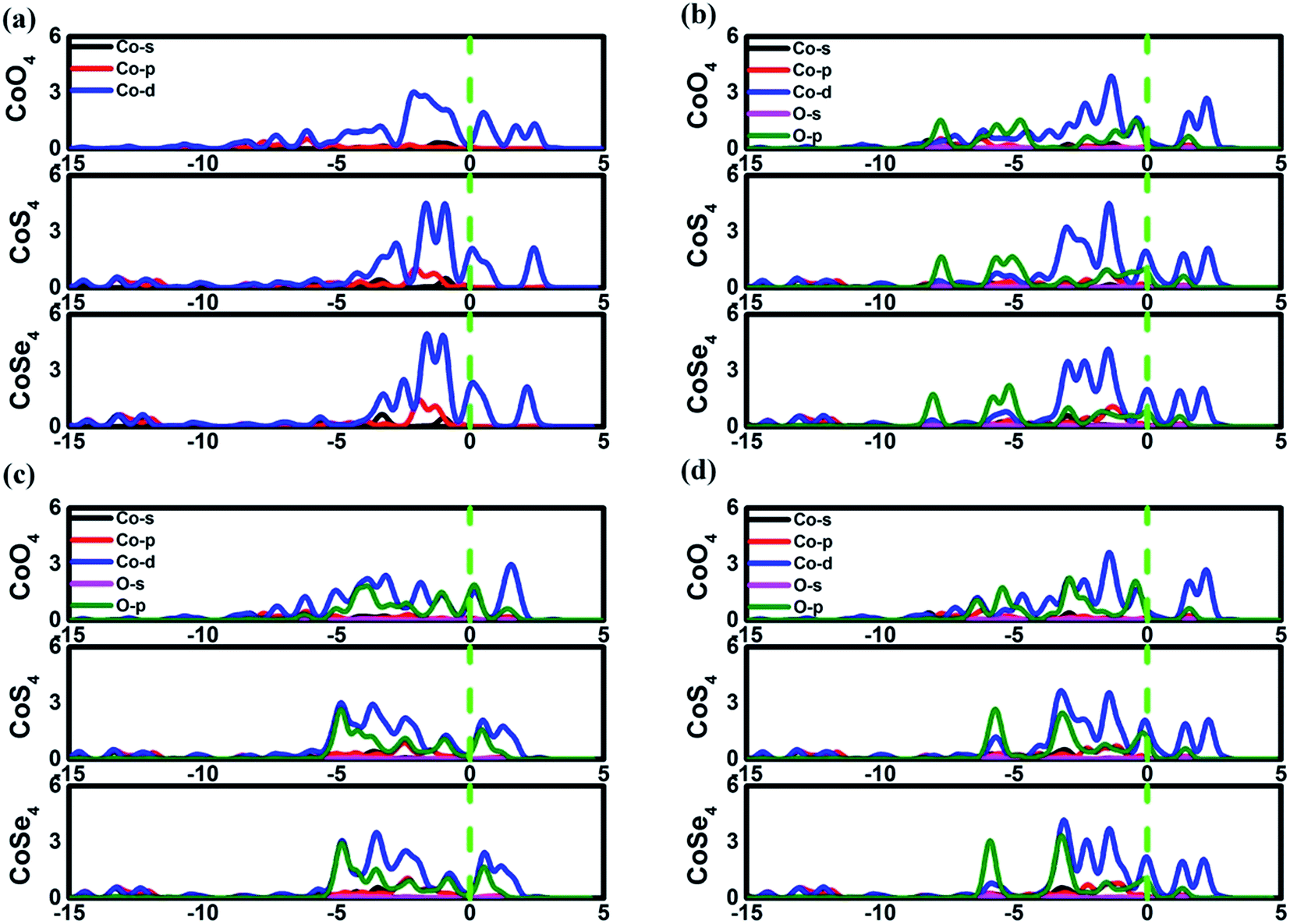
The favorable adsorption properties of the ORR/OER intermediates are the prerequisite for the reaction proceeding. The corresponding adsorption energies are tabulated in Table 1. It should be point out that the values do not signify the absolute strength of the intermediates adsorption. As shown, the adsorption energy decreases monotonically with increasing the d-electron in the valence shell of the metal center, which could be accounted by the d-band model.34 That is, the adsorption ability is tuned by the variation of the metal center, following the order of Fe > Co > Ni.2,15,35 In order to reveal the ligand effect, the corresponding Mulliken charges of the TM active center as well as the ORR intermediates are plotted in Fig. 2 where positive and negative represent charge depletion and accumulation, respectively. As shown in Fig. 2(a), the charge is transferred from the TM atom to the C-based skeleton for the TMO4 systems without adsorption, indicating the charge depletion of the TM active center for NiO4, CoO4 and FeO4, respectively meanwhile the charge accumulation is found for the other two systems, especially for CoS4 and CoSe4. On the other hand, interacted with the ORR intermediates, the phenomenon of the charge depletion for NiO4, CoO4 and FeO4 in combination with the charge accumulation of CoS4 and CoSe4 are remained, being similar compared with the un-adsorbed systems. However, for NiS4, NiSe4, FeS4 and FeSe4, the generally tendency of the charge accumulation before adsorption changed toward to the charge depletion with ORR intermediates adsorption is observed. Herein, the ligand effect of the charge distribution possesses the similar behavior on the certain degrees for the TMS4 and TMSe4, as implied by Fig. 2(a). The point is further supported by the partially density of states (PDOS). For CoX4 as shown in Fig. 3, the appearance of the p-orbital ranged from −3 eV to the Fermi energy is observed for CoS4 and CoSe4, which is missed for the CoO4. Besides, the sharp double d-peaks are located at ∼−1.6 eV and −1.0 eV for the former systems, being different from the d-peaks of the CoO4, which confirms the variation of the d-orbital as the ligand charged. The similarity is found for NiX4 and FeX4, where the detail PDOS plots are shown in Fig. S1 and S2 of the ESI.† As discussed, the charge analysis as well as PDOS morphology demonstrates the complex ligand effect on the subtle electronic structure of the TM active center, being dependent on the TM element selection, which would lead to the variation of the adsorption energy. Taken Co combination as an example, the neglect variation is found for EORR(O) as the ligand changes. However, the EORR(OOH) and EORR(OH) are significantly weakened as the ligand varied from O/S to Se. The situation is completely different for the Ni/Fe combination. As shown in Fig. 2, due to the charge transfer, the ionic bonding between the O-containing species is formed.36 The charge accumulations of the Co active center and the O-containing species are observed, implying the presence of electrostatic repulsion, which is different from the situations between Ni/Fe and the ORR intermediates with the electrostatic attraction.37,38 It is plausibly accounted for the mentioned weakening phenomenon for CoX4. On the other hand, it should been noted that the covalent bonds are dominated by the interaction of the O-p orbital and the TM-d orbital, in combination with the minor contribution of the p–p orbital overlap demonstrated in Fig. 3 as well as Fig. S1 and S2.† Herein, both covalent bonds due to the overlap of orbitals and ionic bonds induced by the charge transfer influence the adsorption.36 Due to the difference of the electronic structures, the general trend of the ligand effect is difficultly summarized.
Table 1 The adsorption energy of ORR intermediates (EORR) and Gibbs free energies of the ORR/OER intermediates. All results are in units of eV
| |
EORR |
GOER |
GORR |
| OOH |
O |
OH |
OOH |
O |
OH |
OOH |
O |
OH |
| Ni–O |
−0.92 |
−1.03 |
−3.64 |
−0.57 |
−0.98 |
−3.34 |
4.35 |
3.94 |
1.58 |
| Co–O |
−1.39 |
−2.43 |
−4.29 |
−1.04 |
−2.38 |
−3.99 |
3.88 |
2.54 |
0.93 |
| Fe–O |
−1.70 |
−3.12 |
−4.65 |
−1.35 |
−3.07 |
−4.35 |
3.57 |
1.85 |
0.57 |
| Ni–S |
−0.67 |
−1.07 |
−3.53 |
−0.32 |
−1.02 |
−3.23 |
4.60 |
3.90 |
1.69 |
| Co–S |
−1.30 |
−2.41 |
−4.23 |
−0.95 |
−2.36 |
−3.93 |
3.97 |
2.56 |
0.99 |
| Fe–S |
−1.68 |
−3.22 |
−4.52 |
−1.33 |
−3.17 |
−4.22 |
3.59 |
1.75 |
0.70 |
| Ni–Se |
−0.73 |
−1.28 |
−3.62 |
−0.38 |
−1.23 |
−3.32 |
4.54 |
3.69 |
1.60 |
| Co–Se |
−0.91 |
−2.45 |
−3.83 |
−0.56 |
−2.40 |
−3.53 |
4.36 |
2.52 |
1.39 |
| Fe–Se |
−1.96 |
−3.37 |
−4.54 |
−1.26 |
−3.32 |
−4.24 |
3.66 |
1.60 |
0.68 |
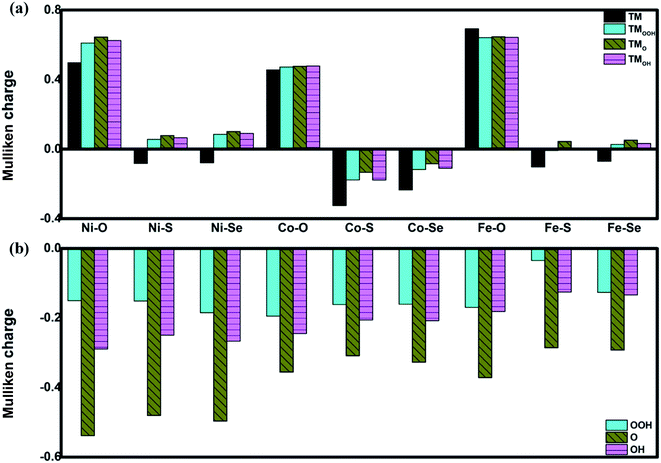 |
| | Fig. 2 The Mulliken charge of the TM active center (a) and the ORR intermediates (b). TM denotes the un-adsorbed TM atom while TMOOH, TMO and TMOH are the OOH, O and OH adsorbed TM atom, respectively. | |
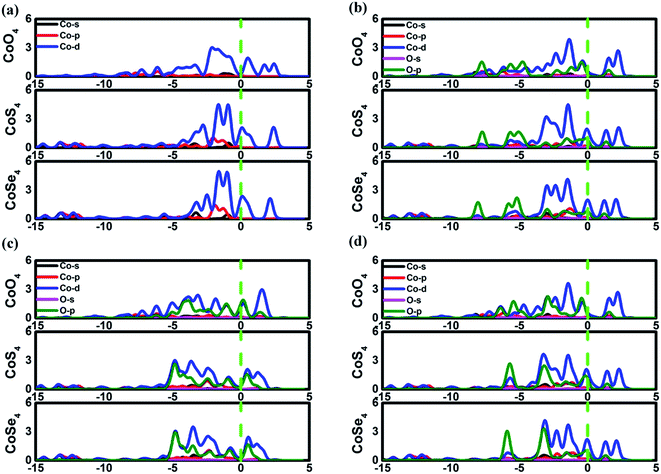 |
| | Fig. 3 The partially density of states of Co3(HHTP)2 and its variations. (a) is the un-adsorbed CoX4. (b), (c) and (d) are OOH, O and OH adsorption systems, respectively. | |
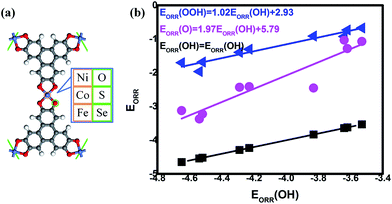
The data could be plotted as a function of OH adsorption, as shown in Fig. 1(b). From our results, the universal linear relationships between the ORR intermediates are clearly observed, which is in agreement with the previous studies.10,15,35,39 That is,
| | |
EORR(OOH) = 1.02EORR(OH) + 2.93
| (6) |
| | |
EORR(O) = 1.97EORR(OH) + 5.79
| (7) |
Compared with the previous data of the porphyrins analogues,10 it is found that the slopes of the fitting lines are comparable. It should be noted that the data deviation of CoSe4 [EORR(O) = −2.45 eV, EORR(OH) = −3.83 eV] from the scaling relation between EORR(O) and EORR(OH) is obvious. Such deviation could be observed in the C-based electrocatalysts,10,15,35 which is caused by the different electron transfer required for ORR intermediates adsorption (formally 2e/1e for O/OH, respectively).10 Herein, our Mulliken charge analysis shows the consistence with the statement where the adsorbed O possesses the more electrons compared with the adsorbed OH, indicated by the values shown in Fig. 2(b). Furthermore, the intercept of EORR(OOH) vs. EORR(OH) generally approaches 3.2 eV, regardless the catalytic materials.10,31,35,39 Herein, it is well-known that the mentioned scaling relations allow the dependence of the ORR activity on the adsorption strength that too strong means the poisoning of the O-containing species whereas too weak implies the insufficient activation ability, both of which is considered as the origin the ORR overpotential.4,40
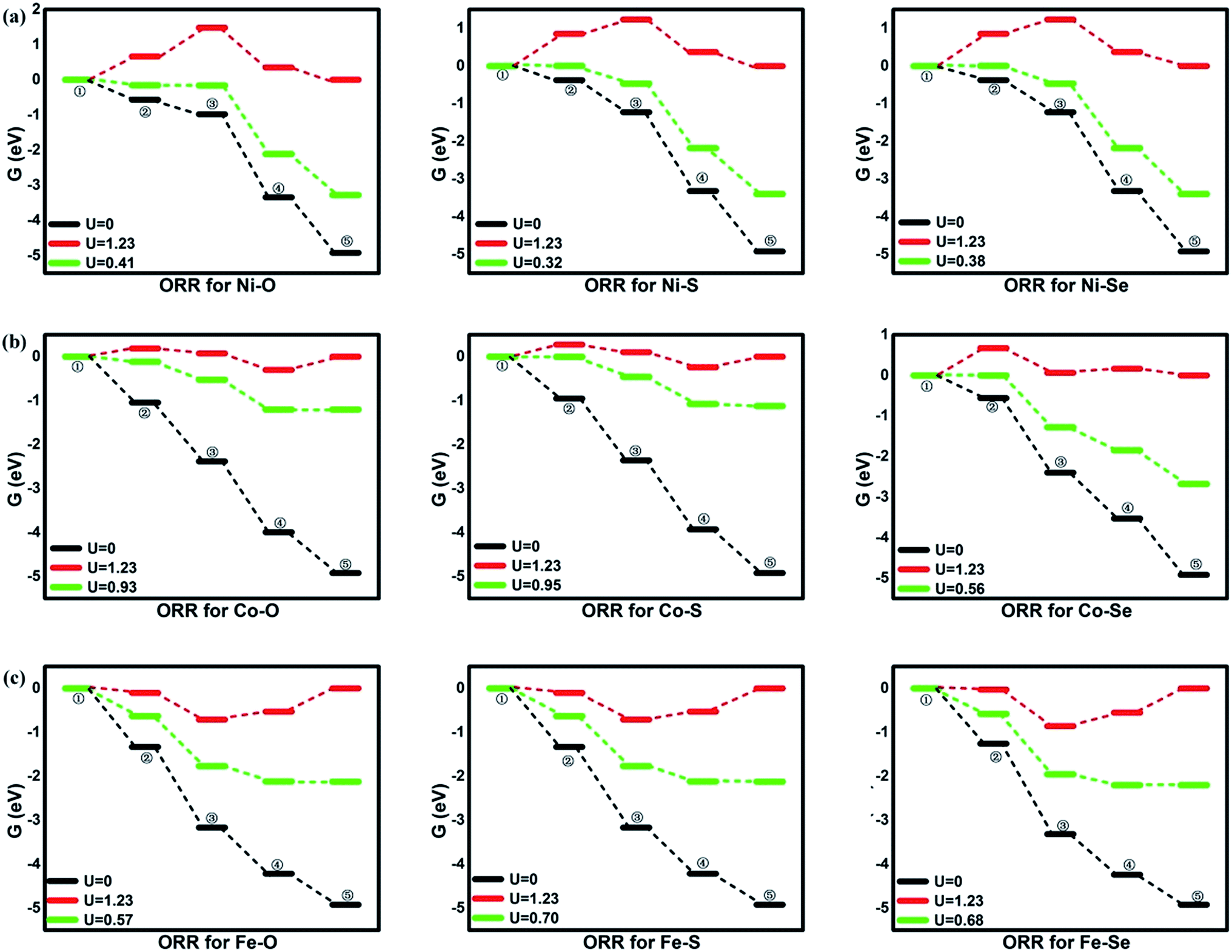
To evaluate the activity of the mentioned systems, the OOH associative mechanisms are taken into consideration with the elemental steps Ri listing in the following,10,35 where asterisks denote active TM sites. Due to the small barrier of proton transfer, which could be ignored at high applied voltages, our attentions are only are focused on the reaction energies.4,15,35,41 The corresponding free energy value G is analyzed and depicted in Fig. 4. As shown by the following equations, the four-electron ORR pathways in OOH association mechanism can proceed through OOH formation (R1), O formation (R2), OH formation (R3) and H2O formation (R4).
| | |
O2(g) + * + (H+ + e−) → OOH*,
| (R1) |
| | |
OOH* + (H+ + e−) → O* + H2O(l),
| (R2) |
| | |
O* + (H+ + e−) → OH* + H2O(l),
| (R3) |
| | |
OH* + (H+ + e−) → 2H2O(l).
| (R4) |
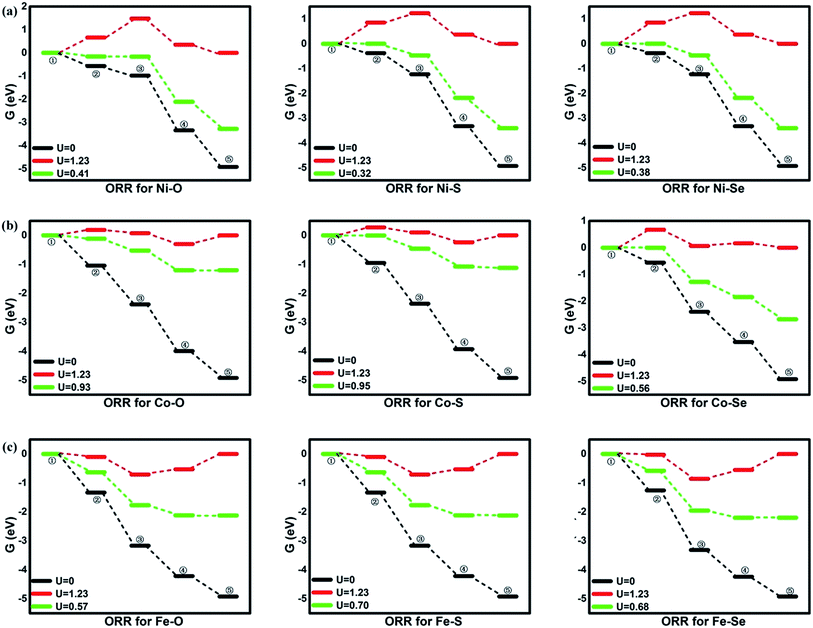 |
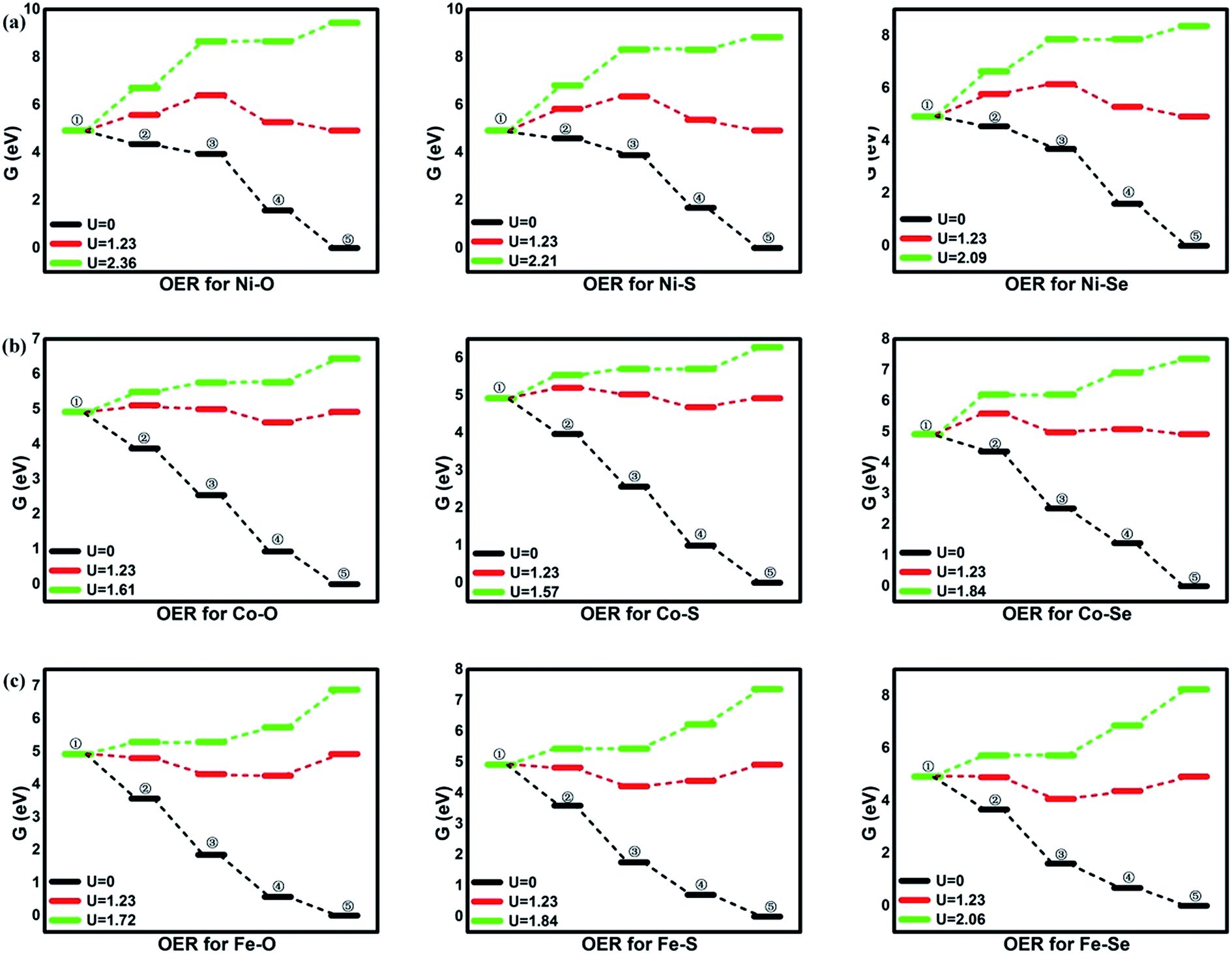
| | Fig. 4 The free energy of ORR. ①: O2 + 4(H+ + e−); ②: *OOH + 3(H+ + e−); ③: *O + H2O + 2(H+ + e−); ④: *OH + H2O + (H+ + e−); ⑤: 2H2O. | |
From the figures, for NiO4 monolayer, the whole elemental steps are exothermic at the potential U of 0 V, indicating the thermodynamic favor. However, as the U is raised to 1.23 V, the situation is changed that (R1) and (R2) become endothermic, respectively, implying the ORR reaction would be not proceeded spontaneously. For clearly observation, the ΔG values are gathered in Table 2. Herein, the rate-determining step (RDS) with the largest ΔGmax value could be acted as a measure of the catalyst activity.4 The RDS is located at (R2) with ΔGmax of 0.82 eV for NiO4 monolayer at 1.23 V. Furthermore, other catalytic materials possess good activity at 0 V without the endothermic reaction steps while the unfavorable thermodynamics are observed at 1.23 V, being similar with NiO4 monolayer. However, it should be noted that two different RDS could be identified. That is, (R1) is for NiS4, NiSe4, CoS4, CoSe4 with the ΔGmax of 0.91, 0.28, 0.85 and 0.67 eV while (R4) is for CoO4, FeO4, FeS4, FeSe4 with the ΔGmax of 0.30, 0.66, 0.53 and 0.55 eV, respectively.
Table 2 Gibbs free energies changes of the elemental steps for ORR at the potential U of 0 and 1.23 V
| |
U = 0 V |
U = 1.23 V |
| R1 |
R2 |
R3 |
R4 |
R1 |
R2 |
R3 |
R4 |
| Ni–O |
−0.57 |
−0.41 |
−2.36 |
−1.58 |
0.66 |
0.82 |
−1.13 |
−0.35 |
| Co–O |
−1.04 |
−1.34 |
−1.61 |
−0.93 |
0.19 |
−0.11 |
−0.38 |
0.30 |
| Fe–O |
−1.35 |
−1.72 |
−1.28 |
−0.57 |
−0.12 |
−0.49 |
−0.05 |
0.66 |
| Ni–S |
−0.32 |
−0.71 |
−2.21 |
−1.69 |
0.91 |
0.52 |
−0.98 |
−0.46 |
| Co–S |
−0.95 |
−1.40 |
−1.57 |
−0.99 |
0.28 |
−0.17 |
−0.34 |
0.24 |
| Fe–S |
−1.33 |
−1.84 |
−1.05 |
−0.70 |
−0.10 |
−0.61 |
0.18 |
0.53 |
| Ni–Se |
−0.38 |
−0.85 |
−2.09 |
−1.60 |
0.85 |
0.38 |
−0.86 |
−0.37 |
| Co–Se |
−0.56 |
−1.84 |
−1.13 |
−1.39 |
0.67 |
−0.61 |
0.10 |
−0.16 |
| Fe–Se |
−1.26 |
−2.06 |
−0.92 |
−0.68 |
−0.03 |
−0.83 |
0.31 |
0.55 |
Based on the free energy profiles, the highest potential for the feasible thermodynamic ORR steps are obtained and its corresponding overpotential μORR are summarized in Table 3. The ORR activity follows the order of CoS4(0.28) ≈ CoO4(0.30) > FeS4(0.53) ≈ FeSe4(0.55) > FeO4(0.66) ≈ CoSe4(0.67) > NiO4(0.82) ≈ NiSe4(0.85) ≈ NiS4(0.91). From the data, the relative minor dependence of the activity on the coordinated ligand of the Ni and Fe active sites is observed. The corresponding μORR changes are less than 0.10 V and 0.15 V, respectively. However, for Co metal center, the μORR of the CoSe4 are much higher than those of CoS4 and CoO4, indicating the importance of the ligand selection. On the other hand, the effect of the metal center on the ORR activity is obvious. Generally, the inferior/moderate activities are found for the Ni/Fe combination. For the Co active centers, the superior performances of the CoO4 and CoS4 are found referred to the current Pt with the μORR of 0.45 V.4
Table 3 The overpotential of the intermediates for ORR and OER
| |
μORR |
μOER |
| Ni–O |
0.82 |
−1.13 |
| Co–O |
0.30 |
−0.38 |
| Fe–O |
0.66 |
−0.49 |
| Ni–S |
0.91 |
−0.98 |
| Co–S |
0.28 |
−0.34 |
| Fe–S |
0.53 |
−0.61 |
| Ni–Se |
0.85 |
−0.86 |
| Co–Se |
0.67 |
−0.61 |
| Fe–Se |
0.55 |
−0.83 |
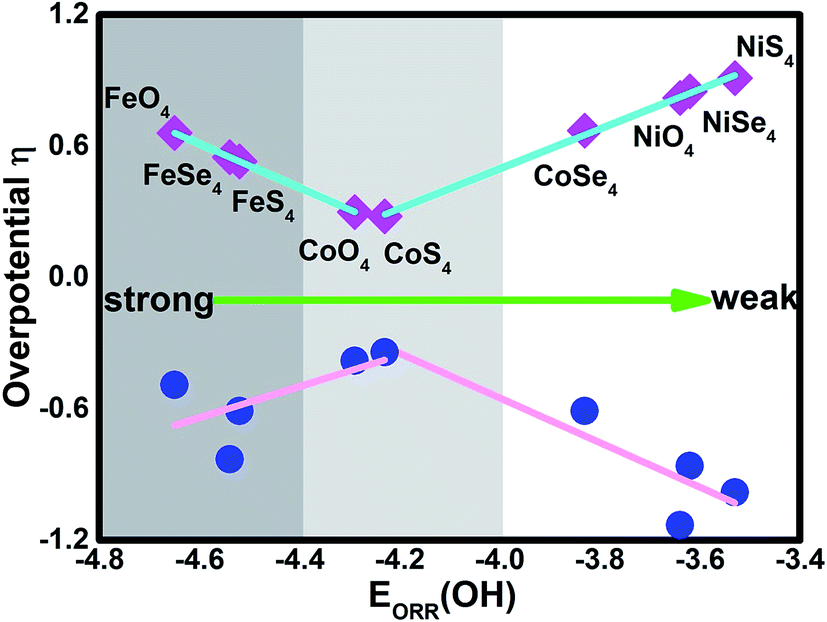
As discussed by the previous reports,10,15,35,39 the ORR activity depends on the adsorption of the intermediates. Due to the linear relationship between the adsorption of ORR intermediates and the OH, the overpotential μORR as a function of the EORR(OH) is described in Fig. 6. As the enhancement of the adsorption ability, the μORR reduces and then increase, demonstrating the classical volcano-shaped activity is found.2,4,10,35 For NiO4, NiS4, NiSe4 and CoSe4 with weak adsorption strength, the high μORR originates from the ineffective weakening the O–O coupling. For Fe combination located at the branch of the strong adsorption, the OH poisoning accounts for the increased μORR. Due to the suitable adsorption ability, the CoO4 and CoS4 are situated at the apex of the volcano curve. Our results are in accordance with the previous reports that the bond strength should be compromised for the effective ORR catalysts on the basis of Sabatier principle.40
Besides the ORR activity, the OER activity is characterized in Fig. 5 where the reversed process of the OOH associative mechanisms is considered. The free energy of OER intermediates are obtained by the eqn (5) and the corresponding data are shown in Table 2. From our results, no OER activity could be found at 1.23 V where the reaction steps are thermodynamically hindered due to the endothermic characterization. The RDS are located at the OH* oxidation (the reserve (R3)) for NiO4, NiS4, NiSe4, CoO4, CoS4 and the OOH* formation from the adsorbed O* (the reserve (R2)) for CoSe4, FeO4, FeS4, FeSe4, respectively. As shown, the unfavorable endothermic steps are changed to be exothermic as the potential U increases. The corresponding equilibrium potentials are in the order of NiO4(2.36) > NiS4(2.21) > NiSe4(2.09) ≈ FeSe4(2.06) > FeS4(1.84) = CoSe4(1.84) > FeO4(1.72) > CoO4(1.61) ≈ CoS4(1.57). Herein, for clear observation, the overpotential μOER are collected in Table 3 and its EORR(OH)-dependence is shown in Fig. 6 where the volcano-curve is roughly observed.2,10 Too weak or too strong EORR(OH) leads to the high μOER and then the inferior activity. The CoO4 and CoS4 possess the superior activity the minimum μOER with the values of 0.38 and 0.34 V, in comparison with the IrO2 of 0.56 V, respectively.5
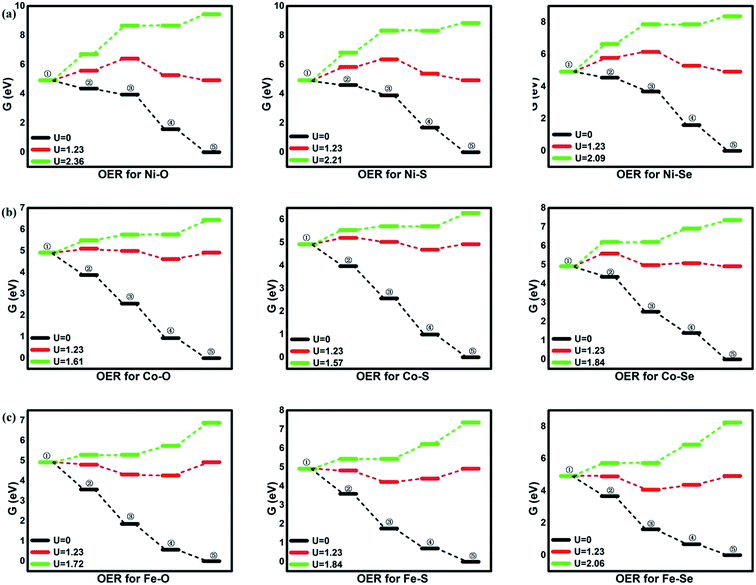 |
| | Fig. 5 The free energy of OER. ①: O2 + 4(H+ + e−); ②: *OOH + 3(H+ + e−); ③: *O + H2O + 2(H+ + e−); ④: *OH + H2O + (H+ + e−); ⑤: 2H2O. | |
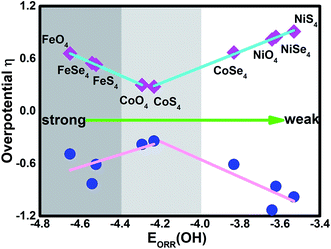 |
| | Fig. 6 The dual volcano plot of the overpotential as a function of the OH adsorption energy for ORR and OER. | |
Based on the mentioned results, CoX4 show the bests activity with the exception of CoSe4 due to the too weak capture ability of the reaction intermediates. Herein, the CoO4 and CoS4 are identified as the high efficient electrocatalysts to replace Pt for ORR and IrO2/RuO2 for OER. Besides, FeX4 provides better activity in comparison with NiX4. Generally, the reversible ORR/OER activity is mainly relied on the selection of the metal center. The different ligand gives slightly tuned. Herein, our stimulation provides the potential candidates for experimental synthesis. However, the structural stability as well as its conductivity are not concerned and out of our scope. Furthermore, due to the structure-dependence property, the model selection leads to distinct possibility for electrocatalysis application. Therefore, due to the limited work, extending our results to the whole MOF systems is not suitable.
4. Conclusion
Based on density functional theory, ORR/OER activity on a TMX4 monolayer has been systematically studied. It is found that the combination of the metal center and the ligand affects the ORR/OER bifunctional activity where the classical volcano-curve as a function of EORR(OH) is roughly observed. Furthermore, based on the overpotential obtained from the free energy profiles, the ORR activity follows the order of CoS4 ≈ CoO4 > FeS4 ≈ FeSe4 > FeO4 ≈ CoSe4 > NiO4 ≈ NiSe4 ≈ NiS4 while the OER activity follows the order of CoS4 ≈ CoO4 > FeO4 > CoSe4 = FeS4 > FeSe4 ≈ NiSe4 > NiS4 > NiO4, suggesting that CoO4 and CoS4 exhibit the superior catalytic activity. These results may serve as guidance for rational material design and synthesis.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We acknowledge the supports from the National Natural Science Foundation of China (No. 21503097, 51631004), the Natural Science Foundation of Jiangsu (No. BK20140518).
References
- Y. Gorlin and T. F. Jaramillo, A Bifunctional Nonprecious Metal Catalyst for Oxygen Reduction and Water Oxidation, J. Am. Chem. Soc., 2010, 132, 13612–13614 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S. Z. Qiao, Molecule-Level g-C3N4 Coordinated Transition Metals as a New Class of Electrocatalysts for Oxygen Electrode Reactions, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- Y. Zhao, K. Kamiya, K. Hashimoto and S. Nakanishi, Efficient Bifunctional Fe/C/N Electrocatalysts for Oxygen Reduction and Evolution Reaction, J. Phys. Chem. C, 2015, 119, 2583–2588 CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist and J. R. Kitchin, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- J. Rossmeisl, Z. W. Qu, H. Zhu, G. J. Kroes and J. K. Nørskov, Electrolysis of water on oxide surfaces, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS.
- F. D. Kong, S. Zhang, G. P. Yin, N. Zhang, Z. B. Wang and C. Y. Du, Pt/porous-IrO2 nanocomposite as promising electrocatalyst for unitized regenerative fuel cell, Electrochem. Commun., 2012, 14, 63–66 CrossRef CAS.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Design of electrocatalysts for oxygen- and hydrogen-involving energy conversion reactions, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- Z. Chen, D. Higgins, A. Yu, L. Zhang and J. Zhang, A review on non-precious metal electrocatalysts for PEM fuel cells, Energy Environ. Sci., 2011, 4, 3167–3192 CAS.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J. J. Zou and Z. L. Wang, Electrocatalytic oxygen evolution reaction for energy conversion and storage: A comprehensive review, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- J. D. Baran, H. Grönbeck and A. Hellman, Analysis of Porphyrines as Catalysts for Electrochemical Reduction of O2 and Oxidation of H2O, J. Am. Chem. Soc., 2014, 136, 1320–1326 CrossRef CAS PubMed.
- A. Zarkadoulas, M. J. Field, V. Artero and C. A. Mitsopoulou, Proton-Reduction Reaction Catalyzed by Homoleptic Nickel–bis-1,2-dithiolate Complexes: Experimental and Theoretical Mechanistic Investigations, ChemCatChem, 2017, 9, 2308–2317 CrossRef CAS PubMed.
- J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Design and synthesis of metal–organic frameworks using metal–organic polyhedra as supermolecular building blocks, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC.
- T. Kambe, R. Sakamoto, K. Hoshiko, K. Takada, M. Miyachi, J. H. Ryu, S. Sasaki, J. Kim, K. Nakazato, M. Takata and H. Nishihara, π-Conjugated Nickel Bis(dithiolene) Complex Nanosheet, J. Am. Chem. Soc., 2013, 135, 2462–2465 CrossRef CAS PubMed.
- A. J. Clough, J. W. Yoo, M. H. Mecklenburg and S. C. Marinescu, Two-Dimensional Metal–Organic Surfaces for Efficient Hydrogen Evolution from Water, J. Am. Chem. Soc., 2015, 137, 118–121 CrossRef CAS PubMed.
- P. Zhang, X. Hou, L. Liu, J. Mi and M. Dong, Two-Dimensional π-Conjugated Metal Bis(dithiolene) Complex Nanosheets as Selective Catalysts for Oxygen Reduction Reaction, J. Phys. Chem. C, 2015, 119, 28028–28037 CAS.
- M. G. Campbell, S. F. Liu, T. M. Swager and M. Dincă, Chemiresistive sensor arrays from conductive 2D metal–organic frameworks, J. Am. Chem. Soc., 2015, 137, 13780–13783 CrossRef CAS PubMed.
- Q. Liu, J. Jin and J. Zhang, NiCo2S4@Graphene as a Bifunctional Electrocatalyst for Oxygen Reduction and Evolution Reactions, ACS Appl. Mater. Interfaces, 2013, 5, 5002–5008 CAS.
- P. Ganesan, M. Prabu, J. Sanetuntikul and S. Shanmugam, Cobalt Sulfide Nanoparticles Grown on Nitrogen and Sulfur Codoped Graphene Oxide: An Efficient Electrocatalyst for Oxygen Reduction and Evolution Reactions, ACS Catal., 2015, 5, 3625–3637 CrossRef CAS.
- J. A. Koza, Z. He, A. S. Miller and J. A. Switzer, Electrodeposition of Crystalline Co3O4—A Catalyst for the Oxygen Evolution Reaction, Chem. Mater., 2012, 24, 3567–3573 CrossRef CAS.
- Y. Liu, H. Cheng, M. Lyu, S. Fan, Q. Liu, W. Zhang, Y. Zhi, C. Wang, C. Xia, S. Wei, B. Ye and Y. Xie, Low Overpotential in Vacancy-Rich Ultrathin CoSe2 Nanosheets for Water Oxidation, J. Am. Chem. Soc., 2014, 136, 15670–15675 CrossRef CAS PubMed.
- B. Delley, An all-electron numerical method for solving the local density functional for polyatomic molecules, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, From molecules to solids with the DMol3 approach, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. Delley, Hardness conserving semilocal pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- T. Todorova and B. Delley, Wetting of paracetamol surfaces studied by DMol3–COSMO calculations, Mol. Simul., 2008, 34, 1013–1017 CrossRef CAS.
- Y. Sha, T. H. Yu, B. V. Merinov, P. Shirvanian and W. A. Goddard, Oxygen Hydration Mechanism for the Oxygen Reduction Reaction at Pt and Pd Fuel Cell Catalysts, J. Phys. Chem. Lett., 2011, 2, 572–576 CrossRef CAS.
- I. E. L. Stephens, A. S. Bondarenko, U. Grønbjerg, J. Rossmeisl and I. Chorkendorff, Understanding the electrocatalysis of oxygen reduction on platinum and its alloys, Energy Environ. Sci., 2012, 5, 6744–6762 CAS.
- D. H. Lee, W. J. Lee, W. J. Lee, S. O. Kim and Y. H. Kim, Theory, Synthesis, and Oxygen Reduction Catalysis of Fe-Porphyrin-Like Carbon Nanotube, Phys. Rev. Lett., 2011, 106, 175502 CrossRef PubMed.
- M. Favaro, L. Ferrighi, G. Fazio, L. Colazzo, C. Di Valentin, C. Durante, F. Sedona, A. Gennaro, S. Agnoli and G. Granozzi, Single and Multiple Doping in Graphene Quantum Dots: Unraveling the Origin of Selectivity in the Oxygen Reduction Reaction, ACS Catal., 2015, 5, 129–144 CrossRef CAS.
- Y. Jia, L. Zhang, A. Du, G. Gao, J. Chen, X. Yan, C. L. Brown and X. Yao, Defect Graphene as a Trifunctional Catalyst for Electrochemical Reactions, Adv. Mater., 2016, 28, 9532–9538 CrossRef CAS PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, Universality in Oxygen Evolution Electrocatalysis on Oxide Surfaces, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- X. Y. Lang, G. F. Han, B. B. Xiao, L. Gu, Z. Z. Yang, Z. Wen, Y. F. Zhu, M. Zhao, J. C. Li and Q. Jiang, Mesostructured Intermetallic Compounds of Platinum and Non-transition Metals for Enhanced Electrocatalysis of Oxygen Reduction Reaction, Adv. Funct. Mater., 2015, 25, 230–237 CrossRef CAS.
- E. M. Miner, T. Fukushima, D. Sheberla, L. Sun, Y. Surendranath and M. Dincă, Electrochemical oxygen reduction catalysed by Ni3(hexaiminotriphenylene)2, Nat. Commun., 2016, 7, 10942 CrossRef CAS PubMed.
- B. Hammer and J. K. Nørskov, Theoretical surface science and catalysis—calculations and concepts, Adv. Catal., 2000, 45, 71–129 CAS.
- F. Calle-Vallejo, J. I. Martínez and J. Rossmeisl, Density functional studies of functionalized graphitic materials with late transition metals for oxygen reduction reactions, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC.
- W. Liu, Y. F. Zhu, J. S. Lian and Q. Jiang, Adsorption of CO on Surfaces of 4d and 5d Elements in Group VIII, J. Phys. Chem. C, 2007, 111, 1005–1009 CAS.
- P. J. Feibelman, d-electron frustration and the large fcc versus hcp binding preference in O adsorption on Pt(111), Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56, 10532 CrossRef CAS.
- M. T. M. Koper and R. A. van Santen, Interaction of H, O and OH with metal surfaces, J. Electroanal. Chem., 1999, 472, 126–136 CrossRef CAS.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, Universality in Oxygen Reduction Electrocatalysis on Metal Surfaces, ACS Catal., 2012, 2, 1654–1660 CrossRef CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Nørskov, Alloys of platinum and early transition metals as oxygen reduction electrocatalysts, Nat. Chem., 2009, 1, 552–556 CrossRef CAS PubMed.
- D. H. Lim and J. Wilcox, Mechanisms of the Oxygen Reduction Reaction on Defective Graphene-Supported Pt Nanoparticles from First-Principles, J. Phys. Chem. C, 2012, 116, 3653–3660 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra09974f |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a,
H. Y. Liua,
X. B. Jiangb,
Z. D. Yua and
Q. Jiang
*a,
H. Y. Liua,
X. B. Jiangb,
Z. D. Yua and
Q. Jiang