
Fabrication of a high-efficiency hydrogen generation Pd/C3N5-K,I photocatalyst through synergistic effects of planar and spatial carrier separation†
Yanan
Gao
a,
Jingjing
Wang
a,
Jingxuan
Yang
a,
Yajie
Wang
a,
Wenjuan
Tian
 *c and
Bin
Liu
*ab
*c and
Bin
Liu
*ab
aInstitute of Molecular Science, Key laboratory of Chemical Biology of Molecular Engineering of Education ministry, Shanxi University, Taiyuan, 030006, China. E-mail: liubin@sxu.edu.cn
bScientific instrument center, Shanxi University, Taiyuan, 030006, China
cInstitute of Molecular Science, Nanocluster Laboratory, Shanxi University, Taiyuan, 030006, China. E-mail: Tianwenjuan@sxu.edu.cn
First published on 28th October 2024
Abstract
Nitrogen-rich graphitic carbon nitride (g-C3N5) has garnered significant attention in photocatalytic hydrogen production due to its unique physicochemical properties. However, g-C3N5 faces persistent challenges stemming from rapid carrier recombination. In this study, we successfully synthesized a Pd/C3N5-K,I photocatalyst through sequential doping of non-metallic heteroatoms and metal nanoparticles. The Pd/C3N5-K,I photocatalyst demonstrates a remarkable enhancement in the photocatalytic H2 evolution rate (2.9 mmol g−1 h−1), approximately 14 times higher than that of pure g-C3N5 (0.2 mmol g−1 h−1). Characterization and calculation analyses reveal that iodine doping into the g-C3N5 skeleton leads to the formation of planar C–I bonds, facilitating C3N5 layer electron–hole pair separation. The subsequent insertion of Pd nanoparticles between the layers leads to electron accumulation on C3N5-K,I, while resulting in hole concentration on Pd nanoparticles, thereby facilitating thorough spatial separation of electron–hole pairs. The strategic co-doping of iodine and palladium nanoparticles effectively restrains carrier recombination of g-C3N5 by connecting intra- and inter-layer interactions. This study constitutes a novel conceptual framework for CN-based photocatalysts, offering a promising approach to improve their performance in hydrogen production applications.
1. Introduction
Hydrogen energy has emerged as a leading alternative to increasingly depleted fossil fuels.1–3 Since the discovery by Fujishima and Honda in 1972 that TiO2 can decompose water to produce hydrogen (H2), extensive research efforts have been dedicated to exploring various photocatalysts, including TiO2, metal-oxo clusters, and carbon nitride.4–7 Among these photocatalysts, graphite-phase carbon nitride (g-C3N4) has attracted widespread attention due to its remarkable economic efficiency, excellent light stability, and tunable bandgap characteristics.8 Wang et al. initially demonstrated the photocatalytic performance of g-C3N4 in producing H2 under visible-light irradiation,9 and since then various modifications have been made to enhance its performance. For instance, Jin et al. developed a g-C3N4/NixMo1−xS2 photocatalyst with anchored Ni–Mo–S nanoparticles, which improved the efficiency of photocatalytic H2 evolution by regulating the charge separation process.10 Lai et al. modified g-C3N4 by S-doping to improve its light absorption ability and carrier separation efficiency.11 Yang et al. achieved an effective improvement in carrier separation efficiency by forming a Schottky barrier through close contact between Cu nanoparticles and g-C3N4.12 Notably, nitrogen-rich g-C3N5, with a narrower bandgap of 2.2 eV, was successfully synthesized by Vinu et al. using 3-amino-1,2,4-triazole as a precursor.13 It has shown enhanced vis-light response compared to g-C3N4, being activatable under ultraviolet, visible, and near-infrared irradiation.14–16 The recently synthesized PtCu–C3N5,17 Pt/C3N518 and CoOOH·CoOx/P-C3N519 have exhibited remarkable H2 production efficiency, highlighting the significant potential of g-C3N5 in photocatalytic applications. However, despite its potential, the inherent low electrical conductivity and suboptimal electronic structure of g-C3N5 impede the transfer of photoinduced charge carriers, limiting its photocatalytic performance.20–22 Therefore, constructing highly efficient g-C3N5-based catalysts to achieve better charge separation efficiency has emerged as a critical focus in the field of photocatalysis.To address the bottleneck in g-C3N5 photocatalysis, doping non-metallic heteroatoms with different electronegativities (e.g., boron, iodine, bromine, and sulfur) has been demonstrated as a vital approach to modulate the electronic states and chemical properties of g-C3N5. These dopants can generate trapping sites for electrons or holes, inhibiting the recombination process.23,24 The charge carrier distribution in heteroatom-doped g-C3N5 can generate an electric field and induce electron rearrangement, facilitating charge migration on the CN layers.25,26 However, while heteroatom doping benefits in-plane charge transfer, it has limited impact on charge transfer between adjacent layers.27 To significantly improve spatial charge transfer and exciton dissociation, incorporating appropriate metal nanoparticles as bridging sites between adjacent CN layers is crucial.28,29 Metal nanoparticles doped on CN supports enable efficient modification of the electronic structure and form directional charge-transfer channels.30,31 Extensive research has shown that introducing metal nanoparticles can establish a build-in electric field towards the metal, leading to efficient spatial light-induced charge separation.32–34 Although these strategies have been explored individually, few researchers have examined the synergistic effect of heteroatoms and metal nanoparticles on planar and spatial charge separation. Resolving both interlayer and in-plane charge transfer in g-C3N5 to achieve superior hydrogen production performance in photocatalysis is the primary motivation of this work.
Here, we designed a modified vis-light-catalyst by loading Pd nanoparticles onto g-C3N5 doped with potassium and iodine. This Pd/C3N5-K,I photocatalyst exhibits excellent hydrogen production properties even with fewer cocatalysts. Characterization results propose a rational chemical structure for the Pd/C3N5-K,I photocatalyst, where stable C–I bonds form in the g-C3N5 skeleton, and Pd nanoparticles are intercalated into the interlayer of C3N5-K,I. By analyzing the calculated electron–hole distribution, we innovatively proposed a synergistic mechanism for distinct charge transfer pathways within Pd/C3N5-K,I, thereby studying the photocatalytic hydrogen evolution reaction (HER) in depth. A small amount of I doped in g-C3N5 significantly improves planar carrier separation. When Pd nanoparticles are further introduced into the space among adjacent layers of C3N5-K,I, significant interlayer electron–hole pair separation is achieved. This mechanism indicates a significant aggregation of electrons on the C3N5-K,I layers, with the interlayer palladium nanoparticles exhibiting a hole-rich state. The synergy between planar and spatial carrier separation remarkably boosts charge carrier dynamics, notably enhancing HER activity.
2. Experimental
2.1. Materials
Triethanolamine (TEOA, ≥99.0%), H2PtCl6·6H2O and 3-amino-1,2,4-triazole (3-AT) were supplied by Aladdin. PdCl2 (AR) was purchased from Changchun Third Party Pharmaceutical Technology Co., Ltd. KI (AR) was obtained from Shanghai Yindian Chemical Co., Ltd. All chemicals were in accordance with the requirement of analytical reagents.2.2. Catalyst preparation
Bulk g-C3N5 was synthesized according to the literature.35 3 g of the 3-AT powder was calcined to 550 °C by 5 °C min−1 for 3 h and then cooled for 12 h in a muffle furnace.C3N5-K,I was synthesized using a thermal polycondensation synthesis method. In detail, 3-AT (200 mg) and KI (10, 60, 100 and 200 mg, respectively) were dispersed in deionized water (20 mL) under magnetic stirring. Next, the suspension was dried at a temperature of 60 °C for a duration of 12 h under vacuum conditions. Following this step, the sample was heated gradually to reach a temperature of 550 °C with an increment rate of 5 °C min−1 and maintained for 3 h to successfully synthesize C3N5-K,I.
As illustrated in Scheme 1, the Pd/C3N5-K,I catalyst was synthesized using low-temperature solvothermal treatment. In detail, C3N5-K,I (80 mg) was added to a solution of ethylene glycol (13 mL), followed by the addition of PdCl2 (3 mg mL−1, 0.7, 0.9 and 1.10 mL, respectively) into the above solution with continuous stirring. The obtained suspension was subsequently heated to 130 °C at a heating rate of 2 °C min−1, and maintained for another 4 h. The solution was thoroughly washed multiple times with absolute ethanol and deionized water after cooling down. Finally, the Pd/C3N5-K,I catalyst was successfully acquired after a 12 h drying process at 60 °C in a vacuum oven. As a control, Pd/C3N5 was synthesis adopting a similar method to the Pd/C3N5-K,I catalyst. C3N5-K,I was substituted with g-C3N5, and the other experimental procedures are identical to the synthesis of Pd/C3N5-K,I.
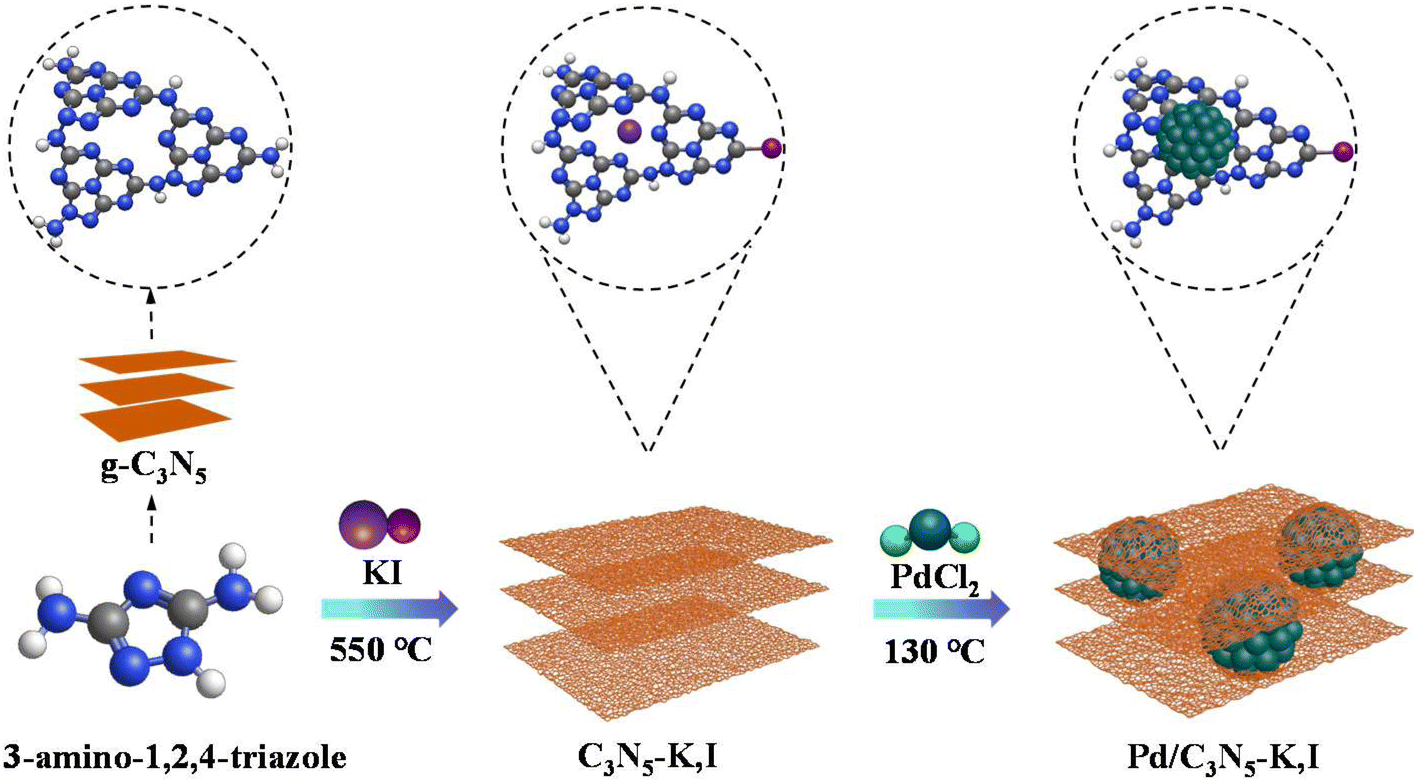 | ||
| Scheme 1 The synthesis process of Pd/C3N5-K,I. Gray, blue, white, pink, purple and dark green represent C, N, H, I, K and Pd. | ||
2.3. Catalyst characterization
The morphologies and structures were observed using a scanning electron microscope (SEM TESCAN MIRA4) and a high-resolution transmission electron microscope (HRTEM, JEOL-JEM 2100). Thickness analysis of the prepared samples was conducted employing an atomic force microscope (AFM Multimode8). The Brunauer–Emmett–Teller (BET) surface areas and pore volumes were determined using a Micrometrics ASAP 2460 automatic surface area and porosity analyser. The X-ray diffraction (XRD) patterns were analysed using a D8 ADVANCE A25 diffractometer. Fourier transform infrared (FT-IR) spectra ranging from 400 to 4000 cm−1 were acquired using an IR Nicolet IS 50 infrared spectrometer. The chemical state of the photocatalyst was investigated using X-ray photoelectron spectroscopy (XPS Thermo Scientific ESCALAB 250Xi). Electronic properties were measured at room temperature using a fluorescence spectrometer (PL F-2700). Time-resolved photoluminescence decay spectra (TRPD, λex = 405 nm, λem = 460 nm) were acquired using an FL920 transient fluorescence spectrophotometer. Electrochemical measurements were performed using the CHI-660E electrochemical workstation, employing 0.1 M Na2SO4 solution as the electrolyte. A dispersion of 5 mg of the photocatalyst in 2 mL of ethyl alcohol was sonicated, and then 40 μL of the resulting solution was dropwise applied onto an FTO substrate (conductive fluorine-doped tin oxide). The substrate served as the working electrode after natural drying. A saturated calomel electrode and platinum sheet were utilized as the reference and counter electrodes, respectively. Electrochemical impedance spectroscopy (EIS) measurements were performed over the frequency range of 0.01–105 Hz. UV-vis absorbance spectra for the samples were measured using a Hitachi UH4150 spectrophotometer. The content of metal Pd was detected by inductively coupled plasma mass spectrometry (ICP-MS, NexION 350X).2.4. Photocatalytic hydrogen evolution experiments
A closed gas circulation system (CEL-PAEM-D6) was employed for the analysis of photocatalytic H2-evolution results. In this experiment, 50 mg of the photocatalyst loaded with H2PtCl6 (45 μL,10 g L−1) were added to a 100 mL mixed solution (10 mL TEOA, 90 mL DI water). The reaction mixture was then transferred to a quartz reactor and maintained at a temperature of 6 °C using a condensate pump while being stirred magnetically. For the light source, we utilized an Xe lamp (300 W, Beijing Perfectlight, PLS–SEX300DUV), which was positioned approximately 15 cm away from the liquid surface to provide visible light irradiation. Throughout the course of the reaction lasting for 6 h and employing N2 as the carrier gas, samples were collected every hour and analysed using gas chromatography (GC-7920) to determine the quantity of produced H2. Additionally, we evaluated the photocatalytic stability over four consecutive reaction cycles. The apparent quantum yield (AQY) was assessed using an Xe lamp equipped with filters at 400, 420, 450 and 500 nm, while the other procedures remained identical to the H2 evolution conditions. The calculations were performed in accordance with eqn (1),6 where n is the amount of H2 production, NA is the Avogadro constant, P is the light intensity, S is the irradiation area, t is the illumination time, λ is the irradiation wavelength, h is the Planck constant, and c is the speed of light. | (1) |
2.5. Theoretical calculations
The geometry optimization and excited state were performed using the Gaussian 16 (Revision A03) software package at the B3LYP-D3(BJ)/Pd/Stuttgart/C,N,H,K,I/6-311G* level. There were no imaginary frequencies in the optimized structures. The implicit solvation model (SMD) was employed to describe the solvation effect, with water as the solvent. And the electron–hole analysis was carried out using Multiwfn software.3. Results and discussion
3.1. Morphology and structure analyses
Fig. 1a–c display SEM images illustrating the microstructures and morphologies of g-C3N5, C3N5-K,I and Pd/C3N5-K,I. Unlike the smooth surface of g-C3N5 (Fig. 1a), C3N5-K,I shows a flat surface with uniform pore distributions (Fig. 1b), likely due to I doping into the g-C3N5 skeleton. This structural change is further supported by BET analysis, which indicates that C3N5-K,I has an increased specific area and pore volume compared with g-C3N5 (Table S1, ESI†). After Pd metalation (Fig. 1c), distinct stratification is observed, attributed to the insertion of Pd into the C3N5-K,I layers. Elemental mapping (Fig. 1d) confirms the existence of all expected elements (C, N, K, I, and Pd) in the Pd/C3N5-K,I sample, demonstrating the uniform distribution of Pd and I species throughout the CN structure. | ||
| Fig. 1 (a)–(c) SEM images of g-C3N5, C3N5-K,I, and Pd/C3N5-K,I. (d) SEM elemental maps of C, N, K, I, and Pd in Pd/C3N5-K,I. (e)–(g) HRTEM images of Pd/C3N5-K,I. (h) AFM images of g-C3N5, C3N5-K,I, and Pd/C3N5-K,I. | ||
As shown in Fig. 1e, the HRTEM image indicates that Pd/C3N5-K,I has a multilayer staggered stack structure. Additionally, small Pd nanoparticles, with an average size of 5.0 nm, are uniformly attached to C3N5-K,I (Fig. 1f). A measured crystal lattice spacing of 0.224 nm is assigned to the (111) lattice plane of Pd (Fig. 1g and Fig. S1a and b, ESI†).36 These results demonstrate that uniform-sized Pd nanoparticles are homogeneously anchored on the unique hierarchical structures of C3N5-K,I.
The AFM topographical views of g-C3N5, C3N5-K,I and Pd/C3N5-K,I were analyzed, and the sample thickness was calculated using Gwyddion, open-source software. The average thicknesses of g-C3N5, C3N5-K,I and Pd/C3N5-K,I are approximately 1.5, 1.6, and 5.3 nm, respectively (Fig. 1h and Fig. S2, ESI†). Combined with TEM results, AFM observations reveal that Pd nanoparticles are successfully intercalated with C3N5-K,I monolayers, resulting in the formation of a multilayer staggered stack structure.
To elucidate the chemical structures, FT-IR spectra of the samples were recorded (Fig. 2a). Characteristic peaks at 810 and 891 cm−1 are associated with the vibration of tris-s-triazine units.37 The absorption band around 1200–1700 cm−1 is attributed to the stretching modes of C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N heterocycles.38 A broad band at 3000–3300 cm−1 is observed due to N–H stretching vibrations of uncondensed amino groups or adsorbed water molecules.39 These spectra confirm that C3N5-K,I and Pd/C3N5-K,I retain the fundamental molecular structure of g-C3N5.
N heterocycles.38 A broad band at 3000–3300 cm−1 is observed due to N–H stretching vibrations of uncondensed amino groups or adsorbed water molecules.39 These spectra confirm that C3N5-K,I and Pd/C3N5-K,I retain the fundamental molecular structure of g-C3N5.
 | ||
| Fig. 2 (a) FT-IR spectra of g-C3N5, C3N5-K,I, and Pd/C3N5-K,I. (b) XRD patterns of g-C3N5, C3N5-K,I, and Pd/C3N5-K,I. (c) XPS survey spectra of g-C3N5 and Pd/C3N5-K,I. (d) The XPS spectra of C 1s regions of g-C3N5 and Pd/C3N5-K,I. (e) The XPS spectra of N 1s regions of g-C3N5 and Pd/C3N5-K,I. (f) The XPS spectra of Pd 3d regions of Pd/C3N5-K,I. | ||
XRD patterns were employed to investigate the crystal structures (Fig. 2b). All materials exhibit two broad peaks at 13° and 27.3°, corresponding to the (100) and (002) planes of g-C3N5, respectively.40 These peaks are attributed to the interlayer stacking of triazine and conjugated aromatic units in graphitic materials,41 corroborating the infrared spectroscopy results. Notably, Pd/C3N5-K,I displays four additional characteristic peaks at 40.1, 46.4, 68.2, and 82°, corresponding to the Pd (111), Pd (200), Pd (220), and Pd (311) planes, respectively. This indicates that the aggregated Pd atoms are in a metallic state.42,43 These findings demonstrate the successful incorporation of Pd nanoparticles into the C3N5-K,I layers.
To investigate the chemical valence states and elemental compositions, XPS analyses of Pd/C3N5-K,I and g-C3N5 were conducted. The binding energies were calibrated by referencing the C 1s peak at 284.8 eV. The presence of C, N, K, I, and Pd in the Pd/C3N5-K,I sample corroborates the EDS elemental analysis results (Fig. 2c). The high-resolution C 1s spectrum (Fig. 2d) of g-C3N5 is deconvoluted into three peaks at 284.8, 286.27, and 288.33 eV, corresponding to C–C bonds,44 the CN bond,45 and sp2-hybridized C bonded to nitrogen atoms within triazine rings,46 respectively. In contrast, the C 1s spectrum of Pd/C3N5-K,I exhibits four individual peaks at 284.8, 285.97, 286.65, and 288.35 eV. The additional peak at 285.97 eV is ascribed to the formation of C–I bonds within the Pd/C3N5-K,I structure. Previous research has shown that specific C atoms in g-C3N5 with low electron density tend to react with negatively charged I atoms to form C–I covalent bonds.47 This is further corroborated by the I 3d XPS spectrum (Fig. S3a, ESI†), where lower C–I peaks appear at 629.95 (I 3d3/2) and 618.51 eV (I 3d5/2) compared with those in iodine salts.48,49 As illustrated in Fig. 2e, the N 1s spectra of both g-C3N5 (398.83/400.17/401.41 eV) and Pd/C3N5-K,I (398.85/400.30/401.30 eV) can be attributed to C–NC, N–(C)3, and C–NH(x) groups, respectively.50 Notably, upon loading Pd nanoparticles onto C3N5-K,I, the C and N atoms shift to higher binding energies, which is favorable for electron transfer in Pd/C3N5-K,I.51 The Pd 3d orbital of Pd/C3N5-K,I was fitted with two components (Fig. 2f). The peaks at 334.67 (Pd 3d5/2) and 340.25 eV (Pd 3d3/2) are assigned to Pd0.52,53 These peaks are accompanied by higher energy satellite peaks at 336.15 and 341.60 eV, respectively. In summary, the XPS results provide strong evidence for the presence of C–I bonds and Pd nanoparticles in the Pd/C3N5-K,I structure.
3.2. Optical and photoelectrochemical properties
Fig. 3a presents PL emission spectra of the samples, which are crucial for understanding carrier trapping and recombination processes, as well as elucidating the photocatalytic mechanism. All samples exhibit a characteristic emission peak at near 460 nm, originating from the intrinsic band gap of g-C3N5. Notably, the PL intensity of C3N5-K,I is quenched compared to that of g-C3N5, suggesting that C–I bond formation suppresses charge recombination. Pd/C3N5-K,I displays the lowest PL intensity, attributed to the strong interaction between Pd nanoparticles and C3N5-K,I, which enhances charge carrier separation.54 | ||
| Fig. 3 (a) PL spectra of samples. (b) TRPL spectra of samples. (c) Photocurrent transient responses of samples. (d) EIS spectra of samples. (e) UV-vis DRS spectra of samples. (f) Band gaps of samples. | ||
TRPD spectra (Fig. 3b) corroborate the PL results, showing an attenuation trend similar to that of the PL spectra. The average fluorescence lifetime (τav) was fitted using a triexponential decay function. The τav values for g-C3N5, C3N5-K,I, and Pd/C3N5-K,I are 1.13, 1.11, and 1.09 ns, respectively. The shortest PL lifetime decay of Pd/C3N5-K,I indicates further enhanced charge carrier separation.
To investigate the charge transfer mechanism in depth, transient photocurrent response experiments were conducted over three on/off cycles under vis-light irradiation. g-C3N5 exhibits very low photocurrent densities, indicating a limited electron migration capacity (Fig. 3c). In contrast, Pd/C3N5-K,I demonstrates a significantly higher photocurrent response intensity, suggesting more efficient electron–hole pair separation. This enhancement can be ascribed to the rapid electron transfer from Pd nanoparticles to the g-C3N5 surface, establishing a Schottky contact.55
EIS was employed to further elucidate charge carrier recombination processes. The radius of the semicircle in the Nyquist plot reflects the interfacial charge transfer resistance, which is related to carrier separation and transfer.56Fig. 3d shows that Pd/C3N5-K,I exhibits the smallest arc radius compared to g-C3N5 and C3N5-K,I, indicating the lowest resistance among these samples. This suggests that Pd/C3N5-K,I possesses superior electroconductivity, with Pd nanoparticles facilitating electron conduction and enhancing electron–hole pair separation efficiency.
The optical absorption characteristics were analyzed using UV-vis DRS, as shown in Fig. 3e. All samples demonstrated vis-light responsiveness, confirming that they are vis-light-driven photocatalysts. The significant red-shift in absorption for C3N5-K,I and Pd/C3N5-K,I was advantageous for improving photocatalytic reactions.57 Notably, Pd/C3N5-K,I exhibited the highest vis-light absorption, attributed to the uniform distribution of Pd nanoparticles on the C3N5-K,I surface. The band gap energies of the samples were determined by applying the Kubelka–Munk equation to the UV-vis DRS spectra (Fig. 3f).58 The band gap (Eg) values of C3N5-K,I (1.66 eV) and Pd/C3N5-K,I (0.90 eV) were narrower compared to that of pure g-C3N5 (1.69 eV), indicating that I and Pd nanoparticles can modulate the electronic structure of g-C3N5.59
The band diagram of the semiconductors was constructed using Mott–Schottky plots and VB-XPS measurements, which are commonly employed to determine the energy levels of the conduction band (CB) and valence band (VB).60 As shown in Fig. 4a–c, all photocatalysts are n-type semiconductors, as indicated by their positive slopes.61 The CB levels of g-C3N5, C3N5-K,I and Pd/C3N5-K,I are determined to be −0.44, −0.53, and −0.55 V, respectively. The VB-XPS measurements in Fig. 4d confirm the VB values of g-C3N5 (1.15 V), C3N5-K,I (1.13 V), and Pd/C3N5-K,I (0.35 V). Consequently, the energy level structures of the photocatalysts can be derived, as illustrated in Fig. 4e. Notably, the CB potential of Pd/C3N5-K,I is more negative than that of g-C3N5, indicating that more photogenerated electrons will accumulate in the CB under vis-light irradiation, which is highly beneficial for the HER.62
 | ||
| Fig. 4 Mott–Schottky plots of g-C3N5 (a), C3N5-K,I (b), and Pd/C3N5-K,I (c). (d) Valence band XPS spectra of samples. (e) Estimated bandgaps of samples. | ||
3.3. Photocatalytic H2 evolution performance
As illustrated in Fig. 5a, pure g-C3N5 exhibits a modest hydrogen production performance of 53 μmol, which is attributed to the rapid recombination of carriers. Upon modification with KI, the photocatalyst displays a significantly higher H2-evolution activity (250.3 μmol, Fig. S4, ESI†), indicating that I plays an essential role in the hydrogen production process. With further introduction of Pd nanoparticles into C3N5-K,I, a substantial enhancement in H2-production performance is observed. Specifically, the Pd/C3N5-K,I photocatalyst with a Pd loading of 2.25% demonstrates an optimal photocatalytic H2-generation activity of 753 μmol (the proportion is determined based on the ICP results, Table S2, ESI†). However, the decline in TEOA, which is critical for maintaining the stability of g-C3N5,63 along with the agglomeration of Pt, may result in a non-linear increase in H2 generation over time (Fig. S5, ESI†). As shown in Fig. 5b and c, the H2 evolution rate and AQY of 2.25% Pd/C3N5-K,I reach 2878 μmol g−1 h−1 and 6.07% (400 nm), respectively, which is relatively high compared to those reported for C3N5-based photocatalysts (more details in Fig. S6 and Table S3, ESI†). In a control experiment, the hydrogen production performance of Pd/C3N5 without I is significantly lower than that of 2.25% Pd/C3N5-K,I (Fig. S7, ESI†), demonstrating the important synergistic role of I and Pd nanoparticles in enhancing photocatalytic performance. The negligible reduction in photocatalytic H2 production of 2.25% Pd/C3N5-K,I after four cycles suggests its exceptional stability (Fig. 5d). The effects of four distinct sacrificial agents on H2 production by g-C3N5 are illustrated in Fig. 5e, demonstrating that TEOA is the most effective photocatalytic sacrificial agent among the tested materials. In Fig. 5f, the addition of a small amount of Pt (0.36 wt%) significantly enhances the H2 production performance for g-C3N5 and Pd/C3N5-K,I. However, further increasing the Pt content to 1 wt% resulted in only marginal improvements due to the formation of agglomerated Pt clusters. Therefore, 0.36 wt% Pt is considered the most suitable option in this study. | ||
| Fig. 5 (a) H2 generation amounts for different samples. (b) Photocatalytic H2 evolution rates of g-C3N5, C3N5-K,I, and Pd/C3N5-K,I. (c) Photocatalytic H2 evolution and AQY (400 nm) of C3N5-based catalysts reported in the literature (more details are given in Table S3, ESI†). (d) Photocatalytic stability tests of Pd/C3N5-K,I. (e) Different sacrificial agents on H2 production of g-C3N5. (f) H2 generation amounts for g-C3N5 and Pd/C3N5-K,I with different Pt contents after 6 hours. | ||
3.4. Photocatalytic reaction mechanism
To elucidate the charge separation process in Pd/C3N5-K,I, we conducted geometry optimization calculations for C3N5-K,I, Pd1/C3N5-K,I and Pd3/C3N5-K,I, where Pd1 and Pd3 represent the single and triple Pd atoms, respectively. Firstly, the electron–hole distributions in the excited states of C3N5-K,I, Pd1/C3N5-K,I and Pd3/C3N5-K,I were calculated. The absence of imaginary frequencies in the optimized structures indicated that all configurations are stable. Subsequently, the structural energies of single Pd atoms at various positions were computed. Although the lowest energy was found at a relatively central position, the energy differences among several positions were not significant (Fig. S8, ESI†). As shown in Fig. 6a, the electron–hole pairs in C3N5-K,I are significantly separated under the S0–S5, S0–S8 and S0–S9 excited states compared to g-C3N5,41 reflecting the pronounced effect of I atoms on in-plane electron migration in g-C3N5. When a single Pd atom is introduced into the interlayer of C3N5-K,I (Fig. 6b), it is evident that the Pd atom acts as an electron donor, facilitating the directed concentration of electrons within C3N5-K,I and achieving efficient spatial electron–hole pair separation. Consistent results were obtained when triple Pd atoms were introduced into the layers (Fig. 6c). In conclusion, the synergistic action of I and Pd nanoparticles effectively enhances the separation efficiency of both planar and spatial carriers. This calculation result supports the observed high photocatalytic H2 production performance. | ||
| Fig. 6 Electron–hole distribution of (a) C3N5-K,I, (b) Pd1/C3N5-K,I, and (c) Pd3/C3N5-K,I in different excited states (Pd1 and Pd3 represent the single atom and triple atoms, respectively). Blue and green isosurfaces represent the hole and electron distributions. | ||
The spatial distribution of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is closely related to the separation of photoexcited electrons and holes, significantly influencing photocatalytic efficiency. Due to the severe overlap of HOMO and LUMO distributions in g-C3N5 (Fig. 7a), photoexcited charges tend to transfer to the same locations, severely hindering electron–hole separation and resulting in poor photocatalytic performance.64 However, the introduction of KI leads to a clear redistribution of the HOMO and LUMO (Fig. 7b), facilitating efficient charge transfer. The electron density of the LUMO for Pd3/C3N5-K,I is concentrated in the C3N5-K,I plane, while in the HOMO, the electron density is primarily distributed around the Pd atoms (Fig. 7c). This confirms that electrons are transferred to the HOMO via Pd nanoparticles.65 Furthermore, the HOMO–LUMO energy (HLG) of g-C3N5 significantly decreases with the introduction of I and Pd, indicating an increase in the electrical conductance of C3N5-K,I and Pd/C3N5-K,I.66 Compared to g-C3N5, Pd/C3N5-K,I exhibits enhanced electron–hole separation and a narrowed HLG, while Pd maintains electron-supplying characteristics, making it highly promising for photocatalytic applications.
 | ||
| Fig. 7 Electronic structure of the optimized HOMO and LOMO of (a) g-C3N5, (b) C3N5-K,I and (c) Pd3/C3N5-K,I (Pd3 represent triple atoms). Blue and green isosurfaces represent the hole and electron distributions. | ||
In this study, we propose a plausible model for the Pd/C3N5-K,I photocatalyst under vis-light illumination to elucidate the reaction mechanism, as depicted in Scheme 2. Taking advantage of the synergistic effect of I and Pd nanoparticles, photogenerated electrons with sufficient energy are excited from the VB to the CB, leaving holes in the VB. Meanwhile, Pd nanoparticles and C3N5-K,I form an ohmic contact with an electric field directed towards Pd, facilitating electron migration from Pd to C3N5-K,I. Consequently, a substantial number of electrons from the Pd nanoparticles are effectively injected into C3N5-K,I, leading to a high concentration of electrons accumulating around C3N5-K,I, facilitated by the synergistic action of I. This enables the H+ (from H2O) adsorbed on the Pd/C3N5-K,I photocatalyst to readily obtain electrons from C3N5-K,I, thereby generating H2.
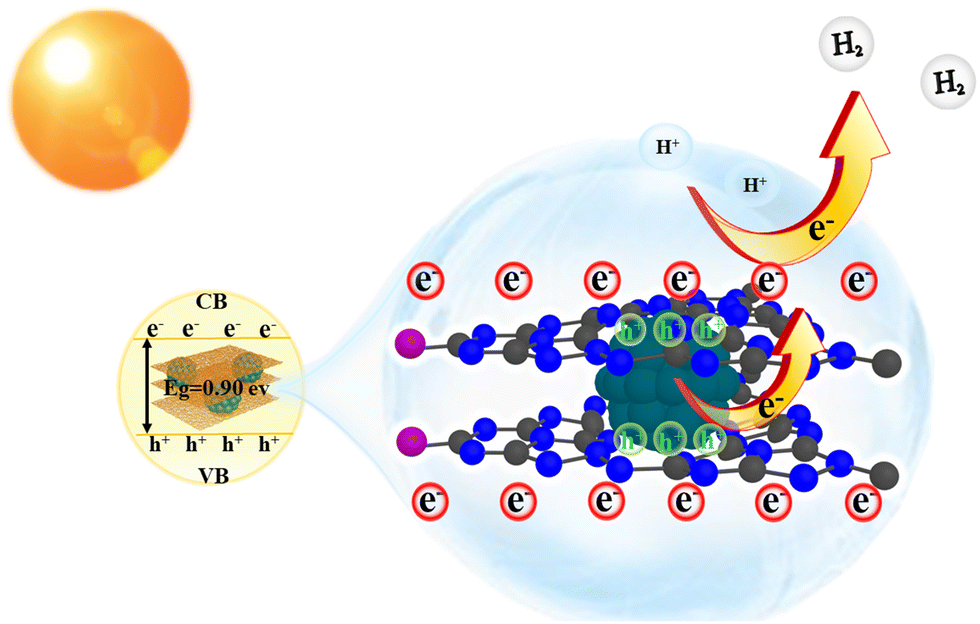 | ||
| Scheme 2 The probable photocatalytic HER mechanism of the Pd/C3N5-K,I photocatalyst. | ||
4. Conclusions
In this study, to solve the problem of rapid carrier recombination of g-C3N5, we successfully synthesized a novel photocatalyst comprising Pd nanoparticles embedded within K,I-doped g-C3N5. This novel conceptual framework exhibits two distinct charge transfer pathways both within the layers and across the interlayers. Our comprehensive characterization and computational analyses demonstrate that the strategic formation of C–I bonds enhances the photocatalytic activity through increased in-plane carrier mobility. Crucially, the incorporation of Pd nanoparticles facilitates the injection of electrons into the C3N5-K,I layer; meanwhile, Pd nanoparticles exhibit a hole-rich state, thereby enhancing the spatial separation of photoinduced charge carriers. The synergistic effect of these modifications optimizes electron–hole pair separation efficiency and enables the Pd/C3N5-K,I photocatalyst to achieve remarkably improved photocatalytic H2 generation performance. This significant improvement underscores the efficacy of our approach in addressing the limitations of conventional CN-based photocatalysts. Furthermore, this approach paves the way for efficient photocatalytic systems with applications in hydrogen production as well as sustainable energy conversion and environmental remediation.Author contributions
Yanan Gao: responsible for designing the study, developing the methodology, conducting investigations, performing formal analysis, and writing and preparing the original draft. Jingjing Wang: involved in calculations and formal analysis. Jingxuan Yang and Yajie Wang: contributed to investigations and formal analysis. Wenjuan Tian: engaged in calculations, formal analysis, and supervision of the project. Bin Liu: oversaw the project, provided guidance on writing and editing, managed the project, and secured funding.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We gratefully acknowledge the Fundamental Research Program of Shanxi Province (Grant No. 202303021221058).References
- W. Li, L. Lu, C. Cheng, N. Ren, S.-T. Yang and M. Liu, Bioresour. Technol., 2022, 364, 128069 CrossRef CAS.
- V. Nikolaou, G. Charalambidis, K. Ladomenou, E. Nikoloudakis, C. Drivas, I. Vamvasakis, S. Panagiotakis, G. Landrou, E. Agapaki, C. Stangel, C. Henkel, J. Joseph, G. Armatas, M. Vasilopoulou, S. Kennou, D. M. Guldi and A. G. Coutsolelos, ChemSusChem, 2021, 14, 961–970 CrossRef CAS.
- N.-N. Vu, S. Kaliaguine and T.-O. Do, ACS Sustainable Chem. Eng., 2019, 8, 853–863 CrossRef.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- Q. Lan, S. Jin, B. Yang, Q. Zhao, C. Si, H. Xie and Z. Zhang, Trans. Tianjin Univ., 2022, 28, 214–225 CrossRef CAS.
- M. Ren, J. Meng, Y. Yang, X. Zhang, G. Yang, L. Qin and Y. Guo, Appl. Catal., B, 2024, 345, 123680 CrossRef CAS.
- Y. Zou, S. Li, D. Zheng, J. Feng, S. Wang, Y. Hou and G. Zhang, Sci. China Mater., 2024, 67, 2215–2223 CAS.
- A. M. Sadanandan, M. Fawaz, N. P. Dharmarajan, M. Huš, G. Singh, C. I. Sathish, B. Likozar, Z. Li, A. M. Ruban, C.-H. Jeon, J.-H. Yang, P. Kumar and A. Vinu, Appl. Catal., B, 2024, 124701 Search PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS.
- H. Yang, Z. Jin, H. Hu, Y. Bi and G. Lu, Appl. Surf. Sci., 2018, 427, 587–597 CrossRef CAS.
- Y. Quan, R. Li, X. Li, R. Chen, Y. H. Ng, J. Huang, J. Hu and Y. Lai, Small, 2024, 2406576 CrossRef CAS.
- P. Song, D. Wang, B. Wang and P. Yang, J. Ind. Eng. Chem., 2024, 138, 472–480 CrossRef.
- G. P. Mane, S. N. Talapaneni, K. S. Lakhi, H. Ilbeygi, U. Ravon, K. Al-Bahily, T. Mori, D.-H. Park and A. Vinu, Angew. Chem., Int. Ed., 2017, 56, 8481–8485 CrossRef CAS PubMed.
- C. Fu, T. Wu, G. Sun, G. Yin, C. Wang, G. Ran and Q. Song, Appl. Catal., B, 2023, 323, 122196 CrossRef CAS.
- H. Li, R. Li, G. Liu, M. Zhai and J. Yu, Adv. Mater., 2023, 36 Search PubMed.
- C. Ma, Z. Yu, J. Wei, C. Tan, X. Yang, T. Wang, G. Yu, C. Zhang and X. Li, Appl. Catal., B, 2022, 319, 121951 CrossRef CAS.
- Q. Liu, X. Du, A. Zhou, J. Chen, X. Wang, R. Wang, M. Cheng, J. Hu, T. Wei, Y. Cui, F. Chen, W. Li, W.-L. Dai and B. Liu, J. Colloid Interface Sci., 2025, 678, 114–124 CrossRef CAS PubMed.
- E. Cui, Y. Lu, Z. Li, J. Sang, Z. Wang, M. Xie, X. Yang, J. Cao and Y. Zhang, Appl. Catal., B, 2024, 347, 123806 CrossRef CAS.
- S. He, G. Wang, Y. Liu, L. Luo, T. Jiang, T. Fan, X. Zhu, Y. Ding, J. Jing and S. Guan, Environ. Sci.: Nano, 2024, 11, 3202–3213 RSC.
- H. S. Gujral, G. Singh, J. H. Yang, C. I. Sathish, J. Yi, A. Karakoti, M. Fawaz, K. Ramadass, A. A. H. Al-Muhtaseb, X. Yu, M. B. H. Breese and A. Vinu, Carbon, 2022, 195, 9–18 CrossRef CAS.
- H. Yin, C. Yuan, H. Lv, K. Zhang, X. Chen and Y. Zhang, Sep. Purif. Technol., 2023, 308, 122815 CrossRef CAS.
- J. Zhang, Z. Li, B. Liu, M. Chen, Y. Zhou and M. Zhu, Appl. Catal., B, 2023, 328, 122522 CrossRef CAS.
- X. Gao, Y. Zhou, Z. Cheng, Y. Tan, S. Liu and Z. Shen, Int. J. Hydrogen Energy, 2019, 44, 27421–27428 CrossRef CAS.
- H. Wang, Y. Ma, S. Tang, Y. Liang, D. Zhang, X. Jin, Q. Wang, W. Sun, L. Zheng and W. Li, Carbon, 2024, 218, 118723 CrossRef CAS.
- Y. Liu, M. Tayyab, W. Pei, L. Zhou, J. Lei, L. Wang, Y. Liu and J. Zhang, Small, 2023, 19, 2208117 CrossRef CAS PubMed.
- H. Long, P. Wang, X. Wang, F. Chen and H. Yu, Appl. Surf. Sci., 2022, 604, 154457 CrossRef CAS.
- S. Cao, H. Li, T. Tong, H.-C. Chen, A. Yu, J. Yu and H. M. Chen, Adv. Funct. Mater., 2018, 28, 1802169 CrossRef.
- G. Dukovic, M. G. Merkle, J. H. Nelson, S. M. Hughes and A. P. Alivisatos, Adv. Mater., 2008, 20, 4306–4311 CrossRef CAS.
- C. Merschjann, S. Tschierlei, T. Tyborski, K. Kailasam, S. Orthmann, D. Hollmann, T. Schedel-Niedrig, A. Thomas and S. Lochbrunner, Adv. Mater., 2015, 27, 7993–7999 CrossRef CAS.
- M. Luna, M. Barawi, S. Gómez-Moñivas, J. Colchero, M. Rodríguez-Peña, S. Yang, X. Zhao, Y.-H. Lu, R. Chintala, P. Reñones, V. Altoe, L. Martínez, Y. Huttel, S. Kawasaki, A. Weber-Bargioni, V. A. de la Peña ÓShea, P. Yang, P. D. Ashby and M. Salmeron, ACS Appl. Mater. Interfaces, 2021, 13, 50531–50538 CrossRef CAS.
- Y. Peng, B. Lu, N. Wang, J. E. Lu, C. Li, Y. Ping and S. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 24707–24714 CrossRef CAS.
- Y. Ben-Shahar, F. Scotognella, I. Kriegel, L. Moretti, G. Cerullo, E. Rabani and U. Banin, Nat. Commun., 2016, 7, 10413 CrossRef CAS PubMed.
- Y. Peng, B. Lu, F. Wu, F. Zhang, J. E. Lu, X. Kang, Y. Ping and S. Chen, J. Appl. Chem. Sci., 2018, 140, 15290–15299 CAS.
- F. Yan, Y. Wu, L. Jiang, X. Xue, J. Lv, L. Lin, Y. Yu, J. Zhang, F. Yang and Y. Qiu, ChemSusChem, 2020, 13, 876–881 CrossRef CAS PubMed.
- J. Yang, Y. Gao, X. Li, Y. Wang, P. Guan and B. Liu, Mater. Sci. Semicond. Process., 2024, 175, 108267 CrossRef CAS.
- W.-W. Liu, J. Pan and R.-F. Peng, Rare Met., 2021, 40, 3554–3560 CrossRef CAS.
- W. Liu, X. Liu, S. Xin, Y. Wang, S. Huo, W. Fu, Q. Zhao, M. Gao and H. Xie, Appl. Energy, 2024, 358, 122552 CrossRef CAS.
- I. Y. Kim, S. Kim, X. Jin, S. Premkumar, G. Chandra, N. S. Lee, G. P. Mane, S. J. Hwang, S. Umapathy and A. Vinu, Angew. Chem., Int. Ed., 2018, 57, 17135–17140 CrossRef CAS PubMed.
- X. Guan, M. Fawaz, R. Sarkar, C.-H. Lin, Z. Li, Z. Lei, P. D. Nithinraj, P. Kumar, X. Zhang, J.-H. Yang, L. Hu, T. Wu, S. Chakraborty, J. Yi and A. Vinu, J. Mater. Chem. A, 2023, 11, 12837–12845 RSC.
- C. Peng, L. Han, J. Huang, S. Wang, X. Zhang and H. Chen, Chin. J. Catal., 2022, 43, 410–420 CrossRef CAS.
- H. Che, J. Wang, X. Gao, J. Chen, P. Wang, B. Liu and Y. Ao, J. Colloid Interface Sci., 2022, 627, 739–748 CrossRef CAS.
- C. Han, W. Yi, Z. Li, C. Dong, H. Zhao and M. Liu, Electrochim. Acta, 2023, 447, 142083 CrossRef CAS.
- S. Lu, X. Li, B. Yu, J. Ding, Y. Zhong and H. Zhang, Ecotoxicol. Environ. Saf., 2022, 246, 114148 CrossRef CAS PubMed.
- J. Shen, C. Luo, S. Qiao, Y. Chen, Y. Tang, J. Xu, K. Fu, D. Yuan, H. Tang, H. Zhang and C. Liu, ACS Catal., 2023, 13, 6280–6288 CrossRef CAS.
- J. Xue, Y.-N. Jing, L.-L. Li, X.-L. Yin, Z.-F. Xu, J. Li and Y.-L. Wang, Colloids Surf., A, 2024, 685, 133158 CrossRef CAS.
- M. Zhai, Y. Zhang, J. Xu, C. Wang and L. Wang, Appl. Surf. Sci., 2024, 652, 159378 CrossRef CAS.
- S. Ma, D. Yang, B. Li, Y. Guan, M. Wu, J. Wu, Y. Guo, L. Sheng, L. Liu and T. Yao, J. Colloid Interface Sci., 2024, 664, 960–971 CrossRef CAS.
- Y. Guo, Q. Liu, Z. Li, Z. Zhang and X. Fang, Appl. Catal., B, 2018, 221, 362–370 CrossRef CAS.
- S. Ma, J. Zhao, Q. Gao, C. Song, H. Xiao, F. Li and G. Li, Angew. Chem., Int. Ed., 2023, 62, e202315564 CrossRef CAS PubMed.
- S. Hu, P. Qiao, X. Liang, G. Ba, X. Zu, H. Hu, J. Ye and D. Wang, Appl. Catal., B, 2024, 346, 123737 CrossRef CAS.
- D. Ramírez-Ortega, D. Guerrero-Araque, P. Acevedo-Peña, E. Reguera, H. A. Calderon and R. Zanella, Int. J. Hydrogen Energy, 2021, 46, 34333–34343 CrossRef.
- R. Chen, S. Chen, L. Wang and D. Wang, Adv. Mater., 2023, 36 CAS.
- C. Wan, L. Zhou, L. Sun, L. Xu, D.-G. Cheng, F. Chen, X. Zhan and Y. Yang, Chem. Eng. J., 2020, 396, 125229 CrossRef CAS.
- N. Ramesh Reddy, U. Bharagav, M. M. Kumari, K. K. Cheralathan, P. K. Ojha, M. V. Shankar and S. W. Joo, Ceram. Int., 2021, 47, 10216–10225 CrossRef CAS.
- Y. Hu, S. Zhang, Z. Zhang, H. Zhou, B. Li, Z. Sun, X. Hu, W. Yang, X. Li, Y. Wang, S. Liu, D. Wang, J. Lin, W. Chen and S. Wang, Adv. Mater., 2023, 35 Search PubMed.
- B. Su, M. Zheng, W. Lin, X. F. Lu, D. Luan, S. Wang and X. W. Lou, Adv. Energy Mater., 2023, 13, 2203290 CrossRef CAS.
- X. Yu, H. Su, J. Zou, Q. Liu, L. Wang and H. Tang, Chin. J. Catal., 2022, 43, 421–432 CrossRef CAS.
- D. Zheng, Q. Wang, Z. Pan, S. Wang, Y. Hou and G. Zhang, Sci. China Mater., 2024, 67, 1900–1906 CrossRef CAS.
- J. Lu, Z. Chen, Y. Shen, H. Yuan, X. Sun, J. Hou, F. Guo, C. Li and W. Shi, J. Colloid Interface Sci., 2024, 670, 428–438 CrossRef CAS.
- J. Li, Z. Huang, C. Wang, L. Tian, X. Yang, R. Zhou, M. N. Ghazzal and Z.-Q. Liu, Appl. Catal., B, 2024, 340, 123181 CrossRef CAS.
- H. Xu, Z. Wang, S. Feng, X. Liu, X. Gong and J. Hua, Int. J. Hydrogen Energy, 2023, 48, 8071–8081 CrossRef CAS.
- X. Guan, X. Zhang, Z. Li, S. Deshpande, M. Fawaz, N. P. Dharmarajan, C.-H. Lin, Z. Lei, L. Hu, J.-K. Huang, P. Kumar, Z. Sun, S. Chakraborty and A. Vinu, Chem. Mater., 2024, 36, 4511–4520 CrossRef CAS.
- M. Wang, S. Shen, L. Li, Z. Tang and J. Yang, J. Mater. Sci., 2017, 52, 5155–5164 CrossRef CAS.
- B. Zhao, J. Xu, D. Gao, F. Chen, X. Wang, T. Liu, X. Wu and H. Yu, Appl. Catal., B, 2024, 355, 124215 CrossRef CAS.
- M. Younas, M. Yar, H. AlMohamadi, T. Mahmood, K. Ayub, A. L. Khan, M. Yasin and M. A. Gilani, Int. J. Hydrogen Energy, 2024, 51, 758–773 CrossRef CAS.
- M. F. Fellah, Int. J. Hydrogen Energy, 2019, 44, 27010–27021 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tc03914a |
| This journal is © The Royal Society of Chemistry 2025 |