
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceControlled polymerization of levoglucosenone-derived enynes to give bio-based polymers with tunable degradation rates and high glass transition temperatures†
Eunsong
Jung‡
 a,
Antonio
Rizzo‡
b,
Hanseul
Ryu
a,
Minyoung
Cho
a and
Tae-Lim
Choi
*a
a,
Antonio
Rizzo‡
b,
Hanseul
Ryu
a,
Minyoung
Cho
a and
Tae-Lim
Choi
*a
aDepartment of Materials, ETH Zürich, Zürich, 8093, Switzerland. E-mail: tae-lim.choi@mat.ethz.ch
bDepartment of Chemistry, the State Key Laboratory of Synthetic Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong 999077, P. R. China
First published on 11th April 2025
Abstract
In recent years, pollution from plastic waste has intensified the demand for sustainable polymers. Hence, biomass-derived degradable polymers offer a promising solution. For example, levoglucosenone, a readily available biomass product from cellulose pyrolysis, is an attractive building block for polymer synthesis. However, the metathesis polymerization of levoglucosenone-derived monomers has been difficult to control due to poor monomer reactivity, requiring an unstable but reactive ruthenium catalyst (C793). To facilitate the polymerization, we introduced a cascade motif to successfully demonstrate controlled polymerization of levoglucosenone-derived enynes using a commercially available 3rd-generation Grubbs catalyst. This living polymerization also enabled block copolymer synthesis. Furthermore, the degradation rates of these polymers can be adjusted over 2 orders of magnitude through monomer structural modifications. Notably, we observed higher glass transition temperatures of 152–198 °C by varying structural parameters.
Introduction
Globally, the demand for degradable polymers is surging due to environmental pollution and climate change.1,2 In particular, typical commodity polymers rely on petroleum resources, warranting the development of more sustainable alternatives. Beyond addressing environmental issues, there is a growing need for degradable polymers in advanced material fields such as bioelectronics and drug release3–5 to avoid accumulation in the body. Degradable polymers possess easily cleavable moieties in their backbones, typically featuring hydrolysable functional groups such as acetals, esters, silyl ethers, and vinyl ethers.6–15 One effective method to introduce degradable moieties into polymer backbones is to utilize renewable bio-based feedstocks that inherently contain these functionalities to reduce synthetic steps to prepare such monomers.16–18 As a result, many novel polymerization methods using lignin, sugar, chitin, and cell walls have been reported.19–22In recent years, our group and the Gutekunst group have developed controlled cascade metathesis polymerization of enyne monomers containing less reactive cycloalkenes such as cyclohexene and sustainable monomers synthesized from carbohydrate-derived scaffolds (Fig. 1A).23–25 The resulting polymers containing acetals degrade under mild acidic conditions to well-defined small aromatic molecules such as furans and pyrroles. In the event, we could roughly tune the degradation rate by adjusting the structure. The relevance of this quantitative and clean degradation is clearly demonstrated in our precise analysis of cascade efficiency during the polymerization of polycyclic enyne monomers.26
 | ||
| Fig. 1 (A) Enyne metathesis polymerization of sugar-based monomers. (B) ROMP of LGO-derived monomers. (C) Enyne metathesis polymerization of LGO-derived monomers (this work). Mes = 2,4,6-trimethylbenzene. | ||
However, these polymers generally exhibit low Tg values and poor stability, commensurate with their rapid degradation under mild conditions. This inevitable trade-off between stability and degradability (i.e. highly stable but poorly degradable) may require harsh conditions, producing various unidentified by-products due to multiple side reactions. Therefore, developing polymers that integrate both stability and degradability remains a challenging task.
To enhance stability while maintaining degradability, a bio-based bicyclic compound such as levoglucosenone (LGO), obtained from the pyrolysis of cellulose, is a promising building block for sustainable polymers (Fig. 1B).27–36 While numerous polymerizations using LGO-derived monomers have been reported, there are only a handful of examples of this LGO incorporation into a polymer backbone by direct ring opening, thereby embedding the 1,3-dioxolane functionality.37,38 Notably, Schlaad and coworkers reported the ring-opening metathesis polymerization (ROMP) of levoglucosenol (LGOH, prepared by LGO reduction) using a less common Ru catalyst called C793, to produce high MW and Tg values around 100 °C (Fig. 1B).37 Additionally, the resulting polymers degraded slowly into oligomers over 40 days due to the slow hydrolysis of stable cyclic acetals compared to acyclic ones.39
Despite these pioneering studies, this ROMP was particularly challenging due to the poor reactivity of the sterically bulky bicyclic alkenes thereby exhibiting a series of drawbacks such as relatively narrow monomer scope, non-controlled polymerization due to low stability of propagating carbenes and initiation rates of the catalyst used. To overcome these limitations, we hypothesize that lowering the kinetic barrier of the LGO derived monomers by introducing an additional alkyne as a cascade motif would vastly improve the polymerization with fast-initiating Grubbs catalysts containing pyridines (Fig. 1C).
Herein, we report the first successful controlled polymerization of LGO-derived monomers to give degradable polymers with Mn up to 65 kDa and low dispersities (Fig. 1C). Furthermore, due to the additional linker, we could greatly broaden the monomer scope and thus systematically investigate structure–reactivity relationship to identify the factors directly influencing the polymerization. Notably, seven resulting polymers showed much higher glass transition temperatures (Tg) of 152–198 °C than those from the previous analogous cascade polymerization due to the bis-cyclic polymer backbone greatly enhancing thermal properties. Nevertheless, these polymers underwent degradation whose rates are systematically modulated with over two orders of magnitudes by the stereoelectronics of the substituents.
Results and discussion
Commercially available LGO was reduced to LGOH, followed by a Mitsunobu reaction to install the N-propargyl moieties, providing the dioxolane monomer M1 in high yield (Fig. 2A) and the regioisomer iso-M1 (Fig. 2B) as a minor product (for the synthesis discussion see Fig. S3 and Table S1,† 71%, 2 steps). The stereochemistry of M1 was confirmed using the X-ray crystal structure of the analogue, M2 (Fig. 2C).40–42 As we have previously demonstrated, the stereochemistry and connectivity in monomers influence polymerization efficiency.26 Therefore, we initially investigated cascade polymerization of M1 and iso-M1 using a conventional third-generation Grubbs catalyst (G3) at an M/I ratio of 50 in THF (0.2 M) at room temperature. Gratifyingly, M1 was completely polymerized to give an Mn of 13 kDa with a relatively low dispersity (Đ = 1.32), showing potential for controlled polymerization. In contrast, polymerization of iso-M1 under the same conditions resulted in only 22% conversion even after a longer reaction time, providing only a 2.2 kDa oligomer (Table S2†). | ||
| Fig. 2 Reaction of cascade enyne metathesis polymerization of (A) M1 and (B) iso-M1. (C) X-ray crystal structure of M2. Plots of conversions and carbene changes monitored by in situ1H NMR during the polymerization of (D) M1 and (E) iso-M1 in THF-d8 (G3, M/I = 10, 0.15 M, RT). | ||
To gain mechanistic insight into how regiochemistry influenced polymerization performance, we monitored the reactions of M1 and iso-M1 with fast initiating G3 by in situ1H NMR (M/I = 10, THF-d8, 0.15 M, RT) (Fig. 2D and E). Indeed, M1 showed 4.3 times faster polymerization than iso-M1 but more importantly, for M1, a high propagating carbene percentage (ca. 90%) was maintained throughout the reaction (Fig. 2D). Conversely, for iso-M1, propagating carbene percentage dropped to lower than 10% (Fig. 2E), suggesting much faster decomposition (or lower stability) of the corresponding Ru carbene. In the case of M1, the propagating carbene has an ether at the β-position, while for iso-M1, an acetal is present, and presumably, higher propensity of β-hydride isomerization activated by the acetal in iso-M1 might facilitate decomposition as proposed by the Schlaad group (Fig. 2A and B).38
Encouraged by these kinetic data, we optimized the polymerization of the more reactive M1 by lowering the temperature to 0 °C to suppress chain transfer and carbene decomposition. With an M/I ratio from 30 to 100, the Mn of P1 increased from 9 to 24 kDa with low dispersity between 1.06 and 1.23 (entries 1–4, Table 1). However, polymerization with an M/I of 150 under the same conditions resulted in lower Mn (29 kDa) than expected with high dispersity (Đ = 1.72), probably due to catalyst decomposition caused by the turnover number limit or ether coordination (entry 5, Table 1). To stabilize the propagating carbene through steric shielding, we switched to the more stable G3-DIPP catalyst containing a bulkier 2,6-diisopropylphenyl (Dipp) group. This resulted in higher Mn of 36 kDa with lower dispersity of 1.45 (entry 6, Table 1). Based on these optimizations, we successfully achieved controlled polymerization of M1 up to an M/I of 150 with a linear increase in molecular weight and low dispersity (Fig. 3A).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | MX | M/I | Temp. (°C) | Conc. (M) | Time (h) | Conv.b (%) | Yieldc (%) | M n (kDa) | Đ |
| a Polymerizations conducted in degassed THF except for M3 (dichloromethane) due to its insolubility. b Determined by 1H NMR analysis of the crude mixture. c Isolated yield. d Determined by THF size exclusion chromatography calibrated using polystyrene standards except for P3 (chloroform SEC). e G3-DIPP was used as a catalyst. f 20 mol% of 3-chloropyridine was added as an additive. | |||||||||
| 1 | M1 | 30 | 10 | 0.2 | 0.5 | >99 | 79 | 9.1 | 1.06 |
| 2 | M1 | 50 | 10 | 0.2 | 1 | >99 | 99 | 14.5 | 1.14 |
| 3 | M1 | 75 | 0 | 0.2 | 3 | >99 | 73 | 19.8 | 1.16 |
| 4 | M1 | 100 | 0 | 0.2 | 4 | >99 | 83 | 24.1 | 1.23 |
| 5 | M1 | 150 | 0 | 0.2 | 6 | 90 | 72 | 29.0 | 1.72 |
| 6e | M1 | 150 | −10 | 0.2 | 16 | 95 | 87 | 35.6 | 1.45 |
| 7 | M2 | 30 | 10 | 0.2 | 0.5 | >99 | 61 | 8.4 | 1.13 |
| 8 | M2 | 50 | 10 | 0.2 | 1 | >99 | 92 | 15.5 | 1.21 |
| 9 | M2 | 75 | 0 | 0.2 | 3 | >99 | 85 | 24.0 | 1.29 |
| 10 | M2 | 100 | −10 | 0.1 | 16 | >99 | 98 | 28.3 | 1.39 |
| 11f | M3 | 30 | RT | 0.2 | 1 | 99 | 91 | 6.8 | 1.21 |
| 12e,f | M3 | 50 | 0 | 0.2 | 8 | >99 | 80 | 11.5 | 1.28 |
| 13e,f | M3 | 75 | 0 | 0.2 | 12 | 89 | 68 | 14.2 | 1.37 |

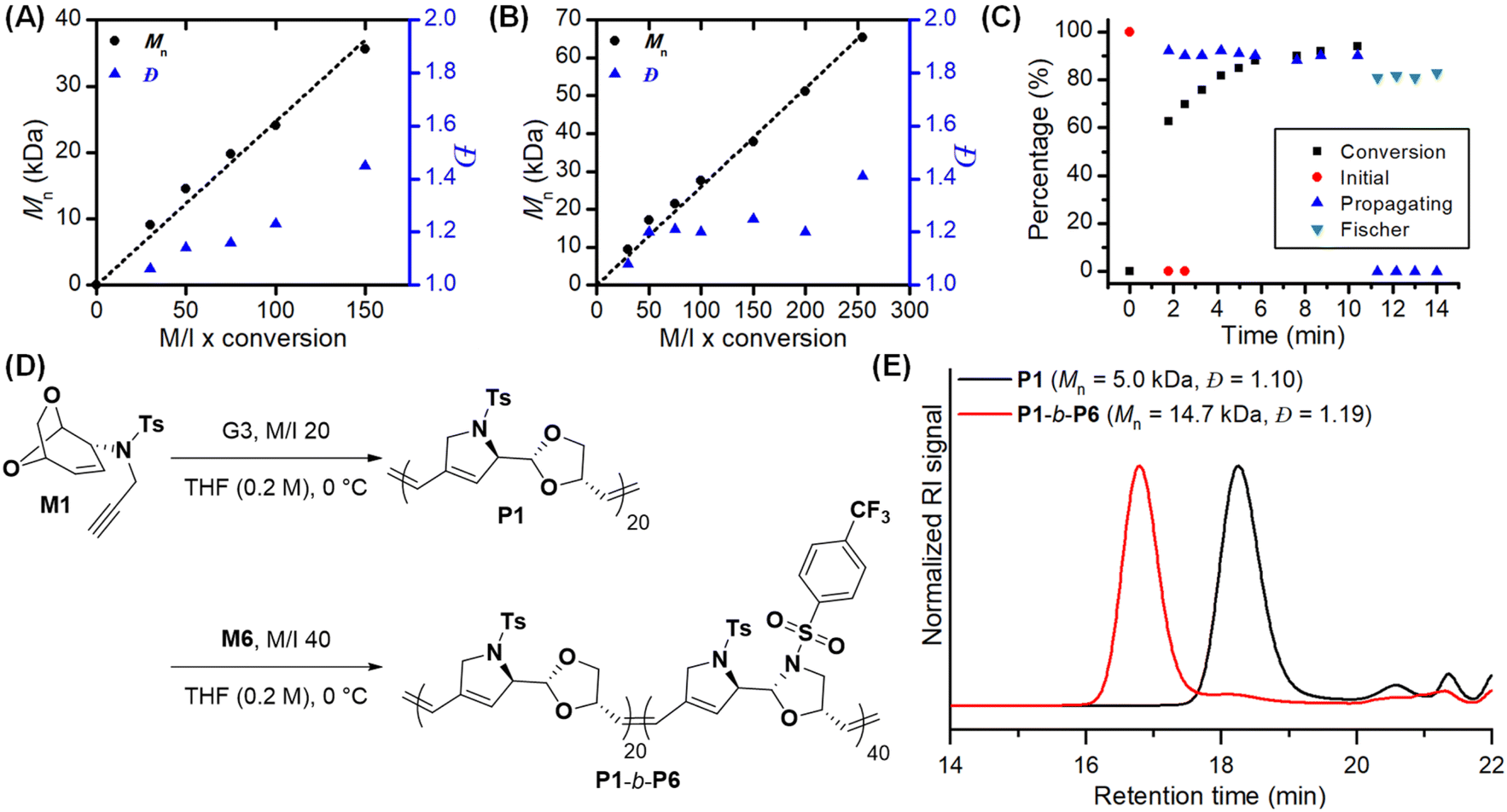 | ||
| Fig. 3 (A) Linear plots of Mnvs. M/I x conversion for (A) P1 and (B) P4. Plots of conversions and carbene changes monitored by in situ1H NMR during the polymerization of (C) M4 in THF-d8 (0.15 M, RT). (D) Synthesis of block copolymers, P1-b-P6, and (E) SEC traces for P1 and P1-b-P6. | ||
Next, to understand the impact of the size of the N-SO2R1 substituent on polymerization, we prepared M2 and M3, containing bigger mesityl (Mes) and smaller methyl groups, respectively. M2 was quantitatively polymerized under the same optimal conditions as M1 at an M/I of 30 to give P2 with Mn of 8 kDa and a low dispersity of 1.13. Further temperature optimization from 10 °C to −10 °C led to another controlled polymerization up to M/I = 100 where Mn increased proportionally from 8 to 28 kDa with low dispersity between 1.13 and 1.39 (entries 7–10, Table 1, Fig. S10†). Unfortunately, increasing to M/I = 150 led to poorly controlled polymerization with Đ greater than 2 even with G3-DIPP (Table S3†).
Since M3 was insoluble in THF, polymerization at an M/I of 30 was conducted in DCM at RT, yielding P3 with an Mn of 5 kDa and a moderate dispersity of 1.30 (Table S3†). To improve the controllability, 3-chloropyridine as an additive was added to further stabilize the Ru carbene and indeed this increased Mn to 7 kDa with a lower Đ of 1.21 (entry 11, Table 1). For the higher M/I of 50, the Mn barely increased to 8 kDa with a higher Đ of 1.43. (Table S3†). Gratifyingly, the bulkier G3-DIPP catalyst resulted in better control with a linear increase in Mn (Mn = 12–14 kDa, Đ = 1.28–1.37) at M/I ratios of 50 and 75 (entries 12 and 13, Table 1, Fig. S10†). Unfortunately, increasing the M/I 100 did not result in a good control even with G3-DIPP (Table S3†), implying that some sterics on the monomers (M1 and M2) improved the stability of the propagating carbenes.
In order to broaden the monomer scope, we modified the 1,3-dioxolane moiety to an oxazolidine moiety. Again from a commercially available biomass-derived triacetyl glucal, we prepared bicyclic oxazolidine monomers M4–M7. We chose the p-tolyl group as the R2 group on the linker, which demonstrated good controllability in dioxolane monomers. Delightfully, polymerization of M4 with the G3 catalyst at room temperature in THF (0.2 M), with an M/I of 30, yielded P4 with an Mn of 9.4 kDa and exceptionally low dispersity of 1.08 (entry 1, Table 2). After a minor optimization, controlled polymerization from M/I ratios of 30 to 150 was achieved with Mns between 9 and 38 kDa and dispersities from 1.08 to 1.25 (entries 1–5, Table 2). For even higher M/I ratios of 200 and 300, further modifying temperature, concentration, and additive (5 mol% of 3,5-dichloropyridine) yielded better control with an Mn of 51 and 65 kDa and a Đ of 1.20 and 1.41, respectively (entries 6 and 7, Table 2, Fig. 3B).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | MX | M/I | Temp. (°C) | Conc. (M) | Time (h) | Conv.b (%) | Yieldc (%) | M n (kDa) | Đ |
| a Polymerizations conducted in degassed THF. b Determined by 1H NMR analysis of the crude mixture. c Isolated yield. d Determined by THF size exclusion chromatography calibrated using polystyrene standards. e 5 mol% of 3,5-dichloropyridine was added as an additive. | |||||||||
| 1 | M4 | 30 | RT | 0.2 | 0.66 | >99 | 85 | 9.4 | 1.08 |
| 2 | M4 | 50 | RT | 0.2 | 1 | >99 | 80 | 17.2 | 1.20 |
| 3 | M4 | 75 | 10 | 0.2 | 4 | >99 | 97 | 21.5 | 1.21 |
| 4 | M4 | 100 | 10 | 0.2 | 5 | >99 | 82 | 27.5 | 1.20 |
| 5 | M4 | 150 | 0 | 0.2 | 15 | >99 | 85 | 37.9 | 1.25 |
| 6e | M4 | 200 | 0 | 0.1 | 20 | 99 | 87 | 51.2 | 1.20 |
| 7e | M4 | 300 | 0 | 0.1 | 30 | 85 | 76 | 65.4 | 1.41 |
| 8 | M5 | 30 | RT | 0.2 | 0.66 | >99 | 99 | 9.6 | 1.06 |
| 9 | M6 | 30 | RT | 0.2 | 0.66 | >99 | 84 | 11.2 | 1.09 |
| 10 | M7 | 30 | RT | 0.2 | 0.66 | 58 | 48 | 11.2 | 1.64 |
| 11 | M7 | 30 | −10 | 0.2 | 12 | >99 | 81 | 10.4 | 1.24 |

Finally, we synthesized M5 and M6 containing electron-donating p-anisyl and electron-withdrawing p-trifluoromethylbenzyl groups as the R2 groups, respectively, for systematic study on the effects on degradation. The resulting P5 and P6 showed Mn values around 10 and 11 kDa with a Đ below 1.10 (entries 8 and 9, Table 2). Meanwhile, the polymerization of M7, containing a smaller and more electron-withdrawing trifluoromethyl group as the R2 group, gave P7 with an Mn of 10 kDa with Đ = 1.24 at −10 °C (entry 11, Table 2).
Taking advantage of good control, we synthesized a block copolymer from dioxolane- and oxazolidine-containing monomers, M1 and M6 (Fig. 3D). After preparing P1 with a Mn of 5 kDa, adding 40 equiv. of M6 yielded P1-b-P6 with a Mn of 15 kDa and Đ of 1.19. A clear shift to the higher molecular weight region in the SEC trace confirmed successful block copolymerization, thereby supporting the living nature of cascade polymerization (Fig. 3E).
With these new polymers, we examined their thermal properties by thermogravimetric analysis which revealed degradation temperatures ranging from 244 to 284 °C. On the other hand, the glass transition temperatures (Tg) measured by differential scanning calorimetry for the dioxolane-containing polymers (P1–P3) were between 152 °C and 175 °C (see the ESI, Section 7†).
Interestingly, the oxazolidine-containing polymers (P4–P7) generally showed higher Tg values (187–198 °C except for P5 of 152 °C) presumably due to additional bulky side-chains compared to the dioxolane-containing polymers. These values are significantly higher than those from the other enyne cascade polymerization of sugar-based monomers (mostly below 80 °C)24–26 and even higher than Tg of levoglucosenone-derived polymers synthesized by ROMP (100 °C).37 This higher Tg is due to the rigid bis-cyclic backbone.
To investigate the impact of structural parameters on the polymer degradability, P1–P7 in a chloroform/MeOH mixture were treated under acidic conditions (0.5 M HCl, at 40 °C) (Fig. 4A). The degradation was monitored over time by taking aliquots for SEC analysis by normalizing Mn,t/Mn,0 values (Mn,0 = initial molecular weight and Mn,t = molecular weight at time t in days) (Fig. 4B). Surprisingly, despite their enhanced thermal properties, dioxolane polymers P1–P3 with a degree of polymerization (DP) of 40 showed substantially faster degradation than the previously reported ROMP polymers with an Mn deduction of more than 80% of their initial molecular weight (approximately Mn,t/Mn,0 = 0.16) after 4 days. On the other hand, the oxazolidine polymer P4 degraded by 30% (Mn,t/Mn,0 = 0.71). Further degradation for 14 days led to a final Mn of 1.2 kDa, 1.4 kDa, and 0.8 kDa for P1 to P3, respectively, while Mn of P4 decreased to half which corresponds to 33-fold slower than P3 (Fig. 4A and C, see the ESI, Section 6†). MALDI-TOF analysis of degraded P3 showed a series of peaks ranging from dimers to pentamers, which aligns with the final Mn from SEC analysis, corresponding to the molecular weight of a trimer (see the ESI, Section 6.1†). The degradation mechanism involves conventional acidic deprotection of cyclic acetal or hemiaminal where MeOH would cleave the polymer backbone. This was observed from the end group analysis from MALDI-TOF, revealing minor peak series corresponding to D3. Interestingly, a major peak of D3′ appeared which is due to another MeOH addition to cleaved alcohol.
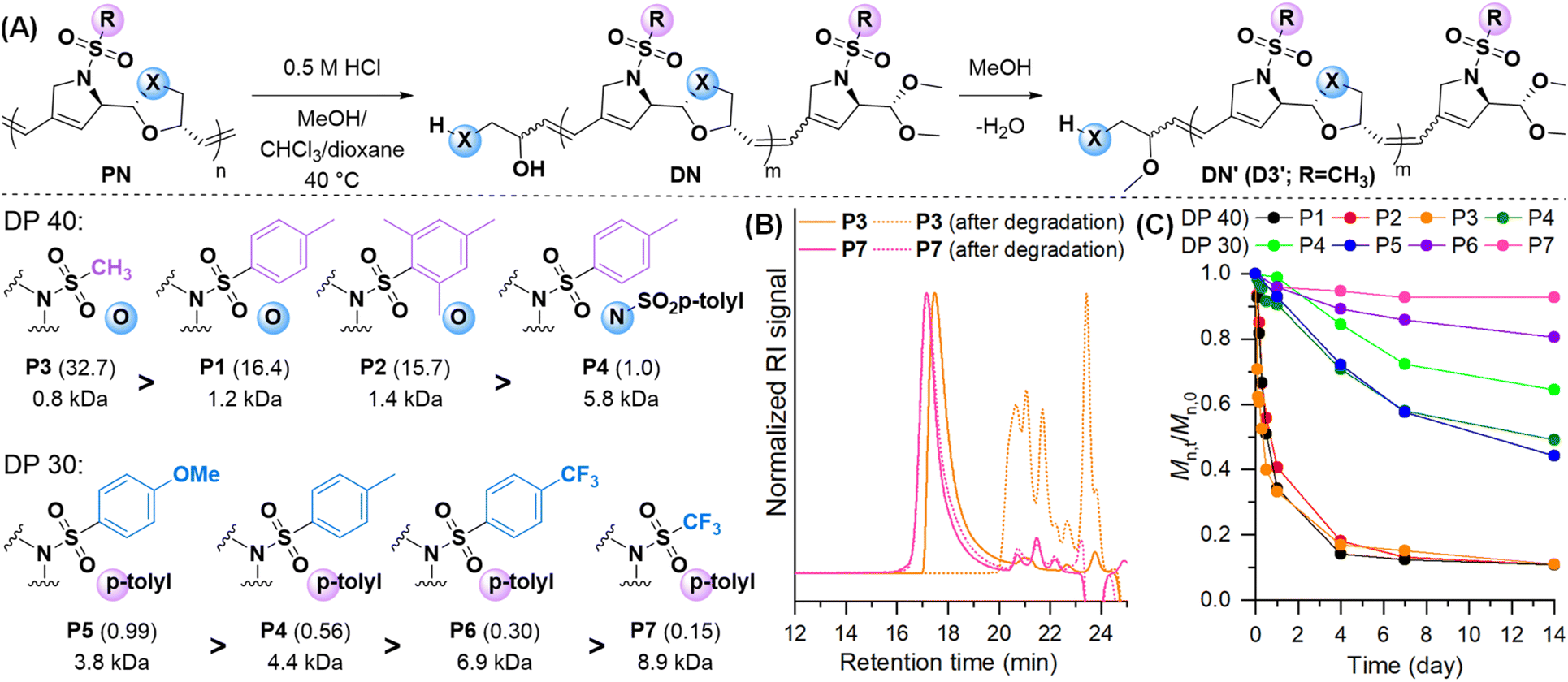 | ||
| Fig. 4 (A) Degradation reaction pathway. Structures of polymers, normalized relative degradation rates (in parenthesis), and Mn of the degraded product after 14 days. (B) SEC traces of P3 and P7 degradation under 0.5 M HCI acidic conditions. (C) Plots of degradation versus time for P1–P7. Mn,0 refers to the initial molecular weight and Mn,t refers to the molecular weight at time t (days). The ratio Mn,t/Mn,0 was normalized to 1 for comparison. | ||
To investigate the substituent effect on oxazolidine on degradation rates, we monitored the degradation of P4–P7 with a DP of 30. P4 degraded to 66% of its initial molecular weight (Mn,t/Mn,0 = 0.66) after 14 days. P5, containing an electron-donating p-anisylsulfonyl group, degraded approximately 1.8 times faster than P4, while P6 containing an electron-withdrawing p-trifluoromethylbenzene sulfonyl group, degraded about 1.9 times slower than P4. The degradation of P7, featuring a stronger electron-withdrawing triflyl group, was 3.7 times slower than P4 and even about 218 times slower than the fastest-degrading dioxolane polymer P3 (Fig. 4B and C). This suggests that the electron-withdrawing group lowered the basicity, thereby slowing both the acid-catalyzed deprotection of the cyclic acetal or hemiaminal and backbone degradation.
Conclusions
In conclusion, we successfully achieved challenging controlled cascade polymerization of levoglucosenone derived monomers using a commercial Grubbs 3rd generation catalyst. We designed various monomers, yielding polymers with Mn up to 65 kDa and low dispersities and block copolymers. Due to rigid bis-cyclic backbones, these polymers showed high Tg values up to 198 °C. Despite this thermal property, polymers underwent degradation under acidic conditions whose rates could be tuned by 200 times with modulating electronic properties on the backbone. This work provides valuable insights on the correlation between structure, polymerization, degradation, and thermal properties.Data availability
All supplementary data for the results of this work are available in the article and its ESI file.† Crystallographic data for M2 and M4 have been deposited at the CCDC 2418634 and CCDC 2418635, respectively, and can be obtained from https://www.ccdc.cam.ac.uk/structures.Author contributions
Conceptualization: A. R. and E. J.; methodology: E. J. and A. R.; investigation: E. J., A. R., H. R., and M. C.; validation: A. R. and T. L. C.; visualization: E. J., A. R., and H. R.; writing: E. J., T. L. C., and A. R.; supervision: A. R. and T. L. C.; funding: T. L. C.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for financial support from the NRF, Korea, through the Creative Research Initiative.Notes and references
- M. Hong and E. Y. X. Chen, Chemically recyclable polymers: a circular economy approach to sustainability, Green Chem., 2017, 19, 3692–3706 RSC.
- P. G. C. Nayanathara Thathsarani Pilapitiya and A. S. Ratnayake, The world of plastic waste: a review, Cleaner Materials, 2024, 11, 100220 CrossRef CAS.
- J. Tropp and J. Rivnay, Design of biodegradable and biocompatible conjugated polymers for bioelectronics, J. Mater. Chem. C, 2021, 9, 13543–13556 RSC.
- S. Lee, S. M. Silva, L. M. Caballero Aguilar, T. Eom, S. E. Moulton and B. S. Shim, Biodegradable bioelectronics for biomedical applications, J. Mater. Chem. B, 2022, 10, 8575–8595 RSC.
- S. Binauld and M. H. Stenzel, Acid-degradable polymers for drug delivery: a decade of innovation, Chem. Commun., 2013, 49, 2082–2102 RSC.
- C. Fraser, M. A. Hillmyer, E. Gutierrez and R. H. Grubbs, Degradable Cyclooctadiene/Acetal Copolymers: Versatile Precursors to 1,4-Hydroxytelechelic Polybutadiene and Hydroxytelechelic Polyethylene, Macromolecules, 1995, 28, 7256–7261 CrossRef CAS.
- Y. Yang, Y. Cho and T.-L. Choi, Designing Degradable Polymers from Tricycloalkenes via Complete Cascade Metathesis Polymerization, Angew. Chem., Int. Ed., 2024, 63, e202400235 CrossRef CAS PubMed.
- D. Moatsou, A. Nagarkar, A. F. M. Kilbinger and R. K. O'Reilly, Degradable precision polynorbornenes via ring-opening metathesis polymerization, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1236–1242 CrossRef CAS.
- B. R. Elling, J. K. Su and Y. Xia, Degradable Polyacetals/Ketals from Alternating Ring-Opening Metathesis Polymerization, ACS Macro Lett., 2020, 9, 180–184 CrossRef CAS PubMed.
- T. Steinbach, E. M. Alexandrino and F. R. Wurm, Unsaturated poly(phosphoester)s via ring-opening metathesis polymerization, Polym. Chem., 2013, 4, 3800–3806 RSC.
- P. Shieh, H. V. T. Nguyen and J. A. Johnson, Tailored silyl ether monomers enable backbone-degradable polynorbornene-based linear, bottlebrush and star copolymers through ROMP, Nat. Chem., 2019, 11, 1124–1132 CrossRef CAS PubMed.
- F. O. Boadi, J. Zhang, X. Yu, S. R. Bhatia and N. S. Sampson, Alternating Ring-Opening Metathesis Polymerization Provides Easy Access to Functional and Fully Degradable Polymers, Macromolecules, 2020, 53, 5857–5868 CrossRef CAS PubMed.
- X. Sui, T. Zhang, A. B. Pabarue, L. Fu and W. R. Gutekunst, Alternating Cascade Metathesis Polymerization of Enynes and Cyclic Enol Ethers with Active Ruthenium Fischer Carbenes, J. Am. Chem. Soc., 2020, 142, 12942–12947 CrossRef CAS PubMed.
- T. An, H. Ryu and T.-L. Choi, Living Alternating Ring-Opening Metathesis Copolymerization of 2,3-Dihydrofuran to Provide Completely Degradable Polymers, Angew. Chem., Int. Ed., 2023, 62, e202309632 CrossRef CAS PubMed.
- P. Shieh, W. Zhang, K. E. L. Husted, S. L. Kristufek, B. Xiong, D. J. Lundberg, J. Lem, D. Veysset, Y. Sun, K. A. Nelson, D. L. Plata and J. A. Johnson, Cleavable comonomers enable degradable, recyclable thermoset plastics, Nature, 2020, 583, 542–547 CrossRef CAS PubMed.
- K. Kümmerer, Sustainable Chemistry: A Future Guiding Principle, Angew. Chem., Int. Ed., 2017, 56, 16420–16421 CrossRef PubMed.
- Y. Liu and S. Mecking, A Synthetic Polyester from Plant Oil Feedstock by Functionalizing Polymerization, Angew. Chem., Int. Ed., 2019, 58, 3346–3350 CrossRef CAS PubMed.
- C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Valorization of Biomass: Deriving More Value from Waste, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- J. Sternberg, O. Sequerth and S. Pilla, Green chemistry design in polymers derived from lignin: review and perspective, Prog. Polym. Sci., 2021, 113, 101344 CrossRef CAS.
- J. L. Shamshina, P. Berton and R. D. Rogers, Advances in Functional Chitin Materials: A Review, ACS Sustain. Chem. Eng., 2019, 7, 6444–6457 CrossRef CAS.
- J. A. Galbis, M. d. G. García-Martín, M. V. de Paz and E. Galbis, Synthetic Polymers from Sugar-Based Monomers, Chem. Rev., 2016, 116, 1600–1636 CrossRef CAS PubMed.
- Y. Zhu, C. Romain and C. K. Williams, Sustainable polymers from renewable resources, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- L. Fu, X. Sui, A. E. Crolais and W. R. Gutekunst, Modular Approach to Degradable Acetal Polymers Using Cascade Enyne Metathesis Polymerization, Angew. Chem., Int. Ed., 2019, 58, 15726–15730 CrossRef CAS PubMed.
- A. Bhaumik, G. I. Peterson, C. Kang and T.-L. Choi, Controlled Living Cascade Polymerization To Make Fully Degradable Sugar-Based Polymers from d-Glucose and d-Galactose, J. Am. Chem. Soc., 2019, 141, 12207–12211 CrossRef CAS PubMed.
- A. Rizzo, G. I. Peterson, A. Bhaumik, C. Kang and T.-L. Choi, Sugar-Based Polymers from d-Xylose: Living Cascade Polymerization, Tunable Degradation, and Small Molecule Release, Angew. Chem., Int. Ed., 2021, 60, 849–855 CrossRef CAS PubMed.
- A. Rizzo, E. Jung, H. Song, Y. Cho, G. I. Peterson and T.-L. Choi, Controlled Living Cascade Polymerization of Polycyclic Enyne Monomers: Leveraging Complete Degradability for a Stereochemical and Structural Investigation, J. Am. Chem. Soc., 2022, 144, 15643–15652 CrossRef CAS PubMed.
- Y. Halpern, R. Riffer and A. Broido, Levoglucosenone (1,6-anhydro-3,4-dideoxy-DELTA.3-beta-D-pyranosen-2-one). Major product of the acid-catalyzed pyrolysis of cellulose and related carbohydrates, J. Org. Chem., 1973, 38, 204–209 CrossRef CAS.
- M. G. Banwell, X. Liu, L. A. Connal and M. G. Gardiner, Synthesis of Functionally and Stereochemically Diverse Polymers via Ring-Opening Metathesis Polymerization of Derivatives of the Biomass-Derived Platform Molecule Levoglucosenone Produced at Industrial Scale, Macromolecules, 2020, 53, 5308–5314 CrossRef CAS.
- S. Fadlallah, A. A. M. Peru, A. L. Flourat and F. Allais, A straightforward access to functionalizable polymers through ring-opening metathesis polymerization of levoglucosenone-derived monomers, Eur. Polym. J., 2020, 138, 109980 CrossRef CAS.
- S. Fadlallah, A. A. M. Peru, L. Longé and F. Allais, Chemo-enzymatic synthesis of a levoglucosenone-derived bi-functional monomer and its ring-opening metathesis polymerization in the green solvent Cyrene™, Polym. Chem., 2020, 11, 7471–7475 RSC.
- S. Fadlallah, A. L. Flourat, L. M. M. Mouterde, M. Annatelli, A. A. M. Peru, A. Gallos, F. Aricò and F. Allais, Sustainable Hyperbranched Functional Materials via Green Polymerization of Readily Accessible Levoglucosenone-Derived Monomers, Macromol. Rapid Commun., 2021, 42, 2100284 CrossRef CAS PubMed.
- M. K. Stanfield, R. S. Terry, J. A. Smith and S. C. Thickett, Levoglucosan and levoglucosenone as bio-based platforms for polymer synthesis, Polym. Chem., 2023, 14, 4949–4956 RSC.
- B. Pollard, M. G. Gardiner, M. G. Banwell and L. A. Connal, Polymers from Cellulosic Waste: Direct Polymerization of Levoglucosenone using DBU as a Catalyst, ChemSusChem, 2024, 17, e202301165 CrossRef CAS PubMed.
- C. M. Warne, S. Fadlallah, F. Allais, G. M. Guebitz and A. Pellis, Controlled Enzymatic Synthesis of Polyesters Based on a Cellulose–Derived Triol Monomer: A Design of Experiment Approach, ChemSusChem, 2024, 17, e202301841 CrossRef CAS PubMed.
- T. Debsharma, Y. Yagci and H. Schlaad, Cellulose-Derived Functional Polyacetal by Cationic Ring-Opening Polymerization of Levoglucosenyl Methyl Ether, Angew. Chem., Int. Ed., 2019, 58, 18492–18495 CrossRef CAS PubMed.
- K. Kaya, T. Debsharma, H. Schlaad and Y. Yagci, Cellulose-based polyacetals by direct and sensitized photocationic ring-opening polymerization of levoglucosenyl methyl ether, Polym. Chem., 2020, 11, 6884–6889 RSC.
- T. Debsharma, F. N. Behrendt, A. Laschewsky and H. Schlaad, Ring-Opening Metathesis Polymerization of Biomass-Derived Levoglucosenol, Angew. Chem., Int. Ed., 2019, 58, 6718–6721 CrossRef CAS PubMed.
- T. Debsharma, B. Schmidt, A. Laschewsky and H. Schlaad, Ring-Opening Metathesis Polymerization of Unsaturated Carbohydrate Derivatives: Levoglucosenyl Alkyl Ethers, Macromolecules, 2021, 54, 2720–2728 CrossRef CAS.
- P. G. Wuts and T. W. Greene, Greene's protective groups in organic synthesis, John Wiley & Sons, 2006 Search PubMed.
- Z. J. Witczak, P. Kaplon and M. Kolodziej, A new approach to isolevoglucosenone via the 2,3-sigmatropic rearrangement of an allylic selenide, J. Carbohydr. Chem., 2002, 21, 143–148 CrossRef CAS.
- M. Jevric, J. Klepp, J. Puschnig, O. Lamb, C. J. Sumby and B. W. Greatrex, Skeletal rearrangement of 6,8-dioxabicyclo[3.2.1]octan-4-ols promoted by thionyl chloride or Appel conditions, Beilstein J. Org. Chem., 2024, 20, 823–829 CrossRef CAS PubMed.
- X. Ma, X. Liu, P. Yates, W. Raverty, M. G. Banwell, C. Ma, A. C. Willis and P. D. Carr, Manipulating the enone moiety of levoglucosenone: 1,3-transposition reactions including ones leading to isolevoglucosenone, Tetrahedron, 2018, 74, 5000–5011 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2418634 and 2418635. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00630a |
| ‡ E. J. and A. R. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |