
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into the visible-light-driven O-arylation of carboxylic acids catalyzed by xanthine-based nickel complexes†
Rafael E.
Rodriguez-Lugo‡
 *a,
Joan
Sander§
ab,
Simon
Dietzmann
a,
Thomas
Rittner
c,
Jannes
Rückel
a,
Vanessa R.
Landaeta‡
a,
Jiyong
Park
bd,
Patrick
Nuernberger
c,
Mu-Hyun
Baik
*bd and
Robert
Wolf
*a
*a,
Joan
Sander§
ab,
Simon
Dietzmann
a,
Thomas
Rittner
c,
Jannes
Rückel
a,
Vanessa R.
Landaeta‡
a,
Jiyong
Park
bd,
Patrick
Nuernberger
c,
Mu-Hyun
Baik
*bd and
Robert
Wolf
*a
aUniversity of Regensburg, Institute of Inorganic Chemistry, 93040 Regensburg, Germany. E-mail: rafael.rodriguez@iccom.cnr.it; robert.wolf@chemie.uni-regensburg.de
bCenter for Catalytic Hydrocarbon Functionalizations, Institute for Basic Science (IBS), Daejeon, 34141, Republic of Korea
cUniversity of Regensburg, Institute of Physical and Theoretical Chemistry, 93040 Regensburg, Germany
dDepartment of Chemistry, Korea Advanced Institute of Science and Technology (KAIST), Daejeon, 43141, Republic of Korea. E-mail: mbaik2805@kaist.ac.kr
First published on 3rd January 2025
Abstract
We present a photocatalytic protocol for the O-arylation of carboxylic acids using nickel complexes bearing C8-pyridyl xanthines. Our studies suggest that the underlying mechanism operates independently of external photosensitizers. Stoichiometric experiments and crystallographic studies characterize the catalytically relevant Ni complexes. Spectroscopic and computational investigations propose a thermally controlled Ni(I)/Ni(III) cycle followed by a photochemical regeneration of Ni(I) species. Furthermore, the pathways leading to the hydrodehalogenation of aryl halides, the comproportionation of Ni(I) and Ni(III) species, the dimerization of Ni(I) intermediates and the influence of the counter ion on the cross-coupling reaction are unveiled. These investigations offer a comprehensive mechanistic understanding of the photocatalytic cross-coupling reaction catalyzed by a single Ni species and highlight key aspects of nickel-catalyzed metallaphotoredox reactions.
Introduction
Metallaphotoredox catalysis effectively merges photoredox catalysis and transition metal catalysis, enabling challenging bond formations and novel chemical reactions under benign conditions.1,2 While a variety of transition metals have been employed, nickel catalysis has enjoyed particular success in facilitating the formation of new C–O, C–N, C–S, and C–C bonds through photoactivated cross-coupling reactions.1,3–11 These dual photocatalytic strategies often use commercially available bipyridine chelates combined with external photosensitizers.12 Among these motifs, 4,4′-di-tert-butyl-2,2′-bipyridine (dtbbpy) usually is the ligand of choice.13–15 In addition, engineered photoactive bipyridine-like scaffolds have also proven effective.16Aryl-esterification is a versatile C–O coupling reaction with wide-ranging applications in fine chemical, pharmaceutical, and natural product synthesis.17–20 Numerous approaches have been investigated for the synthesis of aryl esters using Ni catalysts, including thermal,15,21 electrochemical,22 dual photocatalytic,13,14,23–28 semi-heterogenous14 and single-atom (heterogeneous) photocatalytic29 protocols. Moreover, most documented examples are limited in substrate scope, primarily focusing on aryl iodides,16,30 or require elevated temperatures.31
Since 2017, several reports have detailed the nickel-catalyzed arylation of carboxylates (Fig. 1).13–15 The group led by MacMillan described that Ni-bipyridine catalysts can facilitate visible-light-driven esterification reactions using an iridium complex as photosensitizer.13 Pieber and Seeberger have used a semi-heterogeneous system combining graphitic carbon nitride photocatalysts and nickel complexes for the C–O coupling reaction.13,14 In addition, Tosoni, Vilé and co-workers recently reported that a Ni atom-supported carbon nitride material effectively catalyzes cross-coupling of carboxylic acids and alkyl halides.29 In early 2020, Nocera and co-workers demonstrated that photoredox-like amination, etherification, and esterification of aryl bromides can be achieved in the dark using a Ni(II)/dtbbpy catalyst in conjunction with zinc as a sacrificial reductant, supporting the hypothesis of a thermally driven catalytic cycle.15 Additionally, there are a few reports where nickel catalysts are employed without external photosensitizers.31 A photoactive bipyridine-like ligand, which combines substituted quinoline and pyridine motifs, has been used for various metallaphotoredox cross-coupling reactions as described by Li and co-workers.16 The Pieber group introduced carbazole-decorated bipyridine ligands, Czbpy (Czbpy = 5,5′-dicarbazolyl-2,2′-bipyridyl and 4,4′-dicarbazolyl-2,2′-bipyridyl), that promote intraligand charge transfer to activate the nickel catalyst.30,32 Recently, Mirica and co-workers developed a single nickel catalyst for C–O cross-coupling reactions using a tridentate pyridinophane ligand.33 These advancements in single-metal photoredox catalysts have significantly enhanced the utility of metallaphotoredox catalysis in C–O cross-coupling reactions.1,16,34
 | ||
| Fig. 1 (a) Ligands previously used in C–O cross-couplings, (b) proposed Ni(I)/Ni(III) mechanism for dual Ni-bpy-catalyzed C–O cross-couplings, (c) principal Ni catalyst activation modes, (d) C–O cross-couplings catalyzed by xanthine-based Ni species. | ||
The mechanism of Ni-catalyzed cross-coupling reactions has been debated in recent literature. Most studies focused on the Ni-bipyridine catalyst system for which several viable catalytic manifolds have been proposed, involving Ni(I)/Ni(III), Ni(0)/Ni(II) and Ni(0)/Ni(II)/Ni(III)/Ni(I) cycles.12,32,33,35–38 From these studies, it has emerged that a Ni(I)/Ni(III) cycle likely operates in most dual Ni-bipyridine-catalyzed C–E cross-couplings (E = C, O, S; Fig. 1b).15,32,39–44 Here, the (light-driven) reduction of the Ni(II) precursor to the catalytically competent Ni(I) species initiates the catalytic cycle, comprising individual steps that are accessible without the involvement of photochemical events. Several studies showed that oxidative addition to Ni(I) polypyridine complexes is thermally accessible.33,39–42,45 Van der Veen, Thomas, and Pieber suggested that direct photoactivation of a Ni(II)-halide complex generates the Ni(I) counterpart. These authors showed that the catalytic Ni(I)/Ni(III) cycle can be initiated by a visible-light-induced homolysis (VLIH).30,46–48 Moreover, synthetic mechanistic studies by Mirica and co-workers suggest a thermal Ni(I)/Ni(III) mechanism in the single Ni-catalyzed etherification of aryl bromides.33 In 2023, Bahamonde and co-workers presented mechanistic evidence of a Ni(0)/Ni(II)/Ni(III) cycle for heterogeneous Ni-photoredox amide arylation reactions.49 Recently, the Pieber group suggested the involvement of a Ni(I)/Ni(III) cycle in C(sp2)–C(sp3) cross-coupling reactions.32 Although the Ni(I)/Ni(III) cycle seemingly operates in many reported protocols, the determination of a specific catalytic mechanism is intricate for any new catalyst system. Indeed, it has been noted by Hadt and co-workers that the population of a specific catalyst manifold is governed by the electronic structure of the Ni catalysts,12 which is highly dependent on the chemical environment of the metal atom and especially the ligands.43 Therefore, the mechanism of Ni-catalyzed cross-couplings must be examined case by case. Previous studies have considered several catalyst activation modes (Fig. 1c). In dual photocatalytic systems, one-electron reduction of Ni(I) complexes may generate reactive Ni(0) species capable of undergoing oxidative addition with aryl-halides.37 Pieber and Seeberger demonstrated that light irradiation can produce photoexcited Ni(II)* species, facilitating the product's reductive elimination.27 In addition, Hess described the photochemical reduction of Ni(II) complexes by amines, presenting an alternative light-driven electron transfer process for activating Ni complexes that does not require an additional photocatalyst.50,51
In this study, we demonstrate that the combination of fluorescent C8-pyridyl xanthines and Ni(II) salts facilitates the light-driven O-arylation of carboxylates, specifically using xanthine derivatives such as N7-substituted theophylline or caffeine. Xanthines are appealing scaffolds because they are naturally abundant compounds found in edible products such as coffee, tea, and chocolate. By chemically modifying the xanthine backbone, it is possible to create bidentate ligand motifs that serve as luminescent molecules.52,53 Our findings indicate that these xanthine-based nickel complexes act as effective catalysts in the light-driven O-arylation of carboxylates. To understand how this xanthine-based nickel system functions, we conducted a mechanistic investigation using both experimental and computational techniques. We reveal the coordination behavior and photophysical properties of selected ligands and complexes, which include the isolation and crystallographic characterization of pyridyl-xanthine Ni(I) complexes. Density Functional Theory (DFT) calculations provide essential insights into the mechanistic pathways, suggesting that a thermally accessible Ni(I)/Ni(III) cycle is involved. This cycle is initiated by the light-promoted reduction of the Ni(II) precursor complexes.
Results and discussion
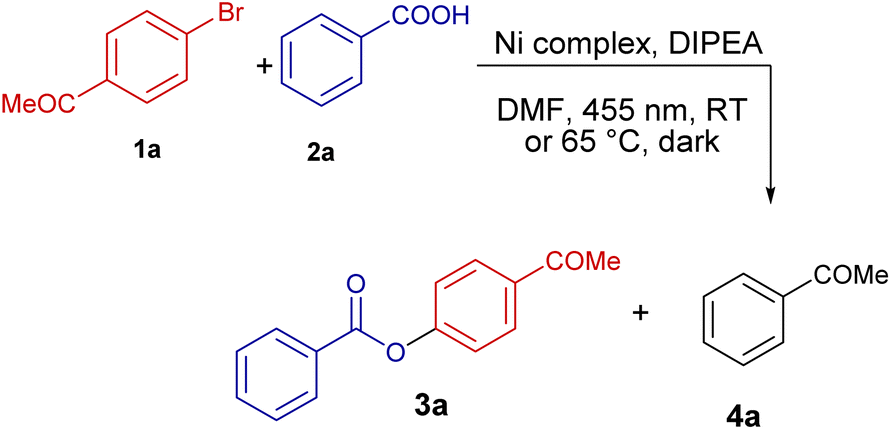
Photocatalytic cross-coupling of carboxylates and aryl halides
A library of several C8-pyridyl xanthine chelate ligands was prepared by modification of established procedures (see ESI for detailed information†).52,54–56 The synthesized ligands and simple Ni(II) salts were used as in situ generated Ni(II) catalysts, which enabled photocatalytic cross-coupling of 4′-bromoacetophenone (1a) with benzoic acid (2a) (Table 1). Among the ligands tested, the para-anisyl substituted xanthine derivative p-AnisNNH (entry 1, p-Anis = p-(OCH3)-C6H4) was most efficient, surpassing the yield and selectivity of 4,4′-di-tert-butyl-2,2′-bipyridine (dtbbpy) which is commonly used for nickel photocatalysis (Table 1, entry 3).1 Typically, alkyl substituents on the N7 atom of the xanthine scaffold, R3 = Me, iPr and Bn, yielded either low catalytic activity or poor selectivity (Table S3, entries 1–4†). In general, aromatic substituents on the xanthine unit, R3 = C6H5, 2-C5H4N, 4-OCH3-C6H4, allowed for high conversions (Table S3, entries 5–8†) except for ligands p-AnisNN6Me and p-OHPhNNH (Table S3, entries 9 and 10†), that gave rise to moderate conversions. The reaction selectivity varies depending on the nature of the aromatic ring on the xanthine N7 atom, with p-OMe or p-OH exhibiting the best product/side product ratios (Table S3, entries 8 and 10†).|
|
|||||
|---|---|---|---|---|---|
| Entry | Metal salt | Ligand | 3a (%) | 4a (%) | Sel.b3a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4a :4a |
| a Standard conditions: 4-bromoacetophenone (39.9 mg, 0.20 mmol), benzoic acid (48.9 mg, 0.40 mmol, 2 equiv.), Ni salt (10.0 μmol, 5 mol%), p-AnisNNH (72.0 mg, 20.0 μmol, 10 mol%), DIPEA (142 μL, 0.81 mmol, 4 equiv.), DMF (0.50 mL, 0.4 mM), 18 h, 40 °C, blue LED (455 nm, 7.0 W, photoreactor TAK 120). b Yield and selectivity (sel.) determined by GC-FID using 1,3,5-trimethoxybenzene as internal standard (8.4 mg, 0.05 mmol) and an appropriate calibration curve. c Average of five independent runs. d No light irradiation. dtbbpy = 4,4′-di-tertbutyl-2,2′-bipyridine. | |||||
| 1c | Ni(OTf)2 | p -AnisNNH | 64 | 32 | 2.00 |
| 2 | NiBr2 | p -AnisNNH | 52 | 35 | 1.49 |
| 3 | Ni(OTf)2 | dtbbpy | 30 | 20 | 1.50 |
| 4 | Ni(OTf)2 | None | 15 | 12 | 1.25 |
| 5 | None | p -AnisNNH | — | 39 | — |
| 6d | Ni(OTf)2 | p -AnisNNH | n.d. | n.d. | — |

Control reactions showed that the lack of an exogenous ligand resulted in a diminished conversion and poor selectivity (Table 1, entry 4). Cross-coupling was not observed in the absence of nickel or without irradiation (Table 1, entries 5 and 6). Details of the optimization steps under different reaction conditions are provided in the ESI,† including the evaluation of the effect of incorporating some common external photosensitizers (Table S2†).
Considering the results obtained in the initial evaluation, we expanded the scope of the reaction to test a range of substrates under the optimized reaction conditions (Fig. 2). Substrates featuring electron-withdrawing groups (EWG) at the o- or p-positions on aryl bromide 1 furnished good product yields (64–74%) (products 3a, 3f, 3h, 3i). The 3,5-(CF3)2-substituted product 3j was obtained in 95% yield. In contrast, substrates with EWGs at the m-position (Br, 3c; CN, 3e) or a heterocyclic substituent (2-py, 3g) afforded low to moderate yields (25–40%). A halide atom (Cl) in p-position resulted in a low yield (12%). Similarly, CN substitution at the o-position significantly reduced the catalytic activity (3d, 13%). Aryl bromides bearing electron-donating groups proved unsuitable as coupling partners for benzoic acid (3k–n). Aryl iodides (3ha) also showed lower product yields, with an increased formation of the dehydrohalogenation product (4h). Aryl chlorides did not undergo the desired transformation (3hc).
 | ||
| Fig. 2 Substrate scope for the nickel catalyzed arylation of carboxylates. Conditions: aryl bromide (0.20 mmol), carboxylic acid (0.40 mmol, 2 equiv.), Ni(OTf)2 (3.6 mg, 10.0 μmol, 5 mol%), p-AnisNNH (7.2 mg, 20.0 μmol, 10 mol%), N,N-diisopropylethylamine (DIPEA) (142 μL, 0.81 mmol, 4 equiv.), DMF (0.50 mL, 0.4 mM), 18 h, 40 °C, blue LED (455 nm, 7.0 W, photoreactor TAK 120). Yields were determined by GC-FID. Yields of isolated product are given in parentheses. | ||
We also screened a series of carboxylic acids 2. Aliphatic R2 substitution of 2 led to moderate yields (44–66%) for aryl esters 3o–s. The use of electron-poor benzoic acids did not significantly influence the product yield. Whereas moderate improvements were observed for compounds 3u and 3v, with yields of 46% and 52%, respectively, the cyano-substituted substrate 3t resulted only in trace amounts of the product. Electron-rich carboxylic acids provided the respective esters in moderate yields, 3w–z (55–66%).
Synthesis and characterization of catalytically relevant species
The coordination behavior of the most effective ligands in the catalytic transformation described above, PyrNNH and p-AnisNNH, was investigated towards Ni(OTf)2 and NiBr2 (Scheme 1). Both ligands form stable and isolable 1:1 complexes [NiBr2(R3NNH)]2 (5Br, R3 = 2-pyr; 6Br, R3 = p-Anis), as well as 2:1 complexes [NiBr2(R3NNH)2] (7Br, R3 = 2-pyr; 8Br, R3 = p-Anis) with NiBr2, depending on the ligand stoichiometry used in the synthesis. In the solid state, complex 6Br adopts a bromide-bridged dimeric structure featuring five-coordinate Ni2+ cations, comprising one bidentate C8-pyridyl xanthine chelate and three bromide ligands (see Scheme 1b). Notably, the solution 1H-NMR spectra of complexes 5Br and 6Br exhibited paramagnetically shifted signals (see the ESI for details†).
 | ||
| Scheme 1 (a) General synthesis of xanthine-based Ni(II) complexes [NiBr2(R3NNH)] (5Br/6Br) and [NiX2(R3NNH)2] (7/8) with solid-state molecular structures of (b) [NiBr(μ-Br)(p-AnisNNH)]2 (6Br), and (c) [NiBr2(PyrNNH)2] (7Br). Displacement ellipsoids are drawn at the 50% probability level. H-atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°] for (6Br)2: Ni–N1 2.052(3), Ni–N2 2.013(3), Ni–Br1 2.4875(7), Ni–Br2 2.5549(7), Ni–Br3 2.4113(7), N1–Ni–N2 80.25(11), N1–Ni–Br1 167.94(9), Br1–Ni–Br3 94.59(2), Br2–Ni–Br3 154.45(3); for 7Br: Ni1–N1 2.284(2), Ni1–N2 2.059(2), Ni1–N3 2.065(2), Ni1–N4 2.244(2), Ni1–Br1 2.504(1), Ni1–Br2 2.500(1), N1–Ni1–N2 76.72(7), N3–Ni1–N4 76.96(7), Br1–Ni1–Br2 100.11(2). | ||
In addition, we isolated 2:1 complexes [Ni(OTf)2(R3NNH)2] (7OTf, R3 = 2-pyr; 8OTf, R3 = p-Anis). Complexes 7 and 8 feature hexacoordinate Ni(II) centers with cis-configuration in the solid state (Scheme 1). In solution, these octahedral Ni(II) complexes are NMR silent.
To investigate the possible role of the synthesized complexes in the catalytic arylation of carboxylates under study, we explored the photoreduction of the Ni(II) compounds. In this regard, we measured the UV-vis absorption spectra of the individual reaction components in the catalysis. Weak d–d transitions were observed in the visible spectrum of the hexacoordinated [Ni(DMF)6]2+ (3A2g → 3T1g(F),1Eg: 700–780 nm; 3A2g → 3T1g(P): 410 nm, Fig. S7 in the ESI†). None of the other components of the reaction mixture absorb light in the spectral range around 450 nm. Nevertheless, we found significant changes in the absorption spectra of the mixtures of NiBr2 or [NiBr2(p-AnisNNH)] (6Br) with N,N-diisopropylethylamine (DIPEA) in DMF after irradiation with blue LED light (455 nm). The resulting spectra showed increased absorption in the spectral regions 370–490 nm and 490–590 nm (Fig. 3). While the absorption below 400 nm stems from p-AnisNNH (Fig. S4 in the ESI†), the new absorption band at 370–490 nm can presumably be attributed to the decomposition of N,N-diisopropylethylamine radical cation (DIPEA˙+) and its reaction with Ni complexes.57 The absorption bands between 490–590 nm are likely associated with the formation of Ni(I) species (vide infra).
 | ||
| Fig. 3 UV-vis absorption spectra of NiBr2 (20 mM) with DIPEA (1.6 M, 80 equiv.) or 6Br with DIPEA (1.6 M, 80 equiv.) after thermal reaction at 65 °C (ΔT), and after 18 h of illumination at 455 nm (hv), respectively. | ||
To gain further insight into this matter, we also prepared low-valent Ni(I) and Ni(0) complexes with the ligand p-AnisNNH (Scheme 2). Commercially available [Ni(cod)2] (9; cod = 1,5-cyclooctadiene) was subjected to ligand exchange with p-AnisNNH (see ESI for details†). Unfortunately, all efforts to isolate and characterize the putative complex [Ni(cod)(p-AnisNNH)] (10) were unsuccessful, possibly due to its high sensitivity to temperature and light. In fact, the reaction mixture forms metallic mirrors alongside the free ligands p-AnisNNH and 1,5-COD above −30 °C or when exposed to daylight.
 | ||
| Scheme 2 (a) Synthesis of low valent Ni species [Ni(cod)(p-AnisNNH)] (10) and [NiX(p-AnisNNH)] (X = Br (11Br), OAr* (11OAr*)), starting from the precursor [Ni(cod)2] (9). (b) Structure of the dimer (11Br)2 calculated with DFT. (c) Solid-state molecular structure of 11OAr*. Displacement ellipsoids are drawn at the 40% probability level. H-atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°]. Ni–N1 1.975(3), Ni–N2 1.935(3), Ni–O4 1.819(7), C10–N2 1.366(4), C1–C2 1.463(4), C8–N1 1.349(4), N1–Ni–N2 82.21(11), N1–Ni–O4 140.59(9), N2–Ni–O4 136.84(8). | ||
To confirm the formation of 10, the related Ni(I) compound [NiBr(p-AnisNNH)] (11Br) was synthesized in two steps from 9. The in situ generated complex 10 was treated with 1 equiv. of complex 6Br to access 11Brvia comproportionation. Complex 11Br forms a precipitate that is insoluble in common organic solvents. Thus, characterization by solution NMR and single-crystal XRD analysis were not viable. The precipitate results from the formation of halide-bridged dimers of complex 11Br. Of note, similar precipitates were reported for Ni(I) complexes with bipyridine (bpy) and phenanthroline (phen) ligands.40,42,44,45,58–60 DFT simulations by Hadt on dimeric dtbbpy complexes of Ni(I) found bent and flat dimer structures with similar energies.45 To better understand the formation of this kind of species in our case, we performed DFT calculations on the structure of dimer (11Br)2. According to these, (11Br)2 exists only as a severely bent structure (spin state S = 1, Scheme 2b). We also found that the dimer formation is energetically uphill by ΔG = 11.2 kcal mol−1 in solution phase. Interestingly, the dimerization is exergonic in gas-phase (ΔG = −8.7 kcal mol−1 and ΔH = −1.3 kcal mol−1), suggesting that the precipitation of the compound is the driving force behind the dimerization.
An aryloxy substituted complex, [Ni(OAr*)(p-AnisNNH)] (11OAr*, Ar* = 2,4,6-(tBu)3-C6H2), closely related to 11Br, was accessed by treating a mixture of [Ni(cod)2] (9) and p-AnisNNH with the aryloxy radical ˙OAr*.61 In contrast to 11Br, complex 11OAr* is soluble in most common organic solvents. The 1H-NMR spectrum exhibits broad high-field shifted signals typical of a paramagnetic species. The molecular structure of 11OAr* was determined by single-crystal X-ray diffraction (Scheme 2c). The N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CN scaffold of the ligand backbone (C–N 1.349(4) and 1.366(4) Å, C–C 1.463(4) Å) remains unaltered relative to the free ligand p-AnisNNH (C–N 1.344(2) Å and 1.342(2) Å, C–C 1.471(2) Å). These findings indicate that the ligand is not reduced.
C–CN scaffold of the ligand backbone (C–N 1.349(4) and 1.366(4) Å, C–C 1.463(4) Å) remains unaltered relative to the free ligand p-AnisNNH (C–N 1.344(2) Å and 1.342(2) Å, C–C 1.471(2) Å). These findings indicate that the ligand is not reduced.
The UV-vis spectrum of 11OAr* supports our initial assignment that the bands between 490–590 nm observed in the photoreduction of the Ni(II) compounds correspond to possible Ni(I) species. In fact, a significant absorption between 400–600 nm was present in the UV-vis spectrum of 11OAr* (Fig. S7 in the ESI†). Moreover, UV-vis monitoring of a diluted and freshly prepared solution of 11Br, obtained in situ by treating [Ni(cod)2] with p-AnisNNH and 4-Br-C6H4-COMe, as shown in Scheme 3a (vide infra), revealed two small absorption bands at 470 and 520 nm (Fig. S10 in the ESI†). Once formed, the isolated material is insoluble in common organic solvents, probably due to the formation of the halide-bridged dimer (11Br)2 (vide supra), hence hampering the measurement of its absorption spectrum. Furthermore, the reduction of NiBr2 with Zn was previously reported to result in increased absorption between 500–600 nm.21
 | ||
| Scheme 3 (a) Oxidative addition of ArBr and transmetalation with benzoic acid from Ni(I) complex 11OAr* as well as decomposition from irradiation. (b) Oxidative addition of ArBr to Ni(0) complex 10. | ||
Additionally, and within our theoretical calculations (using DFT), we explored possible pathways for the reduction of Ni(II) (Scheme S11 in the ESI†). The spectroscopic observations and DFT calculations are consistent with the photoreduction of Ni(II) by DIPEA, proceeding initially to a Ni(I) species via light-induced single electron transfer (SET). The photoreduction most likely results from a Ni2+-centered d–d transition. Further reduction to Ni(0) species can occur as a side reaction. However, our calculations show that these unstable Ni(0) complexes undergo comproportionation with Ni(II) to Ni(I) (see Scheme S11†).
Finally, our efforts to isolate and characterize Ni(III) compounds bearing the ligand p-AnisNNH have been unsuccessful to date (vide infra and see ESI†).
Synthetic model reactions
Once the low-valent nickel species 10 and 11OAr* were identified, and 11OAr* was characterized, we tried to understand whether the Ar–Br bond in the aryl halide substrate can be activated under thermal conditions. In this regard, we examined the oxidative addition of aryl bromide ArBr to the in situ generated species 10 and to complex 11OAr*. By treating 11OAr* with ArBr (Ar = 3,5-(CF3)2-C6H3), we observed the formation of [NiBr2(p-AnisNNH)] (6Br) and [Ni(OAr*)2(p-AnisNNH)] (6OAr*) alongside Ar–Ar and Ar–H (Scheme 3a). The formation of Ar–Ar and Ar–H was detected by 19F-NMR spectroscopy. This is consistent with the oxidative addition of ArBr to 11OAr* forming intermediate 12OAr*,Br, followed by Ni–Ar bond homolysis to 13OAr*,Br and ligand exchange of the complexes. Alternatively, the presumed Ni(III) species 12OAr*,Br can comproportionate with 11OAr* forming Ni(II) compounds. Upon ligand redistribution, reductive elimination from the assumed binary complex [Ni(Ar)2(p-AnisNNH)] (6Ar) would lead to Ar–Ar, while HAT from DIPEA˙+ to any Ni–Ar species will lead to Ar–H. Further evidence of the Ni–Ar bond homolysis was obtained by treating the Ni(II) complex [Ni(Ar)Br(PPh3)2] (14) with p-AnisNNH (one equiv.), affording the Ni(I) complex [NiBr(p-AnisNNH)(PPh3)] (11Br·PPh3), which was isolated and characterized by 1H-NMR spectroscopy and single crystal XRD analysis (see the ESI for further details†).Similarly, upon treatment of the in situ generated Ni(0) complex 10 with 4-bromoacetophenone (Scheme 3b), the Ni(I) complex 11Br was formed along with the biphenyl (Ar–Ar) and the hydrodehalogenation (Ar–H) side products (Ar = 4-(COMe)-C6H4). The formation of Ar–Ar was observed by GC-MS analysis of the reaction mother liquor and confirmed by 1H- and 13C{1H}-NMR spectroscopy (see Scheme S8 and Fig. S83 in the ESI†). Likely, Ni–Ar bond homolysis took place, thus reducing Ni(II) to Ni(I), as reported by Doyle.47 Again, another scenario to explain this reactivity would be the comproportionation of the presumed Ni(II) species [Ni(Ar)(Br)(p-AnisNNH)] with 10, thus forming Ni(I) compounds.
Our synthetic studies therefore suggested that low-valent nickel species 10 and 11OAr* can activate the Ar–Br bond under thermal conditions. Additionally, we treated 11OAr* with PhCOOX (X = H, K) to simulate the transmetalation reaction. Instead of the putative Ni(I) complex [Ni(O(CO)Ph)(p-AnisNNH)] (11OBAc), the Ni(II) complex [Ni(O(CO)Ph)2(p-AnisNNH)] (6OBAc) was formed alongside a metallic mirror of Ni(0). This indicates a disproportionation of the Ni(I) species 11OBAc driven by precipitation of Ni(0). Disproportionation can also be observed when irradiating complex 11OAr* with blue light. These results underline the susceptibility of the Ni(I) and Ni(III) species towards deactivation via comproportionation, driven either thermally or by light.
Furthermore, we aimed to identify viable catalytic intermediates among the abovementioned Ni species. Specifically, we repeated the cross-coupling of benzoic acids with aryl bromides using stoichiometric amounts of the nickel complexes obtained in different formal oxidation states. Complexes 6Br, 10 and 11OAr* were reacted with 1a in a 1:1 ratio, either under light irradiation (455 nm) or in the dark (thermally). The results are summarized in Table 2. Here, the cross-coupling reaction was not observed under thermal conditions (Table 2, entries 1, 3, 5), regardless of the nickel complex and its formal oxidation state. Instead, we observed hydrodehalogenation of 1a promoted by Ni(0) and Ni(I) complexes (compounds 10 and 11OAr*, entries 1 and 3). Note that the Ni(II) complex 6Br was not able to produce cross-coupling or hydrodehalogenation product under thermal conditions (entry 5). However, substantial amounts of hydrodehalogenation product 4a were formed under 455 nm LED light irradiation (entry 6). These findings agree with the reduction of 6Br to Ni(I) complex(es) under irradiation, that can promote hydrodehalogenation. Similarly, more hydrodehalogenation occurred in the presence of the Ni(0) species 10 under irradiation. Strikingly, we observed full conversion of the starting material and significant formation of the cross-coupling product 3a for the reaction with complex 11OAr* under blue light irradiation (entry 4). The thermal process only delivered a relatively small amount of hydrodehalogenation (entry 3).
|
|
|||||
|---|---|---|---|---|---|
| Entry | [Ni] | T | λ | 3a (%) | 4a (%) |
| a Conditions: 4-bromoacetophenone (3.9 mg, 20.0 μmol), benzoic acid (4.9 mg, 40.0 μmol, 2 equiv.), Ni species (20.0 μmol, 1 equiv.), DIPEA (14.2 μL, 80.0 μmol, 4 equiv.), DMF (0.50 mL, 0.04 mM), 18 h, 40 °C or 65 °C, blue LED (455 nm, operating power: 7.0 W, photoreactor TAK 120) or dark. b Yield and selectivity were determined by GC-FID using 1,3,5-trimethoxybenzene as internal standard (8.4 mg, 50.0 μmol). c Complex 10 was generated in situ by mixing [Ni(cod)2] (20.0 μmol, 1 equiv.) and p-AnisNNH (200 μmol, 1 equiv.). | |||||
| 1 | Ni(0) 10c | 65 °C | — | — | 70 |
| 2 | Ni(0) 10c | 40 °C | 455 nm | — | 90 |
| 3 | Ni(I) 11OAr* | 65 °C | — | — | 21 |
| 4 | Ni(I) 11OAr* | 40 °C | 455 nm | 50 | 50 |
| 5 | Ni(II) 6Br | 65 °C | — | — | — |
| 6 | Ni(II) 6Br | 40 °C | 455 nm | — | 57 |
In conclusion, the model cross-coupling reactions show that only the Ni(I) species 11OAr* facilitates the O-arylation of benzoic acid 2a with aryl bromide 1a. The lack of product formation under thermal conditions with 11OAr* can be explained by the high concentration of Ni(I) species in the reaction mixture and a higher energy barrier for rate-limiting oxidative addition or reductive elimination steps compared to 11Br (see Computational section). Ni(III) species formed by oxidative addition of aryl bromides are more likely to comproportionate with the abundant Ni(I) rather than following a productive reaction pathway under these conditions. Likewise, the same argument applies to the lack of product formation under photochemical conditions with 6Br, in contrast with the catalytic experiment shown in Table 1, entry 2. Again, under the relatively high nickel concentration in the model stoichiometric reaction, the comproportionation of 11Br and 12X,Br (X = Br, OBAc) kinetically competes with the productive reaction step, the reductive elimination from Ni(III).
Computational investigations of the mechanistic pathway
The results obtained, including the isolation of possible catalytic intermediates, stoichiometric model reactions and UV-vis monitoring experiments, were taken into consideration to perform a detailed computational mechanistic investigation using density functional theory (DFT) calculations. This allowed for the construction of a complete catalytic cycle in atomistic detail, including all intermediate and transition state structures.We performed DFT calculations to compare competing mechanistic pathways. Technical details of these calculations are given in the ESI.†Scheme 4 shows the most plausible reaction energy profile of the proposed thermal catalytic cycle, which initiates from Ni(I) species 11Br. We selected complex 11Br as the model system for the calculations and compared two scenarios where either the oxidative addition of aryl bromide (path A) or the transmetalation of carboxylate (path B) initiates the catalytic cycle. We found that both pathways are equally viable. In path A, the oxidative addition of the aryl bromide to generate complex 12Br,Br traverses a concerted transition state TS(11Br-12Br,Br), followed by the transmetalation of the carboxylate leading to 12OBAc,Br. Notably, ion pairing between a bromide anion and the cationic amine reductant (DIPEA-H+Br−)43 facilitates the removal of the anion. In pathway B, the order of these steps is reversed: the transmetalation of carboxylate leads to 11OBAc, followed by the oxidative addition TS(11OBAc-12OBAc,Br). The activation energies of the first steps of the two pathways are 14.7 and 14.3 kcal mol−1 respectively. These energies suggest that both pathways are likely operative in parallel. These two initial steps result in a common Ni(III) intermediate 12OBAc,Br. The reductive elimination passes through a concerted transition state TS(12OBAc,Br-3a), which is the most challenging transformation among the computed reaction steps, with an activation energy of 22.3 kcal mol−1. This barrier is sufficiently low to enable a fast reaction under the experimental conditions, indicating that additional energy from photoexcitation is unnecessary for the elementary steps in the catalytic cycle. For instance, when the reaction is performed without irradiation, but using zinc powder as the sacrificial reductant instead, 25% of the O-aryl ester was obtained (Table S7, entry 8†). Moreover, only 15% of product was obtained using a liquid-cooled photoreactor, which keeps the reaction temperature at 18 °C (Table S7, entry 6†), in strong contrast with the 66% yield obtained at 40 °C (Table S7, entry 1†). This evidence is consistent with a significant contribution of thermally driven reaction steps to product formation. Notably, the Pieber group has recently reported a C–C cross-coupling system that operates through a Ni(I)/Ni(III) manifold, without involving photoredox reactivity, according to the mechanistic evidence.32
 | ||
| Scheme 4 Core mechanism of the cross-coupling reaction. (A) Oxidative addition-first mechanism; (B) transmetalation-first mechanism. Ar = 4-(COMe)-C6H4. | ||
Our calculations also explain the influence of the different counter-anions (X = Br, OTf− or OAr*−) on the reaction (see Scheme S14 in the ESI†). The Ni(I) triflate complex OTf− (11OTf) is readily converted into 11Br, which is part of the core mechanism of the cross-coupling reaction (Scheme 4). When OAr*− is used, the activation barrier of the oxidative addition (ΔG‡ = 23.7 kcal mol−1, Scheme S14c†) is higher than that of the reductive elimination commencing from 11Br (TS(12OBAc,Br-3a) in Scheme 4, ΔG‡ = 22.3 kcal mol−1). This barrier is consistent with the formation of cross-coupling product under blue-light irradiation (vide supra, Table 2 entry 4). The lacking success of the thermal reaction under the same conditions (Table 2 entry 3) is likely attributed to the rapid synproportionation of Ni(III) intermediates with 11OAr* (vide supra).
The complete mechanism of Ni-catalyzed aryl esterification reaction is summarized in Scheme 5. The catalytic cycle is initiated via the light-promoted reduction of Ni(II) complexes of the type [NiX2(p-AnisNNH)] (X = Br, OTf−, OAr*−, PhCOO−) to Ni(I) complexes of the type [NiX(p-AnisNNH)] (11X), which is described in detail in the ESI.† Starting from 11X, two alternative mechanistic pathways are feasible. In the first scenario, 11X undergoes oxidative addition of aryl bromide forming the Ni(III) intermediate 12X,Br. Subsequently, transmetalation with benzoic acid generates intermediate 12X,OBAc or 12OBAc,Br. In the second scenario, the transmetalation takes place before oxidative addition, forming the benzoate complex 11OBAc. Then, oxidative addition of aryl bromide yields the same type of intermediate 12OBAc,Br. The catalytic cycle is closed by the reductive elimination of the cross-coupling product 3, which regenerates the Ni(I) intermediate 11X.
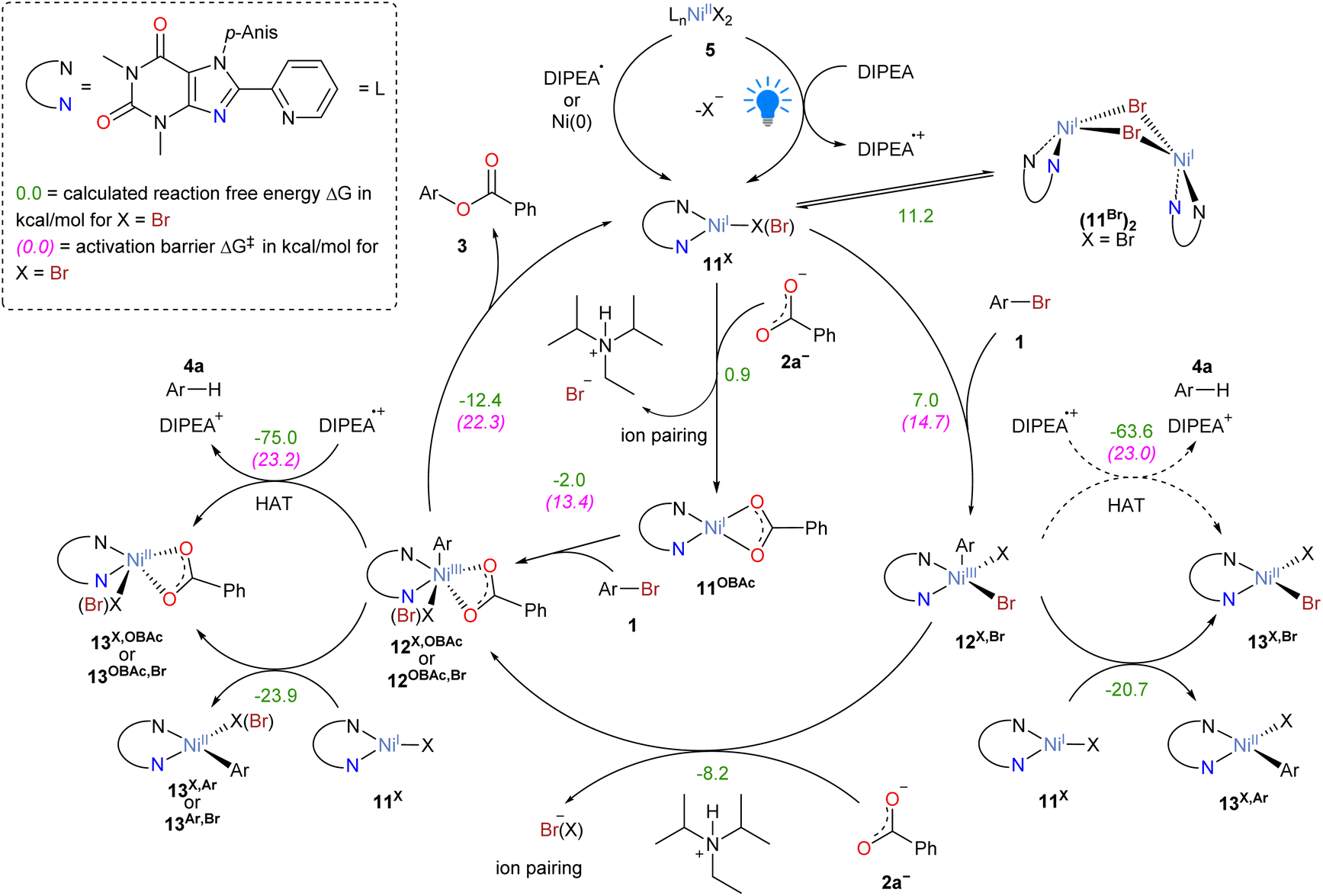 | ||
| Scheme 5 Complete mechanistic proposal of the visible-light-activated Ni-catalyzed O-arylation reaction. The energy unit is kcal mol−1. X = Br, OTf, OAr*. | ||
We note that Ni(III) species 12 can participate in side-reactions, which limit the selectivity and continuity of the catalytic processes (Scheme S13 in the ESI†). Our computational study suggests that 12OBAc,Br can abstract a hydrogen atom from DIPEA˙+, with an activation energy of ΔG‡ = 23.2 kcal mol−1 (TS(12OBAc,Br-13OBAc,Br)), indicating that hydrogen atom transfer (HAT) is thermally viable, but slower than the reductive elimination (ΔG‡ = 22.3 kcal mol−1, TS(12OBAc,Br-3a)). As a result, the hydrodehalogenation side product 4 occurs as a side product in the catalytic reaction. It is also noted that the thermal activation of HAT from intermediate 12Br,Br is not feasible as the overall barrier of such reaction is 30.0 kcal mol−1, associated with TS(12Br,Br-6Br).
In addition, comproportionation of Ni(III) species 12 with complex 11 results in catalytically inactive Ni(II) compounds. These Ni(II) species can reenter the catalytic cycle upon light-activated reduction. In fact, the redox couple Ni(II)/Ni(I) is reversible, as confirmed by cyclic voltammetry measurements with 11OAr* (Fig. S84 in the ESI†). Furthermore, two complexes of type 11Br can be deactivated by the formation of a bent dimer (11Br)2 as reported by Hadt.45 The dimerization is uphill in energy (ΔG = 11.2 kcal mol−1), but is driven by the precipitation of the insoluble dimer.
Conclusions
Xanthine-based chelate motifs can act as supporting ligands in nickel-catalyzed O-arylation of carboxylic acids with aryl bromides, achieving up to 95% product yields under blue light irradiation, while also facilitating the isolation of well-defined Ni(I) and Ni(II) complexes for mechanistic studies. Through UV-vis absorption spectroscopy and DFT calculations, we identified a thermally controlled Ni(I)/Ni(III) mechanism in C–O cross-coupling reactions. Furthermore, the sequence of the oxidative addition and transmetalation steps is variable and depends on the counter-ion present in the reaction. This observation may have implications for other Ni-catalyzed cross-coupling reactions. We also studied the hydrodehalogenation side-reaction and catalyst deactivation in our system. The computational investigations suggested that HAT using DIPEA˙+ as the hydrogen atom source is a viable minor pathway. This might explain the hydrodehalogenation under our reaction conditions. In fact, the process has an activation energy similar to the reductive elimination of the cross-coupling product. Concerning catalyst deactivation, the comproportionation of Ni(I) and Ni(III) species generates inactive Ni(II) compounds which require reactivation by light-induced reduction.DIPEA can play three possible independent roles in the catalytic cycle: First, it acts as a sacrificial reducing agent, producing Ni(I) species and generating the DIPEA˙+ radical, which triggers HAT as a collateral effect. Second, DIPEA functions as a Brønsted base by deprotonating both the carboxylic acid and DIPEA˙+ radical cation. Lastly, the protonated form of DIPEA (DIPEA-H+) facilitates the dissociation of Br− anions and enables transmetalation.
This study underscores the effectiveness of xanthine-based ligands in facilitating nickel-catalyzed C–O couplings. Notably, these ligands eliminate the need for an external photosensitizer and enable detailed mechanistic analyses. The combination of experimental and computational methods supports a mechanistic scenario in which the cross-couplings occur via a thermal mechanism, without requiring photochemical processes during the crucial reductive elimination step. Instead, visible light serves to sustain the thermal nickel(I)/nickel(III) cycle by regenerating the active nickel(I) species.
Data availability
The data supporting the findings of this study are available within the paper and its ESI† files. Crystallographic Information Files (CIFs) have been deposited at the CCDC under the deposition numbers 2354527–2354534 (see ESI†). These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.Author contributions
Rafael E. Rodriguez-Lugo: conceptualization, investigation (synthetic, mechanistic and catalytic studies), writing – original draft, review and editing. Joan Sander: investigation (quantum chemical studies), writing – original draft, review and editing. Simon Dietzmann: investigation (synthetic and mechanistic studies). Thomas Rittner: investigation (spectroscopic investigations). Jannes Rückel: investigation (synthetic and mechanistic studies). Vanessa R. Landaeta: writing – original draft, review and editing. Jiyong Park: supervision, writing – review and editing. Patrick Nuernberger: supervision, writing – review and editing, funding acquisition. Mu-Hyun Baik: supervision, writing – review and editing. Robert Wolf: conceptualization, writing – review and editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Hannes Schneider and David Preitschaft for valuable experimental assistance, Sabrina Dinauer for performing the EPR measurements and the research group of Prof. Burkhard König for technical support. We also acknowledge the support of the Elite Network of Bavaria. This work was funded by the Deutsche Forschungsgemeinschaft (CRC 325 Assembly Controlled Chemical Photocatalysis – 444632635, projects A4 and A5) and the Alexander von Humboldt Stiftung (Georg Förster Research Fellowship, VEN – 1186623 – GF-E). We thank the Institute for Basic Science in Korea for financial support (IBS-R010-A1).References
- A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Chem. Rev., 2022, 122, 1485 CrossRef CAS.
- C. Förster and K. Heinze, Chem. Soc. Rev., 2020, 49, 1057 RSC.
- O. S. Wenger, Chem.–Eur. J., 2021, 27, 2270 CrossRef CAS PubMed.
- M. Marchi, G. Gentile, C. Rosso, M. Melchionna, P. Fornasiero, G. Filippini and M. Prato, ChemSusChem, 2022, 15, e202201094 CrossRef CAS PubMed.
- C. Zhu, H. Yue, J. Jia and M. Rueping, Angew. Chem., Int. Ed., 2021, 60, 17810 CrossRef CAS PubMed.
- K. L. Skubi, T. R. Blum and T. P. Yoon, Chem. Rev., 2016, 116, 10035 CrossRef CAS PubMed.
- H. Luo, Y. Feng and L. Lin, ChemCatChem, 2023, 15, e202300303 CrossRef CAS.
- L. Zou, R. Sun, Y. Tao, X. Wang, X. Zheng and Q. Lu, Nat. Commun., 2024, 15, 5245 CrossRef CAS PubMed.
- A. Y. Chan, A. Ghosh, J. T. Yarranton, J. Twilton, J. Jin, D. M. Arias-Rotondo, H. A. Sakai, J. K. McCusker and D. W. C. MacMillan, Science, 2023, 382, 191 CrossRef CAS.
- B. Ling, S. Yao, S. Ouyang, H. Bai, X. Zhai, C. Zhu, W. Li and J. Xie, Angew. Chem., Int. Ed., 2024, 63, e202405866 CrossRef CAS.
- H. Wang and T. XU, Chem Catal., 2024, 4, 100952 CrossRef CAS.
- D. A. Cagan, D. Bím, N. P. Kazmierczak and R. G. Hadt, ACS Catal., 2024, 14, 9055 CrossRef CAS.
- E. R. Welin, C. Le, D. M. Arias-Rotondo, J. K. McCusker and D. W. C. MacMillan, Science, 2017, 355, 380 CrossRef CAS.
- B. Pieber, J. A. Malik, C. Cavedon, S. Gisbertz, A. Savateev, D. Cruz, T. Heil, G. Zhang and P. H. Seeberger, Angew. Chem., Int. Ed., 2019, 58, 9575 CrossRef CAS PubMed.
- R. Sun, Y. Qin and D. G. Nocera, Angew. Chem., Int. Ed., 2020, 59, 9527 CrossRef CAS PubMed.
- J. Li, C.-Y. Huang and C.-J. Li, Chem, 2022, 8, 2419 CAS.
- D. G. Brown and J. Boström, J. Med. Chem., 2016, 59, 4443 CrossRef CAS PubMed.
- A. Parenty, X. Moreau and J.-M. Campagne, Chem. Rev., 2006, 106, 911 CrossRef CAS PubMed.
- W. Cai, B. Chekal, D. Damon, D. LaFrance, K. Leeman, C. Mojica, A. Palm, M. St Pierre, J. Sieser, K. Sutherland, R. Vaidyanathan, J. van Alsten, B. Vanderplas, C. Wager, G. Weisenburger, G. Withbroe and S. Yu, Transition metal-catalyzed couplings in process chemistry, Case studies from the pharmaceutical industry, Wiley-VCH, Weinheim, Germany, OP, 2013 Search PubMed.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- L. A. Wolzak, F. J. de Zwart, J.-P. H. Oudsen, S. A. Bartlett, B. de Bruin, J. N. H. Reek, M. Tromp and T. J. Korstanje, ChemCatChem, 2022, 14, e202200547 CrossRef CAS.
- Y. Mo, Z. Lu, G. Rughoobur, P. Patil, N. Gershenfeld, A. I. Akinwande, S. L. Buchwald and K. F. Jensen, Science, 2020, 368, 1352 CrossRef CAS PubMed.
- H. Luo, G. Wang, Y. Feng, W. Zheng, L. Kong, Y. Ma, S. Matsunaga and L. Lin, Chem.–Eur. J., 2023, 29, e202200547 Search PubMed.
- X. Zhu, Q. Fan, W. Luo, D. Wang, Y. Jia, H. Li, Z. Wang, Q. Xiao and X. He, Chin. J. Chem., 2023, 41, 411 CrossRef CAS.
- M. He, S. Yang, X. Yu and M. Bao, Synlett, 2022, 33, 1189 CrossRef CAS.
- W. Zu, C. Day, L. Wei, X. Jia and L. Xu, Chem. Commun., 2020, 56, 8273 RSC.
- J. A. Malik, A. Madani, B. Pieber and P. H. Seeberger, J. Am. Chem. Soc., 2020, 142, 11042 CrossRef CAS PubMed.
- J. Lu, B. Pattengale, Q. Liu, S. Yang, W. Shi, S. Li, J. Huang and J. Zhang, J. Am. Chem. Soc., 2018, 140, 13719 CrossRef CAS.
- M. A. Bajada, G. Di Liberto, S. Tosoni, V. Ruta, L. Mino, N. Allasia, A. Sivo, G. Pacchioni and G. Vilé, Nat. Synth., 2023, 2, 1092 CrossRef CAS.
- C. Cavedon, S. Gisbertz, S. Reischauer, S. Vogl, E. Sperlich, J. H. Burke, R. F. Wallick, S. Schrottke, W.-H. Hsu, L. Anghileri, Y. Pfeifer, N. Richter, C. Teutloff, H. Müller-Werkmeister, D. Cambié, P. H. Seeberger, J. Vura-Weis, R. M. van der Veen, A. Thomas and B. Pieber, Angew. Chem., Int. Ed., 2022, 61, e202211433 CrossRef CAS PubMed.
- T.-T. Zhao, H.-N. Qin and P.-F. Xu, Org. Lett., 2023, 25, 636 CrossRef CAS PubMed.
- L. Anghileri, H. Baunis, A. R. Bena, C. Giannoudis, J. H. Burke, S. Reischauer, C. Merschjann, R. F. Wallik, G. Simionato, S. Kovalenko, L. Dell'Amico, R. M. van der Veen and B. Pieber, ChemRxiv, 2024, preprint, DOI:10.26434/chemrxiv-2024-896n0.
- H. Na and L. M. Mirica, Nat. Commun., 2022, 13, 1313 CrossRef CAS PubMed.
- I. Ghosh, N. Shlapakov, T. A. Karl, J. Düker, M. Nikitin, J. V. Burykina, V. P. Ananikov and B. König, Nature, 2023, 619, 87 CAS.
- N. Sanosa, P. Ruiz-Campos, D. Ambrosi, D. Sampedro and I. Funes-Ardoiz, Int. J. Mol. Sci., 2023, 24, 9145 CrossRef CAS PubMed.
- S. Gisbertz, S. Reischauer and B. Pieber, Nat. Catal., 2020, 3, 611 CrossRef CAS.
- E. B. Corcoran, M. T. Pirnot, S. Lin, S. D. Dreher, D. A. DiRocco, I. W. Davies, S. L. Buchwald and D. W. C. MacMillan, Science, 2016, 353, 279 CrossRef CAS PubMed.
- J. A. Terrett, J. D. Cuthbertson, V. W. Shurtleff and D. W. C. MacMillan, Nature, 2015, 524, 330 CrossRef CAS PubMed.
- C. H. Chrisman, M. Kudisch, K. O. Puffer, T. K. Stewart, Y. M. L. Lamb, C.-H. Lim, R. Escobar, P. Thordarson, J. W. Johannes and G. M. Miyake, J. Am. Chem. Soc., 2023, 145, 12293 CrossRef CAS PubMed.
- S. I. Ting, W. L. Williams and A. G. Doyle, J. Am. Chem. Soc., 2022, 144, 5575 CrossRef CAS PubMed.
- Y. Qin, R. Sun, N. P. Gianoulis and D. G. Nocera, J. Am. Chem. Soc., 2021, 143, 2005 CrossRef CAS.
- N. A. Till, S. Oh, D. W. C. MacMillan and M. J. Bird, J. Am. Chem. Soc., 2021, 143, 9332 CrossRef CAS.
- N. A. Till, L. Tian, Z. Dong, G. D. Scholes and D. W. C. MacMillan, J. Am. Chem. Soc., 2020, 142, 15830 CrossRef CAS PubMed.
- R. Sun, Y. Qin, S. Ruccolo, C. Schnedermann, C. Costentin and D. G. Nocera, J. Am. Chem. Soc., 2019, 141, 89 CrossRef CAS PubMed.
- D. A. Cagan, D. Bím, B. J. McNicholas, N. P. Kazmierczak, P. H. Oyala and R. G. Hadt, Inorg. Chem., 2023, 62, 9538 CrossRef CAS.
- D. A. Cagan, D. Bím, B. Silva, N. P. Kazmierczak, B. J. McNicholas and R. G. Hadt, J. Am. Chem. Soc., 2022, 144, 6516 CrossRef CAS PubMed.
- S. I. Ting, S. Garakyaraghi, C. M. Taliaferro, B. J. Shields, G. D. Scholes, F. N. Castellano and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 5800 CrossRef CAS PubMed.
- L. Yang, H.-H. Lu, C.-H. Lai, G. Li, W. Zhang, R. Cao, F. Liu, C. Wang, J. Xiao and D. Xue, Angew. Chem., Int. Ed., 2020, 59, 12714 CrossRef CAS PubMed.
- R. D. Bradley, B. D. McManus, J. G. Yam, V. Carta and A. Bahamonde, Angew. Chem., 2023, 135, e202310753 CrossRef.
- M. Grübel, I. Bosque, P. J. Altmann, T. Bach and C. R. Hess, Chem. Sci., 2018, 9, 3313 RSC.
- R. Lauenstein, S. L. Mader, H. Derondeau, O. Z. Esezobor, M. Block, A. J. Römer, C. Jandl, E. Riedle, V. R. I. Kaila, J. Hauer, E. Thyrhaug and C. R. Hess, Chem. Sci., 2021, 12, 7521 RSC.
- D. Zhao, W. Wang, F. Yang, J. Lan, L. Yang, G. Gao and J. You, Angew. Chem., Int. Ed., 2009, 48, 3296 CrossRef CAS.
- D. Zhao, W. Wang, S. Lian, F. Yang, J. Lan and J. You, Chem.–Eur. J., 2009, 15, 1337 CrossRef CAS.
- M. Mokfi, J. Rust, C. W. Lehmann and F. Mohr, Molecules, 2021, 26, 3705 CrossRef CAS.
- R. A. Altman, E. D. Koval and S. L. Buchwald, J. Org. Chem., 2007, 72, 6190 CrossRef CAS PubMed.
- D. Xue, Z.-H. Jia, C.-J. Zhao, Y.-Y. Zhang, C. Wang and J. Xiao, Chem.–Eur. J., 2014, 20, 2960 CrossRef CAS PubMed.
- A. Böhm and T. Bach, Chem.–Eur. J., 2016, 22, 15921 CrossRef PubMed.
- E. L. B. Johnson Humphrey, A. R. Kennedy, S. Sproules and D. J. Nelson, Eur. J. Inorg. Chem., 2022, 2022, e202101006 CrossRef CAS.
- Q. Lin and T. Diao, J. Am. Chem. Soc., 2019, 141, 17937 CrossRef CAS PubMed.
- M. Mohadjer Beromi, G. W. Brudvig, N. Hazari, H. M. C. Lant and B. Q. Mercado, Angew. Chem., Int. Ed., 2019, 58, 6094 CrossRef CAS PubMed.
- A. Bismuto, P. Müller, P. Finkelstein, N. Trapp, G. Jeschke and B. Morandi, J. Am. Chem. Soc., 2021, 143, 10642 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic and catalytic experimental procedures, detailed spectral and crystallographic characterization, DFT calculations. CCDC 2354527–2354534. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc04257c |
| ‡ Current (permanent) address: Istituto di Chimica dei Composti Organometallici, Consiglio Nazionale delle Ricerche, Via Madonna del Piano 10, Sesto Fiorentino, 50019, Italy. |
| § Current address: Department of Chemistry, Johannes Gutenberg University, Duesbergweg 10-14, 55128 Mainz, Germany. |
| This journal is © The Royal Society of Chemistry 2025 |