
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of mono- and dual-labelled antibody fragment conjugates via reversible site-selective disulfide modification and proximity induced lysine reactivity†
Ioanna A.
Thanasi
a,
Nathalie
Bouloc
b,
Clíona
McMahon
a,
Ning
Wang
a,
Peter A.
Szijj
a,
Tobias
Butcher
a,
Léa N. C.
Rochet
 a,
Elizabeth A.
Love
b,
Andy
Merritt
b,
James R.
Baker
*a and
Vijay
Chudasama
*a
a,
Elizabeth A.
Love
b,
Andy
Merritt
b,
James R.
Baker
*a and
Vijay
Chudasama
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: v.chudasama@ucl.ac.uk; j.r.baker@ucl.ac.uk
bLifeArc, Accelerator Building Open Innovation Campus, Stevenage SG1 2FX, UK
First published on 6th January 2025
Abstract
Many protein bioconjugation strategies focus on the modification of lysine residues owing to the nucleophilicity of their amine side-chain, the generally high abundance of lysine residues on a protein's surface and the ability to form robustly stable amide-based bioconjugates. However, the plethora of solvent accessible lysine residues, which often have similar reactivity, is a key inherent issue when searching for regioselectivity and/or controlled loading of an entity. A relevant example is the modification of antibodies and/or antibody fragments, whose conjugates offer potential for a wide variety of applications. Thus, research in this area for the controlled loading of an entity via reaction with lysine residues is of high importance. In this article, we present an approach to achieve this by exploiting the quantitative and reversible site-selective modification of disulfides using pyridazinediones, which facilitates near-quantitative proximity-induced reactions with lysines to enable controlled loading of an entity. The strategy was appraised on several clinically relevant antibody fragments and enabled the formation of mono-labelled lysine-modified antibody fragment conjugates via the formation of stable amide bonds and the use of click chemistry for modular modification. Furthermore, through the use of multiple cycles of this novel strategy, an orthogonally bis-labelled lysine-modified antibody fragment conjugate was also furnished.
Introduction
Over the past few decades, there has been growing interest in the field of protein modification owing to the opportunities it enables in therapeutics, diagnostics, imaging, etc.1–5 Consequently, a plethora of protein modification strategies have been reported in the literature as valuable tools that have found use in multiple applications.6–9 Notably, and most directly related to this work, antibody and antibody fragment modification/bioconjugation has enabled crucial breakthroughs in cancer therapeutic treatments such as antibody–drug conjugates (ADCs),10–13 diagnostic tools for biomarker detection,14 imaging of tumours (e.g., positron emission tomography (PET) techniques),15–18 drug delivery (e.g., antibody-conjugated nanoparticles),19etc. As substantiated by prior studies in the field, site-selective antibody modification is key to attaining antibody conjugates with predictable and optimal pharmacokinetic/in vivo profiles.20–23 It also minimises issues associated with having antibody conjugate mixtures where there is a wide distribution of loadings (e.g., batch-to-batch variability, no- or low-loaded conjugates limiting efficiency, high-loaded conjugates being ineffective due to them being rapidly cleared from the body, etc.).11,24Several methodologies have been developed to accomplish site-selective antibody modification, including genetic engineering for the incorporation of unnatural amino acids, cysteine mutants24–26 or through the use of enzymatically activatable tags.27 Undoubtedly, these strategies have paved the way for a high degree of site-selective modification of antibodies. However, none of them employ a native antibody scaffold and are thus limited by the generally lower protein expression yields and higher costs/extended periods of time required to produce the desired antibody mutants. Other limitations also include (in some cases) the greater potential for protein aggregation and unfavourable/undesired disulfide scrambling (especially when using cysteine mutants).28,29
An interesting alternative is the modification of native antibodies, which is a rapidly growing area of interest due to its advantages including generally low cost, better scalability and better applicability of conditions to multiple proteins without the need for bespoke optimisation. Several groups have reported using reagents for the site-selective modification of the native interstrand disulfide bond(s) on an antibody (or relevant antibody fragment) by reduction followed by reaction with electrophiles bearing one or two electrophilic centres.30–48 Crucially, the antibody's structural integrity can remain intact when using certain disulfide rebridging reagents such as certain PD reagents under certain reaction conditions that obviate disulfide scrambling issues.49,50 However, when modifying individual cysteine residues liberated from disulfide reduction with electrophiles bearing only one electrophilic centre,51 this leads to a loss of a covalent link between the antibody chains and can result in in vivo instability.52 The modification of native antibodies via the primary amino group of lysine residues has also been extensively investigated. This is especially owing to the diverse array of acylating agents that can be employed for the formation of robustly stable amide-bonded conjugates; many of these conjugates have been clinically validated.53 However, the high abundance of accessible lysine residues on an antibody's surface represents a double-edged sword. While offering multiple available sites for modification, regio-selectivity is very difficult to achieve as many of the lysines harbour similar reactivity. This leads to the formation of highly heterogenous conjugates when targeting a conjugate loading lower than the total number of solvent accessible lysines, which is almost always the case as modifying all the lysines would cause significant issues to the protein (e.g., solubility). Moreover, by the very nature of the modification strategy, it results in a highly varied and difficult to predict pharmacokinetic profile for the plethora of conjugates formed in the product mixture.54 More recently, and to obviate some of the aforementioned issues, several efforts toward enabling site-selective lysine modification and/or lysine-modified mono-labelling have been explored, these include: kinetically controlled labelling,55–63 affinity binding-directed modification,64–80 cooperative stapling81–84 (involving a lysine residue with another residue such as cysteine, arginine, tyrosine etc.),85,86 automated synthesis of stoichiometrically conjugated antibodies,87,88 linchpin-based strategies89–92 and modified group transfer strategies (rapid and reversible modification of a nearby amino acid (typically cysteine) to transfer reactive moieties to lysine(s)).93–95 These methods aim to achieve site-specific modification of lysine(s) with direct applications in the areas of ADCs,96 peptide–drug conjugates and the development of covalently lysine modified drugs.64,65,97,98 Despite many advancements, challenges still exist, e.g., substrate tolerance (some of the proposed methods are limited to application to specific proteins or peptides, and thus lack versatility), reaction efficiency (low yields, long reaction times, complicated synthesise of key reagents, the need for bespoke protein-specific optimisation (time-consuming and costly) and/or high concentrations of reagents, which can limit practical utility), operational complexity (complex procedures can affect scalability), etc. These general issues necessitate the further refinement of strategies for site-selective lysine modification and/or lysine-modified mono-labelling of proteins to enable broader applications to be realised.
In this article, we overcome some of the aforementioned limitations (see Fig. 1) by exploiting (i) the controlled reversible site-selective modification of disulfides with dibromopyridazinediones (PDs) and (ii) proximity induced reactions with proximal lysines. Through careful optimisation of conjugation to disulfides and neighbouring lysines by limiting side-reactions, our methodology achieves lysine-modified mono-labelling of various proteins via amide bond formation reactions. The use of a click handle on the newly formed amide bond enables facile modular modification. The strategy was exemplified on multiple clinically relevant antibody Fabs with evidence of proximity induced reactivity (distal from the antibody fragment complementarity-determining region (CDR)), retention of the native disulfide bond, and retention of binding of the antibody conjugates to their native target verified by MS/MS, SDS-PAGE, LC-MS and/or ELISA analysis.
 | ||
| Fig. 1 Existing strategies for site-selective lysine modification, and a newly proposed strategy to enable selective lysine modification on proteins via a combination of quantitative and reversible disulfide modification and proximity-induced intramolecular reactivity for the formation of mono-labelled lysine-modified protein conjugates, which can then be re-subjected to the reaction protocol using a different functional modality to yield a dually-labelled lysine-modified conjugates. | ||
Results and discussion
Our study began by selecting an appropriate protein to appraise our proposed methodology. We chose the Fab fragment of Ontruzant (a biosimilar of Herceptin™/trastuzumab) as Herceptin™/trastuzumab has been used in numerous applications such as drug delivery and imaging (thus highlighting its clinical importance),2,12,18,51,99–103 it can be obtained in excellent quality and in high quantity (to facilitate thorough analysis, see ESI† for details), and as it contains a single disulfide bond and 26 lysine residues (thus enabling thorough appraisal/investigation of the viability of our proposed strategy). Computational analysis of crystallographic data derived from PDB file 6BAE (trastuzumab Fab) and PDB file 1HZH (human IgG1 against HIV-1, containing the final four amino acids DKTH on the heavy chain where K is Lys-225) identified the lysine residues in the region of the disulfide bond of the Fab. As shown in Fig. 2A, there are three lysines on the heavy chain (K136, K221, K225) and four lysines on the light chain (K126, K190, K183, K207) that are in relatively close proximity to the disulfide bond on the Fab – it is acknowledged that these distances are approximate/dynamic as the protein is flexible. | ||
| Fig. 2 (A) Overlaid Fab crystal structure, derived from PDB files 1HZH (Human IgG against HIV-1, lacking four amino acids DKTH, K225) and 6BAE (trastuzumab Fab); interstrand disulfide bond cysteine side-chains are coloured in red, and lysine side-chains are coloured in blue. The distance (measured in Å) from the disulfide bond to the nitrogen of the side chain of the lysine are displayed to indicate relative proximity, while acknowledging the flexibility of the protein – it is acknowledged that these distances are approximate/dynamic as the protein is flexible. (B) Schematic representation of the proposed proximity induced lysine transfer strategy on Ontruzant Fab 1. Lysines on the light or heavy chain may be modified; the above graphical representation has been simplified as it is difficult to depict all possible regioisomers graphically. | ||
We next needed to appraise what disulfide modification technology would be appropriate. The proposed strategy (see Fig. 2B) requires a reagent that enables chemoselective disulfide rebridging and can be quantitatively removed with a specific trigger to re-liberate the native disulfide bond. An appropriate class of reagent are PDs, which are excellent candidates for re-bridging a reduced disulfide and can also be quantitatively removed with an excess of thiol to restore a native disulfide linkage.104 As such, as shown in Fig. 2B, a general method could be envisaged for the aforementioned strategy to mono-label Ontruzant Fab 1via lysine-based modification; it is based on: (i) a PD unit (to chemoselectively react with the reduced disulfide of a Fab and then be selectively cleaved in the final step to restore the native disulfide bond); (ii) a lysine-reactive moiety (that should only react with a lysine via proximity induced intramolecular reactivity); and (iii) a flexible linker to connect the two reactive groups (this can potentially be used to tune the length/rigidity of the molecule to enable efficient proximity induced reactivity depending on the protein system that is being used).
Phenyl esters and ortho-modified derivatives thereof have been reported in the literature as reagents that can be used for lysine modification including applications in labelling,105–107 as well as in peptide and antibody modification.108–111 Therefore, we initially focused on making constructs based on having a phenolic ester as our lysine reactive group. That being said, it was appreciated from the outset that phenyl esters are prone to hydrolysis and that this was a potential limitation that we may need to address. Reaction of 4-(aminomethyl)phenol with a carboxylic acid-bearing PD led to the formation of a PD featuring a phenol moiety. This molecule was subsequently coupled with various carboxylic acids to form PD-ester derivatives 2, including PD-ester azides 2b–d which would enable subsequent and facile modular Cu-free strain-promoted azide–alkyne click chemistry (SPAAC) functionalisation. The preparation of various PD-ester-azides would enable investigation into how the sterics and distance between the electron-withdrawing azide112 may affect conjugation and/or hydrolysis.
To appraise the feasibility of the strategy, and optimise the reaction conditions, non-azide bearing PD-ester 2a was initially reacted with Ontruzant Fab 1 (reagent 2a, Fig. 3C). As mentioned earlier, the strategy consists of the following steps: (i) conjugation of PD to the reduced disulfide, (ii) proximity induced reaction of the ester with a lysine residue, (iii) removal of the PD and restoration of the native disulfide bond. A series of reaction parameters for the conjugation and lysine reaction step were appraised (e.g., pH, duration of reaction, temperature, etc.). The most optimal conditions were found to be the use of 10 eq. of reagent 2a in PBS (pH 7.4, 2 mM EDTA) for 4 h for the conjugation step, and the use of BBS (pH 8.5, 2 mM EDTA) for 24 h for the proximity induced lysine reaction step. The conditions for the removal of the PD were found to be the use of dithiothreitol (DTT) at 175 eq. in BBS (pH 8.0, no EDTA). Finally, we discovered that the restoration of the native disulfide bond can be achieved via two simple options: oxidation could be achieved by incubation in an EDTA-free buffer at 37 °C for 3 h, or it could be carried out using 200 equivalents of dehydroascorbic acid (DHA, 50 mM in DI H2O) in BBS (pH 8.0, no EDTA) for 2 h.
 | ||
| Fig. 3 (A) A general PD-ester scaffold highlighting its two key features and the specific DiBrPD-ester reagents 2a–d that were made. (B) Reaction scheme for conversion of Ontruzant Fab 1 to mono-labelled lysine-modified conjugates 3a–d: (a) reaction conditions using 2-step PD conjugation and lysine transfer protocol: (i) TCEP (10 eq., 20 mM in DI H2O), 90 min and then addition of compound 2a–2d (10 eq., 10 mM in MeCN) in PBS (pH 7.4, 2 mM EDTA), 4 h, 37 °C, (ii) incubation in pH 8.5 (2 mM EDTA), 24 h, (iii) DTT (175 eq., 50 mM in DI H2O), 1 h; followed by oxidation in BBS (pH 8.0, no EDTA). (b) Reaction conditions using 1-step PD conjugation and lysine transfer protocol: (i) TCEP (10 eq., 20 mM in DI H2O), 90 min and then addition of compound 2d (2.5 eq., 10 mM in MeCN) in PBS (pH 7.4, 2 mM EDTA), 16 h, 37 °C, (iii) DTT (175 eq., 50 mM in DI H2O), 1 h; followed by oxidation in BBS (pH 8.0, no EDTA). All reactions were conducted at 37 °C. (C) Table depicting the % abundance of mono-labelled final conjugates 3a–d, equivalents used, and what protocol was employed. Lysines on the light or heavy chain may be modified; the above graphical representation has been simplified as it is difficult to depict all possible regioisomers graphically. | ||
Initial studies using reagent 2a suggested near quantitative conjugation of the PD onto Fab as determined by LC-MS and SDS-PAGE analysis (see ESI† for details). In the conjugation step, the formation of a side product (ca. 10%) resulting from the hydrolysis of the ester moiety was observed (see Fig. 2B, red arrow). Upon buffer swapping to pH 8.5, the desired proximity induced lysine reaction was achieved whilst the hydrolysed ester conjugate persisted (ca. 11%). Upon addition of DTT and then subsequent purification into EDTA-free water to restore the native disulfide bond, LC-MS analysis revealed the formation of desired conjugate 3a in 89% conversion, with the remaining 11% abundance of native Fab presumably stemming from the hydrolysis side-product.
The preliminary studies on reagent 2a demonstrated the feasibility of the strategy, resulting in the successful formation of mono-labelled lysine-modified Fab conjugate 3a in high conversion, but it also highlighted a hydrolysis side-reaction (see Fig. 2B, red). We next appraised PD-ester-azide reagents 2b–2d in the aforementioned optimised reaction conditions (Fig. 3B). We anticipated that PD-ester-azide 2b, which harbours the shortest alkyl chain length from ester to azide, would have the most reactive ester group. This hypothesis was soon confirmed, as when conjugating PD-ester-azide 2b to reduced Ontruzant Fab 1, additional acylations were observed. This indicated that unselective intermolecular reactions were taking place between the Fab and the ester component of reagent 2b. Substantial hydrolysis of the ester was also observed when using this reagent (ca. 55%). Reagent 2c, bearing a geminal dimethyl group to confer improved hydrolytic stability in the PD conjugation step, was next to be appraised.106 Whilst efficient PD disulfide re-bridging was observed with a small amount of hydrolysis (ca. 10%), lysine transfer was substantially inhibited (only ca. 33% conversion to desired conjugate 3c was observed).
The application of reagent 2d, which has a longer chain length, in the aforementioned reaction sequence yielded the highest abundance of desired singly modified Fab 3d for all the azide-based reagents (ca. 85%) with only ca. 15% of native Fab (see Fig. 3C). The longer alkyl chain length proved to be optimal in achieving a good rate of lysine transfer vs. unwanted hydrolysis whilst also facilitating the attachment of an azide click handle. Having identified reagent 2d, which facilitated high formation of the desired mono-labelled lysine-modified conjugate, it was decided to investigate the feasibility of achieving conjugation and lysine reaction processes in a single step reaction. Such an approach would enhance efficiency and may result in a higher conversion. As described in the ESI (Section 2.4),† a series of reactions were conducted involving the reaction of reagent 2d with reduced Fab using 1-step for PD conjugation and lysine reaction – it was concluded that such a procedure could be enabled when the conjugation and lysine transfer reactions took place at pH 7.4 for 16 h. This simpler and faster procedure resulted in an improved 90% abundance of mono-labelled conjugate 3d as determined by LC-MS. Moreover, in the course of these experiments, it was determined that only 2.5 eq. of reagent 2d was needed to achieve ca. 89% of mono-labelled conjugate 3d.
We next turned our attention to the phenolic component to try to fine tune the reaction further. Inspired by reports on the use of ortho-substituted di-halogen phenyl esters for the generation of protein conjugates modified at lysine(s), due to apparent increased efficiency in terms of lysine reactivity and/or increased hydrolytic stability of the ester,106 it was decided to create a library of ortho-substituted di-halogen analogues of reagent 2d, i.e., ortho-dibromophenolate 4, ortho-dichlorophenolate 5 and ortho-difluorophenolate 6. The experimental pKa values of ortho-dibromophenol, ortho-dichlorophenol and ortho-difluorophenol and phenol are 6.67, 6.78, 7.51 and 9.90 respectively.113 Whilst the dibromo and dichloro reagents could be expected to be the most reactive on the basis of this data, the opposing effect of sterics made it difficult to prima facie predict what reagent would fare best. The study commenced with ortho-dibromo reagent 4 following the protocol described in Fig. 4A, based on reports of the increased hydrolytic stability of this moiety.90,105 Use of 2.5 eq. of reagent 4 and incubation for only 6 h (cf. 16 h) in PBS (pH 7.4, 2 mM EDTA) to achieve PD conjugation and lysine modification (presumably due to the ester being more reactive) was enough to achieve a ∼91% abundance percentage of final, mono-azide-labelled conjugate 3d. Reagents 5 and 6 were also appraised and whilst ortho-dichloro reagent 5 (2.5 eq.) resulted in a similar outcome (∼90% mono-labelled Fab conjugate 3d), the most reactive difluoro ester reagent 6 afforded double and triple labelled products that were almost certainly derived from additional intermolecular reactions between the ester component of reagent 6 and lysine residues on the Fab (Fig. 4D and E). This hypothesis was supported by control reactions of PD reagents 4–6 with native Fab under the reaction conditions showing reactivity only when using PD reagent 6 (see ESI† for details).
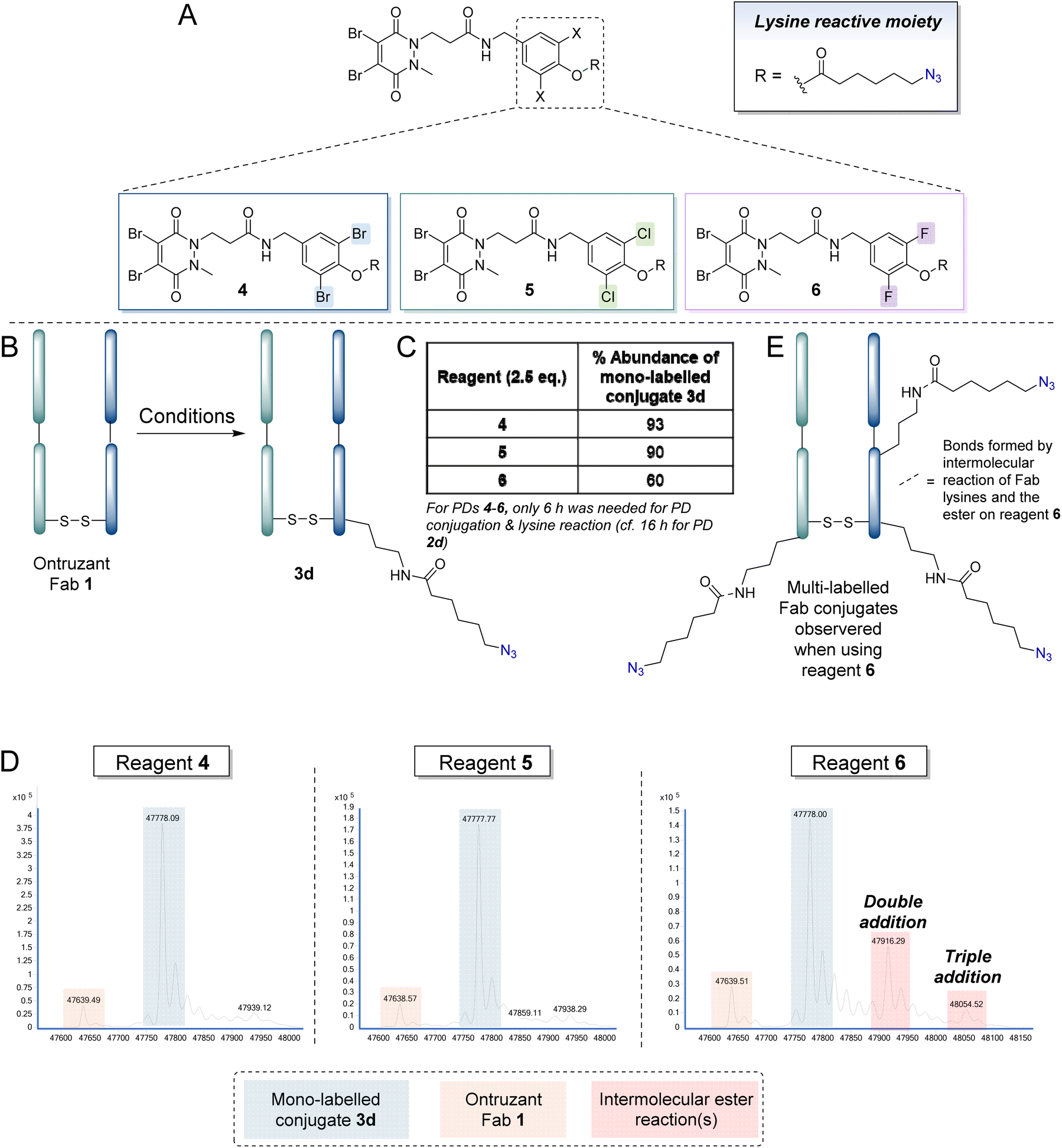 | ||
| Fig. 4 (A) Structures of reagents 4–6. (B) Reaction of reagents 4–6 in the following protocol: (i) TCEP (10 eq., 20 mM in DI H2O), 90 min and then addition of compound 4, 5 or 6 (2.5 eq., 10 mM in MeCN) in PBS (pH 7.4, 2 mM EDTA), 6 h, (iii) DTT (175 eq., 50 mM in DI H2O), 1 h; followed by incubation in BBS (pH 8.0, no EDTA). All reactions were conducted at 37 °C. Lysines on the light or heavy chain may be modified; the above graphical representation has been simplified as it is difficult to depict all possible regioisomers graphically. (C) Table depicting the % abundance of mono-labelled final conjugate 3d for each reagent used. (D) Deconvoluted MS data for after the final step to make conjugate 3d (zoom in of mass range of Fab region) when using reagents 4, 5 and 6. (E) Multi-labelled Fab conjugates observed when using reagent 6. Lysines on the light or heavy chain may be modified; the above graphical representation has been simplified as it is difficult to depict all possible regioisomers graphically. | ||
In order to determine the rate of inherent ester hydrolysis of the reagent 4 in aqueous buffer conditions alone, a stability study on reagent 4 was conducted via NMR spectrometry analysis. In order to mimic the bioconjugation reaction conditions, reagent 4 (5 mM final solution) was incubated in PBS (pH 7.4, 2 mM EDTA)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CD3CN (50:50) at 37 °C (300 rpm). Time points at t = 0, 1, 6, 24 and 48 h were collected (see ESI† for details). Based on the experimental data, reagent 4 exhibited no hydrolysis up to the 48 h time point, demonstrating hydrolytic stability in the buffer (see ESI† for details). This study suggested that the premature hydrolysis may be due to the protein microenvironment.
:CD3CN (50:50) at 37 °C (300 rpm). Time points at t = 0, 1, 6, 24 and 48 h were collected (see ESI† for details). Based on the experimental data, reagent 4 exhibited no hydrolysis up to the 48 h time point, demonstrating hydrolytic stability in the buffer (see ESI† for details). This study suggested that the premature hydrolysis may be due to the protein microenvironment.
We next set out to explore the effect of the flexible linker component of the scaffold on the strategy. We wanted to investigate whether adjusting the length/rigidity of the molecule would result in more, less or similarly efficient proximity-induced modification vs. competing ester hydrolysis. To appraise this, reagents 7 and 8 (see Fig. 5B) were synthesised (see ESI† for details). The library of reagents 4, 7 and 8 would thus enable us to investigate the relationship between linker length and the effectiveness of the protocol described above. According to the LC-MS results, mono-azide-labelled lysine conjugate 3d was detected with abundance of ∼89% and ∼90% when using reagents 7 and 8 (respectively), suggesting they had a similar reactivity profile to reagent 4 (see ESI† for details). Nonetheless, we assumed that the shorter linker species may result in only the most proximal lysines being modified and we next appraised if this assumption would be borne out by carrying out the click reaction prior to the final PD removal step as this would exacerbate any potential differences in sterics (see Fig. 5A). Moreover, performing the click reaction prior to PD removal (see Fig. 5A) is ideal (more generally) due to the potential issues of DTT compromising the stability of certain azides114,115 and/or other click handles (e.g. strained alkynes, etc.) if they were to be used in the linker. In view of this, a simple BCN-PEG2-amine 9 (10 eq., 10 mM in DMSO) was added after the lysine transfer step and the reaction was incubated in the dark for 16 h at 37 °C prior to PD removal and disulfide bond restoration. According to LC-MS, all the click reactions proceeded in quantitative conversion, but a difference was observed in the following DTT-based PD removal step where a longer time was needed for the conjugate made using the short linker (PD 7, 5 h was required compared to 2 h for the medium and longer linkers PDs 4 and 8). Nonetheless, as shown in Fig. 5B, the final mono-clicked lysine modified Fab conjugate 10 was obtained in conversions of 90–93% for reagents 4, 7 and 8.
 | ||
| Fig. 5 (A) General scheme for the conversion of Ontruzant Fab 1 to conjugate 10 using the following conditions: (i) TCEP (10 eq., 20 mM in DI H2O), 90 min and then addition of compound 4, 7 or 8 (2.5 eq., 10 mM in MeCN) in PBS (pH 7.4, 2 mM EDTA), 6 h, (ii) BCN-PEG2-amine 10 (10 eq., 10 mM in DMSO) in BBS (pH 8.0, no EDTA), 16 h, (iii) DTT (175 eq., 50 mM in DI H2O), 2 h for reagents 4 and 8, 5 h for reagent 7 followed by oxidation in BBS (pH 8.0, no EDTA). All reactions were conducted at 37 °C. Lysines on the light or heavy chain may be modified; the above graphical representation has been simplified as it is difficult to depict all possible regioisomers graphically. (B) Table displaying the structures of reagents 4, 7 and 8, as well as (for each reagent) the % abundance of conjugate 10 and its analysis by LC-MS and SDS-PAGE (L: Ladder, 1: Ontruzant Fab 1, 2: Reduction of Fab, 3: PD 4, 7 or 8 conjugation, 4: Click reaction, 5: PD removal, 6: Restoration of disulfide bond). | ||
To appraise the exact sites of modification, the final conjugates 10 obtained from reaction with reagents 4, 7 and 8 were subjected to chymotryptic digestions and analysed by LC-MS/MS analysis (see ESI† for details). Consistent with our expectations (see Fig. 2A), four lysine residues on the light chain (K126, K183, K190, K207) and three on the heavy chain (K136, K221, K225) were modified with the presumably most proximal/reactive lysine (K225) being modified most (ca. 25%) in all cases (see ESI† for details). Thus, all modifications occurred distant from the CDR region and proximal to the disulfide, but, and perhaps somewhat surprisingly, a significant difference in the distributions of the lysines modified in relation to the three different length linkers was not observed. Nonetheless, as expected, since the modification took place distal from the CDR, the final lysine-modified conjugates (10) formed from using reagents 4, 7 and 8 demonstrated full retention of binding activity relative to the Ontruzant Fab 1 (see ESI† for details). Based on the optimisation studies on the flexible linker outlined earlier, it was concluded that the linker length had no significant impact on proximal lysine reaction vs. hydrolysis. Due to the extended time required for removal of the PD when using reagent 7, the potential risks in vivo (i.e. immunogenicity) associated with the PEG chain employed for reagent 8,116 the higher complexity of the multi-step synthesis required of both reagents 7 and 8, and the higher conversion when using reagent 4, it was decided to proceed with reagent 4 for the remainder of the studies. Nonetheless, these results do demonstrate that the strategy is amenable to using linkers with different lengths and for systems where a lysine is not proximal to a disulfide (or cysteine) modification site, a longer length linker entity could be used.
To appraise the broader application of the proposed strategy, it was decided to investigate the application of PD reagent 4 on other readily available antibody Fabs of therapeutic relevance such as FabCD20 and FabCD3.117–119 Rituximab is a chimeric mAb against CD20, primarily located on the surface of B cells and is widely employed to treat cancer and autoimmune diseases.118 Muromonab, or Orthoclone OKT3, is a murine anti-CD3 antibody and was the first monoclonal antibody approved for preventing organ transplant rejection.120 Both Rituximab and OKT3 fragments have been engineered and utilized in various bispecific antibodies to overcome resistance mechanisms.121 FabCD20 and FabCD3 fragments were obtained via enzymatic digestions of commercially available native anti-CD20 (rituximab) and anti-CD3 (OKT3) respectively, adhering to previously outlined methods.122,123 As depicted in Fig. 6, clicked mono-labelled lysine modified FabCD20 conjugate 11 and FabCD3 conjugate 12 were both formed in 90% abundance percentage as determined by LC-MS and SDS-PAGE when using the optimised reaction protocol (see Fig. 6 caption).
 | ||
| Fig. 6 Application of the optimised protocol for mono-labelling and clicking of a Fab to FabCD20 and FabCD3 – “optimised protocol”: (i) TCEP (10 eq., 20 mM in DI H2O), 90 min and then addition of compound 4 (2.5 eq., 10 mM in MeCN) in PBS (pH 7.4, 2 mM EDTA), 6 h, (iii) BCN-PEG2-amine 10 (10 eq., 10 mM in DMSO) in BBS (pH 8.0, no EDTA), 16 h, (iv) DTT (175 eq., 50 mM in DI H2O), 2 h; followed by oxidation in BBS (pH 8.0, no EDTA). (A) Structure of clicked mono-labelled lysine modified FabCD2011. Lysines on the light or heavy chain may be modified; the above graphical representation has been simplified as it is difficult to depict all possible regioisomers graphically; (B) LC-MS analysis of FabCD20 conjugate 11 formed in the final step; (C) SDS-PAGE analysis (L: Ladder, 1: FabCD20, 2: Reduction of Fab, 3: PD 4 conjugation, 4: Click reaction, 5: PD removal, 6: Restoration of disulfide bond); (D) structure of clicked mono-labelled lysine modified FabCD312. Lysines on the light or heavy chain may be modified; the above graphical representation has been simplified as it is difficult to depict all possible regioisomers graphically; (E) LC-MS analysis of FabCD3 conjugate 12 formed in the final step, (F) SDS-PAGE analysis (L: Ladder, 1: FabCD3, 2: Reduction of Fab, 3: PD 4 conjugation, 4: Click reaction, 5: PD removal, 6: Restoration of disulfide bond). Note that in lane 2 of both SDS-PAGE some oxidised Fab is observed, this is likely a feature of oxidation taking place when preparing the sample. | ||
With the above discoveries in mind, we attempted to form a dually modified Fab bearing two different fluorophores by using cycles of the optimised reaction protocol. To the best of our knowledge, this would be the first example of controlled differential modification of the solvent accessible lysines on a native antibody fragment. The first stage involved application of the proposed strategy on Ontruzant Fab 1 using a click reaction with 5-FAM-PEG3-BCN (exo) 13, as shown in Fig. 7A. LC-MS and SDS-PAGE analysis confirmed the formation of the mono-labelled BCN-fluorescein clicked conjugate 14 in ca. 94% abundance (Fig. 7B). We next repeated two cycles of the optimisation protocol on Ontruzant Fab 1 using 5-FAM-PEG3-BCN (exo) 13 for the first click reaction and BP-Fluor568-DBCO 15 for the second click reaction. We chose to use two different strained alkynes to demonstrate tolerability. To our delight, LC-MS and SDS-PAGE analysis of the final conjugate revealed the formation of dually modified Fab 16 in ca. 85% abundance.
 | ||
| Fig. 7 (A) Application of the optimised protocol for mono-labelling and clicking of a Fab (see Fig. 6 for details) on Ontruzant Fab 1 using 5-FAM-PEG3-BCN (exo) 13 as the strained alkyne component to afford mono-labelled Fluorescein Fab conjugate 14. Lysines on the light or heavy chain may be modified; the above graphical representation has been simplified as it is difficult to depict all possible regioisomers graphically; (B) LC-MS and SDS-PAGE analysis of mono-labelled fluorescein Fab conjugate 14. The final % abundance of conjugate 14 was determined by analysis of a previous conjugate in the sequence as the disulfide bond of the Fab did not appear to be fully reformed in this particular experiment (see ESI† for details). (C) Application of the optimised protocol for mono-labelling and clicking of a Fab (see Fig. 6 for details) on Ontruzant Fab 1 in two cycles using 5-FAM-PEG3-BCN (exo) 13 (in cycle 1) and BP-Fluor568-DBCO 15 (in cycle 2) as the strained alkyne components and bypassing the disulfide restoration step at the end of cycle 1 to afford dually-labelled Fab conjugate 16. Lysines on the light or heavy chain may be modified; the above graphical representation has been simplified as it is difficult to depict all possible regioisomers graphically; (D) LC-MS analysis of dually-labelled Fab conjugate 16, (E) ELISA study on a dually-labelled Fab conjugate 17 prepared using BCN-PEG2-amine 9 and a DBCO-biotin (see ESI† for details), (F–H) SDS-PAGE analysis of protocol (L: Ladder, 1: Ontruzant Fab 1, 2: Reduction of Fab, 3: PD 4 conjugation, 4: Click reaction with 5-FAM-PEG3-BCN (exo) 13, 5: PD removal, 6: PD 4 conjugation, 7: Click reaction with BP-Fluor-DBCO 15, 8: PD removal, 9: Restoration of disulfide bond) using various light sources for analysis. (I) Functional modalities present on dual clicked Fab conjugate 16. | ||
To further exemplify the technology and to enable ELISA analysis (i.e., to prevent UV-vis absorption interference with the ELISA analysis), a further dually-modified Fab conjugate (17) was prepared using BCN-PEG2-amine 9 and DBCO-biotin (see ESI† for details) as the click partners. Pleasingly, this conjugate was also made in 85% abundance, and the ELISA assay indicated no decrease in binding relative to native Fab (Fig. 7E). As with the previous reactions, SDS-PAGE analysis corroborated these findings (Fig. 7F). As shown in Fig. 7, visualisation of the gel using a light source of 472 nm (Fig. 7G) and 302 nm + 365 nm (Fig. 7H) showed differentiation between the fluorescein only modification and when both fluorophores were attached to the Fab (see ESI† for further details). It is acknowledged that the signal intensity of the mass spectra of the conjugates decreases as one goes through the reaction sequence – we believe this is due to the loss of material over the sequence of steps.
We finally appraised our optimised protocol on the full antibody of Ontruzant, a multi-disulfide system, by scaling the amount of PD 4 per disulfide bond. However, use of these conditions resulted in a complex mixture of products (see ESI† for details) that could perhaps be anticipated/rationalised by competing thiolate-based reaction of the ester moiety of PD 4 post-conjugation to a reduced disulfide bond, i.e., once the first PD reacts with a reduced disulfide bond, the ester reacts (intramolecularly) with a reduced disulfide bond thiolate faster than the thiolate conjugates to another molecule of PD 4 (intermolecularly). As such, there is still some work to be carried out to adapt this strategy to multi-disulfide systems.
Conclusion
Bioconjugation to native solvent-accessible lysines on antibodies (and other proteins) has been utilized for many years, however, numerous limitations have been identified owing to lack of selective and/or controlled modification (e.g., batch-to-batch variability, propensity for aggregation, reduced protein function, etc.). Whilst strategies have been developed to try and address this, in this manuscript, we provide a new platform to help overcome some of these limitations by presenting a novel strategy to obtain lysine-modified mono-labelled Fab fragments via the formation of stable amide bonds. The selective modification of lysine residues is based on exploiting the quantitative and reversible site-selective modification of disulfides by using pyridazinediones that can enable near-quantitative proximity induced reactions with lysines on several antibody Fab fragments. The strategy was shown to be amenable to modular Cu-free click chemistry, proven to only modify lysines that are proximal to the native single disulfide bond (and thus distal from the CDR), and the final conjugates were shown to retain the original disulfide bond. The strategy was successfully demonstrated on three clinically relevant antibody Fab fragments (FabHER2, FabCD20 and FabCD3). Moreover, through the use of multiple cycles of the aforementioned strategy, we were able to furnish a dually-labelled lysine-modified Fab fragments bearing two different functional modalities (using BCN and DBCO derivatives) in excellent conversion. The Fab conjugates were analysed via LC-MS, SDS-PAGE and ELISA studies and MS–MS analysis to determine the exact sites of modification and this verified the principle design of the strategy. It is envisaged that this methodology, which facilitates the straightforward formation of mono- and dual-labelled lysine-modified Fab conjugates in excellent conversions, will have broad ranging applications owing to the need for novel site-selective lysine modification strategies to enable novel applications in therapeutics, diagnostics, imaging and related fields.Data availability
Synthetic chemistry experimental details, including synthetic procedures and compound characterizations, i.e., NMR, IR & MS spectra, chemical biology experimental details, including bioconjugation procedures, details on antibody digestion, LC-MS methodology, full LC-MS spectra including TIC and raw data, and LC-MS/MS have been included in the Electronic Supplementary Information (ESI) file.†Author contributions
I. A. T. synthesised the small molecules. I. A. T. carried out the bioconjugation experiments on the Fabs. T. B. carried out some of the experiments on small molecules re. stability studies by NMR. P. A. S. generated the FabCD20 fragment. L. N. C. R. generated the FabCD3 fragment. C. M., N. W. and I. A. T. conducted the ELISA studies. I. A. T. conducted the Fab digestion protocols N. B. analysed the LC-MS/MS data. N. B., E. A. L. and A. M. helped to support I. A. T. in their time at LifeArc and during their PhD. I. A. T., J. R. B. and V. C. conceived and designed the project/experiments. I. A. T. and V. C. co-wrote the manuscript. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare, but we highlight that V. C. and J. R. B. are directors of UCL spin-out ThioLogics.Acknowledgements
The authors would like to acknowledge the EPSRC for funding I. A. T. (EP/T517793/1), C. M. (EP/T517793/1) and T. B. (EP/W524335/1), the BBSRC for funding N. W. (BB/Y002180/1), and the EC for funding L. N. C. R. (Project No. 859458). P. A. S. was funded by the Wellcome Trust (grants no. 175282 and 214941/Z/18/Z) I. A. T and C. M. are also partly funded by LifeArc and MSD, respectively. The authors also gratefully acknowledge the UCL NMR facility (Dr A. Aliev), and the UCL Chemistry Mass Spectrometry Facility (Dr K. Karu).References
- J. T. Senders, I. S. Muskens, R. Schnoor, A. V. Karhade, D. J. Cote, T. R. Smith and M. L. D. Broekman, Acta Neurochir., 2017, 159, 151–167 CrossRef.
- P. Gogia, H. Ashraf, S. Bhasin and Y. Xu, Cancers, 2023, 15, 3886 CrossRef CAS PubMed.
- S. Jin, Y. Sun, X. Liang, X. Gu, J. Ning, Y. Xu, S. Chen and L. Pan, Signal Transduction Targeted Ther., 2022, 7, 39 CrossRef CAS PubMed.
- J. T. W. Tong, P. W. R. Harris, M. A. Brimble and I. Kavianinia, Molecules, 2021, 26, 5847 CrossRef CAS PubMed.
- P. Adumeau, S. K. Sharma, C. Brent and B. M. Zeglis, Mol. Imaging Biol., 2016, 18, 1–17 CrossRef CAS PubMed.
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- L. Xu, S. L. Kuan and T. Weil, Angew. Chem., Int. Ed., 2021, 60, 13757–13777 CrossRef CAS.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- Z. Fu, S. Li, S. Han, C. Shi and Y. Zhang, Signal Transduction Targeted Ther., 2022, 7, 1–25 CrossRef PubMed.
- P. Dennler, E. Fischer and R. Schibli, Antibodies, 2015, 4, 197–224 CrossRef.
- J. T. W. Tong, P. W. R. Harris, M. A. Brimble and I. Kavianinia, Mol., 2021, 26, 5847 CrossRef CAS.
- Y. Cao, J. Y. Axup, J. S. Y. Ma, R. E. Wang, S. Choi, V. Tardif, R. K. V. Lim, H. M. Pugh, B. R. Lawson, G. Welzel, S. A. Kazane, Y. Sun, F. Tian, S. Srinagesh, T. Javahishvili, P. G. Schultz and C. H. Kim, Angew. Chem., Int. Ed., 2015, 54, 7022–7027 CrossRef CAS PubMed.
- M. R. Kierny, T. D. Cunningham and B. K. Kay, Nano Rev., 2012, 3, 17240 CrossRef CAS.
- G. A. M. S. van Dongen, G. W. M. Visser, M. N. Lub-de Hooge, E. G. de Vries and L. R. Perk, Oncologist, 2007, 12, 1379–1389 CrossRef CAS PubMed.
- A. M. Wu, Methods, 2014, 65, 139–147 CrossRef CAS.
- F. Kraeber-Bodéré, C. Rousseau, C. Bodet-Milin, C. Mathieu, F. Guérard, E. Frampas, T. Carlier, N. Chouin, F. Haddad, J.-F. Chatal, A. Faivre-Chauvet, M. Chérel and J. Barbet, Int. J. Mol. Sci., 2015, 16, 3932–3954 CrossRef.
- A. Creamer, A. Lo Fiego, A. Agliano, L. Prados-Martin, H. Høgset, A. Najer, D. A. Richards, J. P. Wojciechowski, J. E. J. Foote, N. Kim, A. Monahan, J. Tang, A. Shamsabadi, L. N. C. Rochet, I. A. Thanasi, L. R. de la Ballina, C. L. Rapley, S. Turnock, E. A. Love, L. Bugeon, M. J. Dallman, M. Heeney, G. Kramer-Marek, V. Chudasama, F. Fenaroli and M. M. Stevens, Adv. Mater., 2024, 36, 2300413 CrossRef CAS PubMed.
- M. Kumari, A. Acharya and P. T. Krishnamurthy, Beilstein J. Nanotechnol., 2023, 14, 912–926 CrossRef CAS.
- S. A. Hurvitz, R. Hegg, W.-P. Chung, S.-A. Im, W. Jacot, V. Ganju, J. W. Y. Chiu, B. Xu, E. Hamilton, S. Madhusudan, H. Iwata, S. Altintas, J.-W. Henning, G. Curigliano, J. M. Perez-Garcia, S.-B. Kim, V. Petry, C.-S. Huang, W. Li, J.-S. Frenel, S. Antolin, W. Yeo, G. Bianchini, S. Loi, J. Tsurutani, A. Egorov, Y. Liu, J. Cathcart, S. Ashfaque and J. Cortés, Lancet, 2023, 401, 105–117 CrossRef CAS.
- S. Jeger, K. Zimmermann, A. Blanc, J. Grünberg, M. Honer, P. Hunziker, H. Struthers and R. Schibli, Angew. Chem., Int. Ed., 2010, 49, 9995–9997 CrossRef CAS.
- D. Y. Jackson, Org. Process Res. Dev., 2016, 20, 852–866 CrossRef CAS.
- M. Dorywalska, P. Strop, J. A. Melton-Witt, A. Hasa-Moreno, S. E. Farias, M. Galindo Casas, K. Delaria, V. Lui, K. Poulsen, J. Sutton, G. Bolton, D. Zhou, L. Moine, R. Dushin, T.-T. Tran, S.-H. Liu, M. Rickert, D. Foletti, D. L. Shelton, J. Pons and A. Rajpal, PLoS One, 2015, 10, e0132282 CrossRef.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. Lee Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- N. Zacharias, V. N. Podust, K. K. Kajihara, D. Leipold, G. Del Rosario, D. Thayer, E. Dong, M. Paluch, D. Fischer, K. Zheng, C. Lei, J. He, C. Ng, D. Su, L. Liu, S. Masih, W. Sawyer, J. Tinianow, J. Marik, V. Yip, G. Li, J. Chuh, J. H. Morisaki, S. Park, B. Zheng, H. Hernandez-Barry, K. M. Loyet, M. Xu, K. R. Kozak, G. L. Phillips, B.-Q. Shen, C. Wu, K. Xu, S.-F. Yu, A. Kamath, R. K. Rowntree, D. Reilly, T. Pillow, A. Polson, V. Schellenberger, W. L. W. Hazenbos and J. Sadowsky, Chem. Sci., 2022, 13, 3147–3160 RSC.
- J. P. M. Nunes, V. Vassileva, E. Robinson, M. Morais, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 24828–24832 RSC.
- P. M. Drake, A. E. Albers, J. Baker, S. Banas, R. M. Barfield, A. S. Bhat, G. W. de Hart, A. W. Garofalo, P. Holder, L. C. Jones, R. Kudirka, J. McFarland, W. Zmolek and D. Rabuka, Bioconjugate Chem., 2014, 25, 1331–1341 CrossRef CAS PubMed.
- S. K. Lyons, D. Plenker and L. C. Trotman, Cancer Drug Resist., 2021, 4, 745–754 CAS.
- S. H. Kang and C.-H. Lee, Biotechnol. Bioprocess Eng., 2021, 26, 295–311 CrossRef CAS PubMed.
- G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed.
- P. Bryant, M. Pabst, G. Badescu, M. Bird, W. McDowell, E. Jamieson, J. Swierkosz, K. Jurlewicz, R. Tommasi, K. Henseleit, X. Sheng, N. Camper, A. Manin, K. Kozakowska, K. Peciak, E. Laurine, R. Grygorash, A. Kyle, D. Morris, V. Parekh, A. Abhilash, J. Choi, J. Edwards, M. Frigerio, M. P. Baker and A. Godwin, Mol. Pharm., 2015, 12, 1872–1879 CrossRef CAS.
- J. P. M. Nunes, M. Morais, V. Vassileva, E. Robinson, V. S. Rajkumar, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC.
- M. Morais, J. P. M. Nunes, K. Karu, N. Forte, I. Benni, M. E. B. Smith, S. Caddick, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2017, 15, 2947–2952 RSC.
- E. Robinson, J. P. M. Nunes, V. Vassileva, A. Maruani, J. C. F. Nogueira, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 9073–9077 RSC.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T.-L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- N. Gupta, J. Kancharla, S. Kaushik, A. Ansari, S. Hossain, R. Goyal, M. Pandey, J. Sivaccumar, S. Hussain, A. Sarkar, A. Sengupta, S. K. Mandal, M. Roy and S. Sengupta, Chem. Sci., 2017, 8, 2387–2395 RSC.
- F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 RSC.
- G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed.
- M. W. Jones, R. A. Strickland, F. F. Schumacher, S. Caddick, J. R. Baker, M. I. Gibson and D. M. Haddleton, Chem. Commun., 2012, 48, 4064 RSC.
- M. Nisavic, G. J. Wørmer, C. S. Nielsen, S. M. Jeppesen, J. Palmfeldt and T. B. Poulsen, Bioconjugate Chem., 2023, 34, 994–1003 CrossRef CAS PubMed.
- C. R. Behrens, E. H. Ha, L. L. Chinn, S. Bowers, G. Probst, M. Fitch-Bruhns, J. Monteon, A. Valdiosera, A. Bermudez, S. Liao-Chan, T. Wong, J. Melnick, J.-W. Theunissen, M. R. Flory, D. Houser, K. Venstrom, Z. Levashova, P. Sauer, T.-S. Migone, E. H. van der Horst, R. L. Halcomb and D. Y. Jackson, Mol. Pharm., 2015, 12, 3986–3998 CrossRef CAS PubMed.
- P. Wilson, A. Anastasaki, M. R. Owen, K. Kempe, D. M. Haddleton, S. K. Mann, A. P. R. Johnston, J. F. Quinn, M. R. Whittaker, P. J. Hogg and T. P. Davis, J. Am. Chem. Soc., 2015, 137, 4215–4222 CrossRef CAS PubMed.
- N. Griebenow, A. M. Dilmaç, S. Greven and S. Bräse, Bioconjugate Chem., 2016, 27, 911–917 CrossRef CAS.
- T. Wang, A. Riegger, M. Lamla, S. Wiese, P. Oeckl, M. Otto, Y. Wu, S. Fischer, H. Barth, S. L. Kuan and T. Weil, Chem. Sci., 2016, 7, 3234–3239 RSC.
- B. Shi, M. Wu, Z. Li, Z. Xie, X. Wei, J. Fan, Y. Xu, D. Ding, S. H. Akash, S. Chen and S. Cao, Cancer Med., 2019, 8, 1793–1805 CrossRef CAS.
- O. Koniev, I. Dovgan, B. Renoux, A. Ehkirch, J. Eberova, S. Cianférani, S. Kolodych, S. Papot and A. Wagner, Medchemcomm, 2018, 9, 827–830 RSC.
- C. E. Stieger, L. Franz, F. Körlin and C. P. R. Hackenberger, Angew. Chem., Int. Ed., 2021, 60, 15359–15364 CrossRef CAS.
- A. L. Baumann, S. Schwagerus, K. Broi, K. Kemnitz-Hassanin, C. E. Stieger, N. Trieloff, P. Schmieder and C. P. R. Hackenberger, J. Am. Chem. Soc., 2020, 142, 9544–9552 CrossRef CAS PubMed.
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC.
- M. T. W. Lee, A. Maruani, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2015, 7, 799–802 RSC.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- C. Bahou, E. A. Love, S. Leonard, R. J. Spears, A. Maruani, K. Armour, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2019, 30, 1048–1054 CrossRef CAS PubMed.
- M. R. Gordon, M. Canakci, L. Li, J. Zhuang, B. Osborne and S. Thayumanavan, Bioconjugate Chem., 2015, 26, 2198–2215 CrossRef CAS.
- P. Agarwal and C. R. Bertozzi, Bioconjugate Chem., 2015, 26, 176–192 CrossRef CAS PubMed.
- G. H. Pham, W. Ou, B. Bursulaya, M. DiDonato, A. Herath, Y. Jin, X. Hao, J. Loren, G. Spraggon, A. Brock, T. Uno, B. H. Geierstanger and S. E. Cellitti, ChemBioChem, 2018, 19, 799–804 CrossRef CAS PubMed.
- A. R. Nanna, X. Li, E. Walseng, L. Pedzisa, R. S. Goydel, D. Hymel, T. R. Burke, W. R. Roush and C. Rader, Nat. Commun., 2017, 8, 1112 CrossRef PubMed.
- D. Hwang, K. Tsuji, H. Park, T. R. Burke and C. Rader, Bioconjugate Chem., 2019, 30, 2889–2896 CrossRef CAS.
- L. Purushottam, S. R. Adusumalli, M. Chilamari and V. Rai, Chem. Commun., 2017, 53, 959–962 RSC.
- H. Sun, M. Xi, Q. Jin, Z. Zhu, Y. Zhang, G. Jia, G. Zhu, M. Sun, H. Zhang, X. Ren, Y. Zhang, Z. Xu, H. Huang, J. Shen, B. Li, G. Ge, K. Chen and W. Zhu, J. Med. Chem., 2022, 65, 11840–11853 CrossRef CAS.
- M. Chilamari, N. Kalra, S. Shukla and V. Rai, Chem. Commun., 2018, 54, 7302–7305 RSC.
- M. Chilamari, L. Purushottam and V. Rai, Chem.–Eur. J., 2017, 23, 3819–3823 CrossRef CAS PubMed.
- M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, A. Guerreiro, P. M. S. D. Cal, J. Bertoldo, M. Maneiro, E. Perkins, J. Howard, M. J. Deery, J. M. Chalker, F. Corzana, G. Jiménez-Osés and G. J. L. Bernardes, J. Am. Chem. Soc., 2018, 140, 4004–4017 CrossRef CAS PubMed.
- L. Chen, L. Wang, H. Shion, C. Yu, Y. Q. Yu, L. Zhu, M. Li, W. Chen and K. Gao, MAbs, 2016, 8, 1210–1223 CrossRef CAS.
- A. Koperniku and N. A. Meanwell, Nat. Rev. Chem., 2024, 8, 235–237 CrossRef CAS.
- F. Shen, W. Yang, K. Zhang, Y. Jiao, J. Cui, Y. Hou and G. Bai, Chin. Chem. Lett., 2023, 34, 108203 CrossRef CAS.
- X. Xin, Y. Zhang, M. Gaetani, S. L. Lundström, R. A. Zubarev, Y. Zhou, D. P. Corkery and Y.-W. Wu, Chem Sci, 2022, 13, 7240–7246 RSC.
- S. Asano, J. T. Patterson, T. Gaj and C. F. Barbas, Angew. Chem., Int. Ed., 2014, 53, 11783–11786 CrossRef CAS PubMed.
- T. Yamaguchi, M. Asanuma, S. Nakanishi, Y. Saito, M. Okazaki, K. Dodo and M. Sodeoka, Chem. Sci., 2014, 5, 1021–1029 RSC.
- Y. Takaoka, Y. Nishikawa, Y. Hashimoto, K. Sasaki and I. Hamachi, Chem Sci, 2015, 6, 3217–3224 RSC.
- M. Kawano, S. Murakawa, K. Higashiguchi, K. Matsuda, T. Tamura and I. Hamachi, J. Am. Chem. Soc., 2023, 145, 26202–26212 CrossRef CAS.
- C. Lian, Y. Li, Z. Hou, W. Zhong, Y. Tian, F. Yin, Z. Li, D. Zhou and R. Wang, Chin. Chem. Lett., 2021, 32, 3623–3626 CrossRef CAS.
- V. Postupalenko, L. Marx, M. Pantin, D. Viertl, N. Gsponer, G. Giudice, N. Gasilova, M. Schottelius, F. Lévy, P. Garrouste, J.-M. Segura and O. Nyanguile, Chem. Sci., 2024, 15, 1324–1337 RSC.
- Y. J. Cao, C. Yu, K.-L. Wu, X. Wang, D. Liu, Z. Tian, L. Zhao, X. Qi, A. Loredo, A. Chung and H. Xiao, Theranostics, 2021, 11, 9107–9117 CrossRef CAS PubMed.
- C. Yu, J. Tang, A. Loredo, Y. Chen, S. Y. Jung, A. Jain, A. Gordon and H. Xiao, Bioconjugate Chem., 2018, 29, 3522–3526 CrossRef CAS PubMed.
- T. Lee, J. H. Kim, S. J. Kwon, J. W. Seo, S. H. Park, J. Kim, J. Jin, J. H. Hong, H. J. Kang, C. Sharma, J. H. Choi and S. J. Chung, J. Med. Chem., 2022, 65, 5751–5759 CrossRef CAS PubMed.
- S. Kim, S. Kim, S. Kim, N. Kim, S. W. Lee, H. Yi, S. Lee, T. Sim, Y. Kwon and H. S. Lee, Adv. Sci., 2024, 11, 2306401 CrossRef CAS PubMed.
- M. Tanriver, M. Müller, M. D. Levasseur, D. Richards, S. Majima, A. DeMello, Y. Yamauchi and J. W. Bode, Angew. Chem., Int. Ed., 2024, 63, e202401080 CrossRef CAS.
- K. I. Taylor, J. S. Ho, H. O. Trial, A. W. Carter and L. L. Kiessling, J. Am. Chem. Soc., 2023, 145, 25056–25060 CrossRef CAS PubMed.
- T. Ueda, T. Tamura, M. Kawano, K. Shiono, F. Hobor, A. J. Wilson and I. Hamachi, J. Am. Chem. Soc., 2021, 143, 4766–4774 CrossRef CAS.
- Y. Zeng, W. Shi, Q. Dong, W. Li, J. Zhang, X. Ren, C. Tang, B. Liu, Y. Song, Y. Wu, X. Diao, H. Zhou, H. Huang, F. Tang and W. Huang, Angew. Chem., Int. Ed., 2022, 61, e202204132 CrossRef CAS PubMed.
- K. Kubota, P. Dai, B. L. Pentelute and S. L. Buchwald, J. Am. Chem. Soc., 2018, 140, 3128–3133 CrossRef CAS.
- J. Ceballos, E. Grinhagena, G. Sangouard, C. Heinis and J. Waser, Angew. Chem., Int. Ed., 2021, 60, 9022–9031 CrossRef CAS PubMed.
- R. Molla, P. N. Joshi, N. C. Reddy, D. Biswas and V. Rai, Org. Lett., 2023, 25, 6385–6390 CrossRef CAS PubMed.
- B. Li, H. Tang, A. Turlik, Z. Wan, X. Xue, L. Li, X. Yang, J. Li, G. He, K. N. Houk and G. Chen, Angew. Chem., Int. Ed., 2021, 60, 6646–6652 CrossRef CAS.
- X. Li, W. D. Tolbert, H.-G. Hu, N. Gohain, Y. Zou, F. Niu, W.-X. He, W. Yuan, J.-C. Su, M. Pazgier and W. Lu, Chem. Sci., 2019, 10, 1522–1530 RSC.
- X. Li, S. Chen, W.-D. Zhang and H.-G. Hu, Chem. Rev., 2020, 120, 10079–10144 CrossRef CAS PubMed.
- I. Dovgan, A. Hentz, O. Koniev, A. Ehkirch, S. Hessmann, S. Ursuegui, S. Delacroix, M. Riomet, F. Taran, S. Cianférani, S. Kolodych and A. Wagner, Chem. Sci., 2020, 11, 1210–1215 RSC.
- V. Lehot, O. Lidický, J. Most, S. Erb, I. Dovgan, A. Osypenko, O. Koniev, S. Kolodych, L. Kotrchová, G. Chaubet, S. Cianférani, T. Etrych and A. Wagner, ACS Omega, 2023, 8, 40508–40516 CrossRef CAS.
- N. C. Reddy, R. Molla, P. N. Joshi, T. K. Sajeev, I. Basu, J. Kawadkar, N. Kalra, R. K. Mishra, S. Chakrabarty, S. Shukla and V. Rai, Nat. Commun., 2022, 13, 6038 CrossRef CAS PubMed.
- S. R. Adusumalli, D. G. Rawale, K. Thakur, L. Purushottam, N. C. Reddy, N. Kalra, S. Shukla and V. Rai, Angew Chem. Int. Ed. Engl., 2020, 59, 10332–10336 CrossRef CAS.
- T. Sahu, M. Chilamari and V. Rai, Chem. Commun., 2022, 58, 1768–1771 RSC.
- S. R. Adusumalli, D. G. Rawale, U. Singh, P. Tripathi, R. Paul, N. Kalra, R. K. Mishra, S. Shukla and V. Rai, J. Am. Chem. Soc., 2018, 140, 15114–15123 CrossRef CAS.
- V. Agouridas, O. El Mahdi, V. Diemer, M. Cargoët, J.-C. M. Monbaliu and O. Melnyk, Chem. Rev., 2019, 119, 7328–7443 CrossRef CAS.
- B. K. Hansen, C. J. Loveridge, S. Thyssen, G. J. Wørmer, A. D. Nielsen, J. Palmfeldt, M. Johannsen and T. B. Poulsen, Angew. Chem., Int. Ed., 2019, 58, 3533–3537 CrossRef CAS PubMed.
- D. Hymel and F. Liu, Org. Lett., 2020, 22, 3067–3071 CrossRef CAS PubMed.
- M. Haque, N. Forte and J. R. Baker, Chem. Commun., 2021, 57, 10689–10702 RSC.
- J. Pettinger, K. Jones and M. D. Cheeseman, Angew. Chem., Int. Ed., 2017, 56, 15200–15209 CrossRef CAS PubMed.
- Y. Ma, Y. Liang, M. Guo, D. Min, L. Zheng, Y. Tang and X. Sun, Chin. Chem. Lett., 2023, 34, 108072 CrossRef CAS.
- Y. Tokuda, Int. J. Clin. Oncol., 2003, 8, 224–229 CrossRef CAS.
- K. P. Garnock-Jones, G. M. Keating and L. J. Scott, Drugs, 2010, 70, 215–239 CrossRef CAS PubMed.
- A. Jeyakumar and T. Younis, Clin. Med. Insights:Oncol., 2012, 6, CMO.S6460 CrossRef.
- C. Dumontet, J. M. Reichert, P. D. Senter, J. M. Lambert and A. Beck, Nat. Rev. Drug Discovery, 2023, 22, 641–661 CrossRef CAS PubMed.
- M. K. Greene, D. A. Richards, J. C. F. Nogueira, K. Campbell, P. Smyth, M. Fernández, C. J. Scott and V. Chudasama, Chem. Sci., 2018, 9, 79–87 RSC.
- C. Bahou and V. Chudasama, Org. Biomol. Chem., 2022, 20, 5879–5890 RSC.
- Y. Takaoka, Y. Nishikawa, Y. Hashimoto, K. Sasaki and I. Hamachi, Chem. Sci., 2015, 6, 3217–3224 RSC.
- K. Lin, C. Hin Lam, X. Lin, J. Hsu, S. Fan, N. K. Gupta, Y. Lin, B. Khoon Tee, J. Li, J. Chen and K. Tan, Chem.–Asian J., 2021, 16, 937–948 CrossRef CAS.
- H. Chan, X. Lin, S. Fan, J. Ru Hwu and K. Tan, Chem.–Asian J., 2020, 15, 3416–3420 CrossRef CAS PubMed.
- M. C. Martos-Maldonado, C. T. Hjuler, K. K. Sørensen, M. B. Thygesen, J. E. Rasmussen, K. Villadsen, S. R. Midtgaard, S. Kol, S. Schoffelen and K. J. Jensen, Nat. Commun., 2018, 9, 3307 CrossRef.
- J. H. Mikkelsen, M. B. F. Gustafsson, T. Skrydstrup and K. B. Jensen, Bioconjugate Chem., 2022, 33, 625–633 CrossRef CAS PubMed.
- K. B. Jensen, J. H. Mikkelsen, S. P. Jensen, S. Kidal, G. Friberg, T. Skrydstrup and M. B. F. Gustafsson, Bioconjugate Chem., 2022, 33, 172–179 CrossRef CAS PubMed.
- H. Wu, Z. Sun and X. Li, Angew. Chem., Int. Ed., 2024, 63, e202403396 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165–195 CrossRef CAS.
- J. Han and F.-M. Tao, J. Phys. Chem. A, 2006, 110, 257–263 CrossRef CAS PubMed.
- N. D. Baker, R. J. Griffin, W. J. Irwin and J. A. Slack, Int. J. Pharm., 1989, 52, 231–238 CrossRef CAS.
- A. L. Handlon and N. J. Oppenheimer, Pharm. Res., 1988, 5, 297–299 CrossRef CAS.
- F. Zhang, M. Liu and H. Wan, Biol. Pharm. Bull., 2014, 37, 335–339 CrossRef CAS.
- H. M. Gürcan, D. B. Keskin, J. N. H. Stern, M. A. Nitzberg, H. Shekhani and A. R. Ahmed, Int. Immunopharmacol., 2009, 9, 10–25 CrossRef PubMed.
- M. D. Pescovitz, Am. J. Transplant, 2006, 6, 859–866 CrossRef CAS PubMed.
- J. P. Van Wauwe, J. R. De Mey and J. G. Goossens, J. Immun., 1980, 124, 2708–2713 CrossRef CAS PubMed.
- P. A. Todd and R. N. Brogden, Drugs, 1989, 37, 871–899 CrossRef CAS PubMed.
- Y. Sun, X. Yu, X. Wang, K. Yuan, G. Wang, L. Hu, G. Zhang, W. Pei, L. Wang, C. Sun and P. Yang, Acta Pharmacol. Sin., 2023, 13, 3583–3597 CrossRef CAS PubMed.
- F. Thoreau, P. A. Szijj, M. K. Greene, L. N. C. Rochet, I. A. Thanasi, J. K. Blayney, A. Maruani, J. R. Baker, C. J. Scott and V. Chudasama, ACS Cent. Sci., 2023, 9, 476–487 CrossRef CAS PubMed.
- A. Maruani, P. A. Szijj, C. Bahou, J. C. F. Nogueira, S. Caddick, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2020, 31, 520–529 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06500j |
| This journal is © The Royal Society of Chemistry 2025 |