DOI:
10.1039/C0PY00222D
(Paper)
Polym. Chem., 2011,
2, 175-180
π-Conjugated polymers with thermocleavable substituents for use as active layers in organic photovoltaics†
Received
18th July 2010
, Accepted 8th September 2010
First published on 4th October 2010
Abstract
The incorporation of tetrahydropyranyl (THP) groups into poly(3-alkylthiophene) offers an opportunity to prepare soluble π-conjugated polymers (πCPs) which, upon thermal annealing, may be rendered insoluble. Recent studies have shown that πCPs that may be rendered insoluble following thin film fabrication have potential use in photovoltaic (PV) applications. Furthermore, inclusion of 3,4-ethylenedioxythiophene (EDOT) into the polymer backbone, which lowers the band gap of the πCPs, provides a system with increased absorption over the solar spectrum. In that regard, the synthesis, absorption profiles, thin film X-ray diffraction and PV properties of poly(3-(6-(2-tetrahydropyranyloxy)hexyl)thiophene), poly(3-(8-(2-tetrahydropyranyloxy)octyl)thiophene) and poly[3,4-ethylenedioxy-3′-(6-(2-tetrahydropyranyloxy)hexyl)-2,2′-bithiophene] are reported herein. In addition to absorption profiles similar to that of poly(3-hexylthiophene) (P3HT), a large degree of order was observed, especially for the octyl derivative; however, upon device fabrication these systems proved to be inferior to that of P3HT:PCBM the current standard for polymeric PVs. This is attributed to a reduction in crystallinity of the conjugated polymer upon: (i) deprotection to the insoluble thin film and (ii) blending with PCBM.
Introduction
π-Conjugated organic polymers (πCPs) have attracted considerable attention over the past decade on account of their tunable optoelectronic properties coupled with their desirable characteristics similar to plastics.1–3 The potential use of these materials in thin-film electronics, particularly photovoltaics (PVs), provides the drive for the development of new and modified π-conjugated polymers.4–12 Polythiophene (PT) has been extensively studied for use in organic electronics; however, its application in thin film devices is limited due to its extremely low solubility in common organic solvents. Increased solubility may be achieved through the introduction of flexible side chains to the thiophene backbone, as is observed for poly(3-alkylthiophene).13,14 In addition to enhanced solubility, the electronic and optical properties of πCPs may be tuned to some degree by variation of the size, structure, and electronic properties of those side chain substituents.14 In regard to organic PVs, bulk heterojunction (BHJ) solar cells based on poly(3-hexylthiophene) (P3HT):[6,6]-phenyl C61-butyric acid methyl ester (PCBM) blends are the current benchmark with power conversion efficiencies (PCEs) of 4–5%.15–19 BHJ PVs based on other electron donating polymers have also been realized with efficiencies approaching 6% and, more recently, a PCE value of 7.4% has been achieved.20–22
Although such PCE values are encouraging, the efficiency is not yet sufficient for large-scale commercialization. Moreover, issues such as stability, operational lifetime and processing require more attention in order for organic PVs to compete with inorganic solar cells. Degradation in polymer solar cells is complex, and many mechanisms act simultaneously, leading to a reduction in device performance. In the absence of oxygen and water, stability can be divided into morphology, physical, chemical, and interface stability.23 The strong immiscibility coupled with the tendency for crystallization of the donor and acceptor components within a BHJ constitutes one of the major challenges associated with long term PV stability. Sivula et al. have addressed this issue and demonstrated that a small decrease in the effective regioregularity of P3HT leads to increased thermal stability of BHJ P3HT:PCBM devices.24 The authors note that the introduction of disorder into the BHJ serves to attenuate the driving force for crystallization (which is the primary cause of phase segregation between donor and acceptor domains) and facilitates the retention of a thermally stable BHJ during annealing.24
Alternatively, decreasing the solubility of the polymer increases its stability by raising the glass transition temperature, thus potentially making the device more stable in terms of the morphology and interfaces.23 One method employed to decrease solubility and, therefore, enhance stability following thin film fabrication is the use of thermal- or photo-crosslinkable polymers. Recently, Png et al. reported that bis(fluorophenyl azide)s can be incorporated into a πCP thin film and, following exposure to deep-ultraviolet light, photo-crosslinking occurs between the side chain substituents on the πCP with the azides thus rendering the thin film insoluble.25 In this manner, BHJ and bilayer PV devices were fabricated, as were FET and LED devices. Similarly, Gholamkhass and Holdcroft have demonstrated that blending azide-functionalized graft copolymers with PCBM preserves the morphology of nanophase separated domains via thermal-crosslinking.26 Using this strategy, the donor (azide-functionalized P3HT) and acceptor (PCBM) moieties are “locked-in” as a result of the covalent linkage formed between the P3HT and PCBM upon thermal treatment. The stability of device performance may also be enhanced via removal of solubilizing groups in the active layer following device fabrication as demonstrated by Gevorgyan and Krebs (illustrated in Fig. 1a).27 In this work, the authors prepared a BHJ between native polythiophene (PT) (an insoluble and intractable material) and either [60]PCBM or [70]PCBM from a soluble polymer precursor (P3MHOCT). Subsequent chemical conversion via thermal processing resulted in the desired native PT thin film blend.27 In all the examples described above, although the observed efficiencies (up to 2%) are lower than that obtained for P3HT based devices, the importance of this work lies in the long term stability of device performance; after an initial decrease in PCE, the device performance levels out whereas under the same conditions the PCE of P3HT:PCBM devices continues to decrease.25–27
 |
| | Fig. 1 Chemical transformation of (a) P3MHOCT to P3CT (at 200 °C) to PT (at 300 °C)27 and (b) tetrahydropyran protected polymers to the corresponding alcohol derivative by thermal treatment at 300 °C. | |
In this report, tetrahydropyranyl (THP) protected poly(3-alkylthiophene)s (4, Scheme 1) were used in an effort to improve stability as they may be rendered insoluble following solution processing thus “locking-in” the morphology of the thin film. In contrast to the work described by Krebs' group, the side chain substituents are not removed in their entirety; instead, upon thermal treatment the THP group is converted to an alcohol (Fig. 1b). This builds on our previous work in which poly(3-(2-(2-tetrahydropyranyloxy)ethyl)thiophene) (4, where n = 2) was rendered insoluble via photochemical conversion of the THP group to a hydroxyl group (5, where n = 2) by acid catalysis.28 Consideration must also be taken in regard to the alkyl chain length as longer alkyl chains (e.g., 4, where n = 11) remain soluble even after removal of the THP group. Since P3HT is the current benchmark for organic PV devices,15–19,24hexyl (n = 6) and octyl (n = 8) chains were chosen. Furthermore, in addition to homopolymers, poly[3,4-ethylenedioxy-3′-(6-(2-tetrahydropyranyloxy)hexyl)-2,2′-bithiophene] (9, Scheme 2) was also prepared as the inclusion of 3,4-ethylenedioxythiophene (EDOT) into the polymer backbone raises the HOMO level of the conjugated system, thereby reducing the band gap.29–32 Absorption profiles, thin film crystallinity and device performance of 4, 5, 9 and 10 blended with PCBM were probed and compared with P3HT based systems.
 |
| | Scheme 1 | |
 |
| | Scheme 2 | |
Results and discussion
Poly(3-(6-(2-tetrahydropyranyloxy)hexyl)thiophene) (4a) was prepared according to the route outlined in Scheme 1,33,34 beginning with the treatment of 6-bromo-1-hexanol with a slight (50%) excess of dihydropyran (DHP) in the presence of p–toluenesulfonic acid monohydrate to afford 6-bromo-1-tetrahydropyranyloxyhexane (1a, where X = Br). Initially, a much larger excess of DHP was used (5 equivalents), however, this resulted in poorer yield of 1a and proved to be more difficult to purify. Treatment of 1a with magnesium metal in the presence of a small amount of iodine at reflux in tetrahydrofuran (THF) for 6 hours provided the Grignard reagent which was slowly added to a THF solution of 3-bromothiophene containing Ni(dppp)Cl2. After refluxing for 16 hours, the desired product 3-(6-(2-tetrahydropyranyloxy)hexyl)thiophene (2a) was isolated. Alternatively, 2a may be prepared from 6-chloro-1-tetrahydropyranyloxyhexane (1a, where X = Cl) providing a more cost effective route as 6-chloro-1-hexanol is significantly cheaper than 6-bromo-1-hexanol. Subsequent treatment of 2a with N-bromosuccinimide (NBS) generated the selectively brominated 2-bromo-3-(6-(2-tetrahydropyranyloxy)hexyl)thiophene (3a). Initially purification by vacuum distillation was employed, however, this led to deprotection of the alkyl chain. Column chromatography was therefore required for purification of 3a. McCullough polymerization35,36 resulted in very low yield of the desired polymer 4a (1–5%) with a regioregularity of ∼69% (obtained from the ratio of the methylene peaks attached to the thiophene moiety). Analogous treatment of 8-chloro-1-octanol led to 3b which, upon McCullough polymerization,35,36 afforded poly(3-(8-(2-tetrahydropyranyloxy)octyl)thiophene) (4b) as a dark red/purple sticky solid with a regioregularity of ∼85%. Interestingly, upon heating 4b above 60 °C the polymer changes color to bright orange then back to red/purple upon cooling to room temperature.
The copolymer poly[(3-(2-(2-tetrahydropyranyloxy)hexyl)thiophene)-(3,4-ethylenedioxythiophene)] (9) was prepared as outlined in Scheme 2. Following the preparation of 3,4-ethylenedioxy-3′-(6-(2-tetrahydropyranyloxy)hexyl)-2,2′-bithiophene (7), obtained via coupling of 3a with the Grignard reagent 6 (prepared from EDOT), selective bromination with NBS at the α-position of 7 followed by condensation should afford the desired polymer 9. Unfortunately purification of monobrominated 7 proved to be a challenge as poor separation of the desired product from by-products viacolumn chromatography led to mixtures of compounds. Attempts to polymerize (via McCullough polymerization)35,36 the material that could be isolated resulted in low molecular weight material and no sign of the desired polymer. To alleviate these issues, other polymerization methods were explored, specifically GRIM polymerization.37 Thus, treatment of 7 with two equivalents of NBS in dimethylformamide (DMF) afforded the desired dibrominated compound 3,4-ethylenedioxy-5,5′-dibromo-3′-(6-(2-tetrahydropyranyloxy)hexyl)-2,2′-bithiophene 8. Purification by flash column chromatography and subsequent treatment of 8 with methylmagnesium bromide in the presence of Ni(dppp)Cl2 afforded the desired polymer 9 as a dark purple solid.
Spectroscopic studies
The optical properties of dichloromethane solutions and thin films of 4a–b, 5a–b, 9, 10, and P3HT spin cast onto glass substrates from concentrated dichlorobenzene solutions (40 mg mL−1) have been investigated by UV-vis spectroscopy. As illustrated in Fig. 2, extension of the alkyl chain length had no effect on the absorption maxima in solution (i.e., λmax = 444 nm for both 4a and 4b) whereas a red shift was observed upon inclusion of EDOT into the polymer backbone (λmax = 479 nm for 9). The absorption profiles of the thin films, however, are influenced by alkyl chain length, as a red shift in the absorption maximum is observed upon extension from a hexyl chain to octyl (e.g., λmax = 465 nm for 4a and 509 nm for 4b). This may be attributed to increased steric interactions in 4a between juxtapositioned thienyl rings possessing bulky THP groups which are alleviated somewhat in 4b by the extension of the alkyl chain.34 Furthermore, a larger degree of co-planarity and ordering is observed in the absorption profile of 4b as indicated by the fine structure observed (with λonset = 658 nm corresponding to Eg = 1.89 eV) which is far less pronounced in the hexyl derivative (small shoulder at ∼600 nm with λonset = 647 nm corresponding to Eg = 1.92 eV). Further evidence of the larger degree of ordering exhibited by 4b (compared to 4a) is observed in the XRD pattern (vide infra). In addition to the expected red shift in the absorption maximum associated with incorporation of EDOT into the polymer backbone (λmax = 525 nm and 465 nm for 9 and 4a, respectively) the UV-vis spectrum also reveals fine structure, indicative of ordering within the thin film. This fine structure is also observed in the solution spectrum which may indicate that the polymer is relatively rigid and may form aggregates in solution as has been reported for πCPs such as polyoctylfuran.34,38,39 Upon thermal conversion (annealing at 300 °C for 5 min) to the insoluble alcohol derivatives 5b and 10, a slight blue shift in the absorption maximum is observed (9 nm and 16 nm, respectively) whereas the hexyl derivative 5a remains essentially unchanged (see Fig. 2 and Table 1). More critical, however, is the loss of fine structure which, coupled with an increase in the optical band gap, does not bode well for its potential use as a photoactive material in PV applications.
 |
| | Fig. 2 Absorbance of dichloromethane solutions (left) and thin films (right) of 4a (red solid line), 4b (blue solid line), 5a (red dotted line), 5b (blue dotted line), 9 (green solid line), 10 (green dotted line), and P3HT (pink solid line) spin cast onto glass substrates from dichlorobenzene solutions (40 mg mL−1). | |
Table 1 Thin filma photophysical and XRD properties of 4, 5b, 9, 10b, and P3HT
| |
4a
|
5a
|
4b
|
5b
|
9
|
10
|
P3HT
|
|
Thin films prepared by spin-casting dichlorobenzene solutions (40 mg mL−1) onto glass substrates.
Deprotected films obtained via thermal annealing at 300 °C for 5 min.
Dilute dichloromethane solutions.
Calculated from λonset.
|
|
M
w/kDa |
13.6 |
— |
7.31 |
— |
6.81 |
— |
6224 |
|
M
n/kDa |
8.98 |
— |
6.09 |
— |
5.45 |
— |
3124 |
| PDI |
1.52 |
— |
1.20 |
— |
1.25 |
— |
2.024 |
|
λ
max/nm |
465 |
465 |
509 |
500 |
525 |
509 |
522 |
|
λ
onset/nm |
647 |
647 |
658 |
642 |
671 |
680 |
653 |
|
λ
max
c/nm |
444 |
— |
444 |
— |
479 |
— |
— |
|
E
g
d/eV |
1.92 |
1.92 |
1.89 |
1.94 |
1.85 |
1.83 |
1.90 |
|
d-Spacing/Å |
23.9 |
— |
26.1 |
— |
— |
— |
16.740–42 |
Thin film morphology
X-Ray diffraction (XRD) experiments were performed on spin cast thin films of 4a–b, 5b, and 9 as well as 4b![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :PCBM (in 1:1 wt ratio) in order to investigate the solid-state microstructure in terms of chain self-assembly and crystallinity. The results, depicted in Fig. 3, show clearly resolved reflections of first, second and third order at 2θ angles of 3.4°, 6.7° and 10.0°, respectively, for 4b. These results resemble the literature data for layers of highly regioregular P3HT and are associated with a film morphology consisting of well-ordered lamellae formed by stacks of planarized thiophene main chains regularly spaced by hexyl substituents.40–42 As expected, the lower angle of the (100) reflection maximum (3.4° for 4b and 5.4° for P3HT) indicates an increased interlamellar spacing in 4b with respect to P3HT.40–42Polymer 4a also exhibits some degree of order with a peak at 2θ = 3.7° although the degree of order is lower compared with 4b, as evidenced by the absence of higher order peaks. Although the absorption profile of 9 revealed fine structure which is generally attributed to ordering within the thin film, the XRD pattern did not exhibit any diffraction peaks. Inclusion of EDOT into the polymer backbone increases the distance between alkyl chains which therefore affects the packing pattern of the resulting polymer. The resulting lower degree of ordering may be a consequence of the high rigidity of the polymer, different sizes of substituents, or variation in interlamellar distances.34,43,44 Similar results (i.e., decrease in thin film crystallinity) are observed for bulk heterojunction blends of 4b with PCBM (in 1:1 wt ratio). As in the case of 9, the inclusion of PCBM appears to disrupt the packing pattern of 4b. Although PCBM is not incorporated into the polymer backbone, a BHJ of 4b with PCBM generates an interpenetrating network of the two materials resulting in a decrease in the degree of order. Deprotection of 4b into the insoluble film 5b also results in a loss of crystallinity as evidenced by the lack of peaks in the XRD pattern. To understand this better, the thin film surface profiles of 4b and 5b were investigated. As shown in Fig. 3f, the images reveal very smooth films for 4b whereas large craters are formed on the surface of the film upon deprotection (5b). Thus, thermal annealing to remove the THP results in significant changes to the surface topology which may account for the loss of crystallinity.
:PCBM (in 1:1 wt ratio) in order to investigate the solid-state microstructure in terms of chain self-assembly and crystallinity. The results, depicted in Fig. 3, show clearly resolved reflections of first, second and third order at 2θ angles of 3.4°, 6.7° and 10.0°, respectively, for 4b. These results resemble the literature data for layers of highly regioregular P3HT and are associated with a film morphology consisting of well-ordered lamellae formed by stacks of planarized thiophene main chains regularly spaced by hexyl substituents.40–42 As expected, the lower angle of the (100) reflection maximum (3.4° for 4b and 5.4° for P3HT) indicates an increased interlamellar spacing in 4b with respect to P3HT.40–42Polymer 4a also exhibits some degree of order with a peak at 2θ = 3.7° although the degree of order is lower compared with 4b, as evidenced by the absence of higher order peaks. Although the absorption profile of 9 revealed fine structure which is generally attributed to ordering within the thin film, the XRD pattern did not exhibit any diffraction peaks. Inclusion of EDOT into the polymer backbone increases the distance between alkyl chains which therefore affects the packing pattern of the resulting polymer. The resulting lower degree of ordering may be a consequence of the high rigidity of the polymer, different sizes of substituents, or variation in interlamellar distances.34,43,44 Similar results (i.e., decrease in thin film crystallinity) are observed for bulk heterojunction blends of 4b with PCBM (in 1:1 wt ratio). As in the case of 9, the inclusion of PCBM appears to disrupt the packing pattern of 4b. Although PCBM is not incorporated into the polymer backbone, a BHJ of 4b with PCBM generates an interpenetrating network of the two materials resulting in a decrease in the degree of order. Deprotection of 4b into the insoluble film 5b also results in a loss of crystallinity as evidenced by the lack of peaks in the XRD pattern. To understand this better, the thin film surface profiles of 4b and 5b were investigated. As shown in Fig. 3f, the images reveal very smooth films for 4b whereas large craters are formed on the surface of the film upon deprotection (5b). Thus, thermal annealing to remove the THP results in significant changes to the surface topology which may account for the loss of crystallinity.
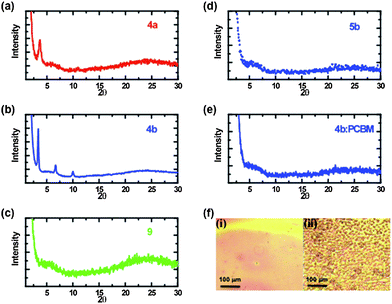 |
| | Fig. 3
X-Ray diffraction pattern of thin films of (a) 4a, (b) 4b, (c) 5b, and (e) 4b:PCBM (1:1 wt ratio 40 mg mL−1) spin cast onto glass substrates from dichlorobenzene solutions (40.0 mg mL−1). (f) Surface profile images of (i) 4b and (ii) 5b. | |
Device fabrication
Photovoltaic devices were fabricated by spin-coating dichlorobenzene solutions of 4a–b and 9 blended with PCBM (1:1 wt ratio, 40 mg mL−1) and their performance compared with P3HT:PCBM (1:1 by wt, 40 mg mL−1) devices. The solutions were cast onto clean tin-doped indium oxide (ITO)-coated glass anodes, modified by first spin-coating PEDOT:PSS [poly(3,4-ethylenedioxythiophene):polystyrene sulfonate] as a hole extraction/electron-blocking layer (∼35 nm). Spin casting (at 100 rpm for 3 seconds, 300 rpm for 5 seconds and 900 rpm for 20 seconds) was followed by solvent annealing for 1 hour in the glove box. After drying, the cells were then completed by thermal vacuum deposition of aluminium as the cathode. In addition to BHJ devices, bilayer systems were also prepared and characterized as it was recently shown that high conversion efficiencies could be obtained for bilayer devices based on P3HT:PCBM (using orthogonal solvents).45 Unfortunately, very poor PV properties (see Table 2) were observed, including 4b even though it exhibited promising thin film and optical properties—i.e., high degree of crystallinity and fine structure in absorption profile. We attribute these findings to loss of crystallinity upon (i) deprotection and (ii) blending with PCBM. In the case of the former (i.e., deprotection), inspection of the thin film surface profiles (Fig. 3f) revealed significantly different surface topologies in which very smooth films were obtained for 4b and, upon deprotection to 5b, large craters are formed on the surface of the film. This indicates that upon deprotection significant morphological changes occur. We therefore require systems in which very little structural rearrangement occurs upon rendering the films insoluble. In the later case, blending with PCBM disrupts the packing pattern of the polymer resulting in a decrease in crystallinity as BHJ generate an interpenetrating network of the two materials. Furthermore, although inclusion of EDOT into the polymer backbone lowered the band gap, the XRD pattern did not reveal an ordered structure thus the poor device performance may be attributed to the high rigidity of the polymer, different sizes of substituents, or variation in interlamellar distances.34,43,44
Table 2
PV properties for 4a–b, 5a–b,a and 10a with PCBM (1:1 wt ratio)
| |
J
SC/mA cm−2 |
V
OC/V |
FF |
PCE (%) |
|
Thermal conversion to the insoluble alcohol derivative obtained via thermal annealing at 300 °C for 5 min prior to thermal deposition of aluminium.
|
|
4a
|
0.28 |
0.68 |
0.30 |
0.064 |
|
5a
a
|
0.04 |
1.13 |
0.24 |
0.012 |
|
4b
|
0.20 |
0.63 |
0.32 |
0.039 |
|
5b
a
|
0.18 |
0.15 |
0.27 |
0.008 |
|
10
|
0.19 |
0.33 |
0.28 |
0.017 |
|
P3HT:PCBM |
7.22 |
0.55 |
0.53 |
2.12 |
Summary and conclusions
Homopolymers poly(3-(6-(2-tetrahydropyranyloxy)hexyl)thiophene) (4a) and poly(3-(8-(2-tetrahydropyranyloxy)octyl)thiophene) (4b) were prepared as well as the copolymer poly[3,4-ethylenedioxy-3′-(6-(2-tetrahydropyranyloxy)hexyl)-2,2′-bithiophene] (9) as possible donor materials for PV applications. Thermal conversion of the THP group to an alcohol rendered the polymers insoluble; thus, they may be used as donor materials for PV applications in an effort to improve the overall stability of PV devices. The absorption profiles were similar to that of P3HT and, even more promising, 4b exhibited highly ordered thin films as evidenced by the XRD pattern. Although such properties are usually associated with relatively efficient solar cells, upon fabrication of PV devices, these systems proved to be inferior to that of P3HT:PCBM. This we attribute to loss of crystallinity upon (i) deprotection (5b) and (ii) blending with PCBM. In the case of deprotection, inspection of the thin film surface profiles revealed very smooth films for 4b whereas large craters are formed on the surface of the film upon deprotection (5b). This leads to the conclusion that, upon deprotection, significant morphological changes occur. We therefore require systems in which very little structural rearrangement occurs upon rendering them insoluble. Similarly, blends of 4b with PCBM disrupt the packing pattern as the BHJ generates an interpenetrating network of the two materials and, in this case, result in a decrease in crystallinity. Furthermore, inclusion of EDOT into the polymer backbone lowered the band gap thus providing a material with better overlap with the solar spectrum. Based on the absorption profile, it appeared the crystallinity was maintained; however, the XRD pattern did not reveal an ordered structure and the device performance was rather poor. These results may be attributed to the high rigidity of the polymer, different sizes of substituents, or variation in interlamellar distances.34,43,44
In conclusion, polymers exhibiting desirable characteristics, such as increased solubility, thermally switched solubility following thin film fabrication, high degree of order, etc., have potential as donor materials in PV devices. Although the polymers currently described exhibited poor PV properties, the concepts are still valid and do not diminish the rationale behind this work. Rather, it reveals that πCP systems are required in which the crystallinity and order of the thin films are maintained upon rendering them insoluble.
Experimental section
General procedures
1H spectra were obtained on a 500 MHz Varian AS500 spectrometer; chemical shifts are reported in parts per million (ppm), referenced to CDCl3 (1H: δ = 7.26). Molecular weights were measured by gel permeation chromatography (GPC) (Waters model 1515 isocratic pump) equipped with µ-styragel columns against polystyrene standards. Polymers were eluted with THF using a flow rate of 1.0 mL min−1 and detected with a UV-vis detector (Waters model 2487) at 254 nm. UV-vis absorption spectra were recorded on a Cary 3EI (Varian) spectrophotometer. Film thickness and surface roughness were measured on a KLA-Tencor Alpha-Step IQ Surface Profiler.
Fabrication of polymer thin films
Polymeric films were fabricated following the “solvent annealing” procedure described by Liet al.46Dichlorobenzene solutions (40 mg mL−1) of 4a, 4b, 9 and blends of 4a, 4b, 9, and P3HT with PCBM at 1:1 weight ratios were spin-coated at 100 rpm for 3 s, 300 rpm for 5 s and 900 rpm for 20 s onto PEDOT coated glass substrates in a glove box under N2-filled atmosphere. Following spin-coating, the substrates were left in a covered glass Petri dish for one hour to allow for slow evaporation of the solvent resulting in 80–100 nm thick films as measured by profilometry.
X-Ray measurements
XRD was performed on a Bruker-AXS D8 Discover High-Resolution Diffractometer system using Cu Kα wavelength (about 1.544 Å). Polymer films were prepared on glass substrates by the same spin casting method described before.
Acknowledgements
We are grateful to the Natural Sciences and Engineering Research Council of Canada (NSERC) for financial support and for an NSERC postdoctoral fellowship to J.L.B. The authors also thank Bobak Gholamkhass (SFU) and Dr Terry Gordon (SFU) for useful discussions.
References
- J. Roncali, Chem. Rev., 1997, 97, 173–205 CrossRef CAS.
- H. Sirringhaus, P. J. Brown, R. H. Friend, M. M. Nielsen, K. Bechgaard, B. M. W. Langeveld-Voss, A. J. H. Spiering, R. A. J. Janssen, E. W. Meijer, P. Herwig and D. M. de Leeuw, Nature, 1999, 401, 685–688 CrossRef CAS.
-
Handbook of Conducting Polymers, Marcel Dekker, Inc., ed. T A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, New York, 2nd edn, 1998 Search PubMed.
-
D. F. Perepichka, I. F. Perepichka, H. Meng and F. Wudl, in Organic Light-Emitting Diodes, ed. Z. R. Li, CRC Press, Boca Raton, FL, 2006 Search PubMed.
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS.
- C. Reese and Z. N. Bao, Mater. Today (Oxford, U. K.), 2007, 10, 20–27 CrossRef CAS.
- A. Facchetti, Mater. Today (Oxford, U. K.), 2007, 10, 28–37 CrossRef CAS.
- E. Bundgaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2007, 91, 954–985 CrossRef CAS.
- H. Hoppe and N. S. Sariciftci, J. Mater. Res., 2004, 19, 1924–1945 CAS.
- Y. J. Cheng, S. H. Yang and C. S. Hsu, Chem. Rev., 2009, 109, 5868–5923 CrossRef CAS.
- H. Yan, Z. H. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–U1 CrossRef CAS.
- F. Silvestri and A. Marrocchi, Int. J. Mol. Sci., 2010, 11, 1471–1508 Search PubMed.
- K. Y. Jen, G. G. Miller and R. L. Elsenbaumer, J. Chem. Soc., Chem. Commun., 1986, 1346–1347 RSC.
-
R. D. McCullough and P. C. Ewbank, in Handbook of Conducting Polymers, ed. T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Marcel Dekker, Inc., New York, 2nd edn, 1998, p. 225 Search PubMed.
- W. L. Ma, C. Y. Yang, X. Gong, K. Lee and A. J. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- M. Reyes-Reyes, K. Kim and D. L. Carroll, Appl. Phys. Lett., 2005, 87, 083506 CrossRef.
- K. Sivula, Z. T. Ball, N. Watanabe and J. M. J. Frechet, Adv. Mater., 2006, 18, 206–210 CrossRef CAS.
- G. Li, V. Shrotriya, J. S. Huang, Y. Yao, T. Moriarty, K. Emery and Y. Yang, Nat. Mater., 2005, 4, 864–868 CAS.
- T. Nishizawa, K. Tajima and K. Hashimoto, J. Mater. Chem., 2007, 17, 2440–2445 RSC.
- Y. Y. Liang, D. Q. Feng, Y. Wu, S. T. Tsai, G. Li, C. Ray and L. P. Yu, J. Am. Chem. Soc., 2009, 131, 7792–7799 CrossRef CAS.
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–U5 Search PubMed.
- Y. Y. Liang, Z. Xu, J. B. Xia, S. T. Tsai, Y. Wu, G. Li, C. Ray and L. P. Yu, Adv. Mater., 2010, 22, E135–E137 CrossRef CAS.
- M. Bjerring, J. S. Nielsen, N. C. Nielsen and F. C. Krebs, Macromolecules, 2007, 40, 6012–6013 CrossRef CAS.
- K. Sivula, C. K. Luscombe, B. C. Thompson and J. M. J. Frechet, J. Am. Chem. Soc., 2006, 128, 13988–13989 CrossRef CAS.
- R. Q. Png, P. J. Chia, J. C. Tang, B. Liu, S. Sivaramakrishnan, M. Zhou, S. H. Khong, H. S. O. Chan, J. H. Burroughes, L. L. Chua, R. H. Friend and P. K. H. Ho, Nat. Mater., 2010, 9, 152–158 CrossRef CAS.
- B. Gholamkhass and S. Holdcroft, Chem. Mater., 2010, 22, 5371–5376 CrossRef CAS.
- S. A. Gevorgyan and F. C. Krebs, Chem. Mater., 2008, 20, 4386–4390 CrossRef CAS.
- T. J. Gordon, J. F. Yu, C. Yang and S. Holdcroft, Chem. Mater., 2007, 19, 2155–2161 CrossRef CAS.
- Y. P. Fu, H. T. Cheng and R. L. Elsenbaumer, Chem. Mater., 1997, 9, 1720–1724 CrossRef CAS.
- S. Akoudad and J. Roncali, Chem. Commun., 1998, 2081–2082 RSC.
- H. Huang and P. G. Pickup, Chem. Mater., 1998, 10, 2212–2216 CrossRef CAS.
- F. Wang, M. S. Wilson, R. D. Rauh, P. Schottland, B. C. Thompson and J. R. Reynolds, Macromolecules, 2000, 33, 2083–2091 CrossRef CAS.
- J. F. Yu and S. Holdcroft, Chem. Commun., 2001, 1274–1275 RSC.
- J. F. Yu and S. Holdcroft, Chem. Mater., 2002, 14, 3705–3714 CrossRef CAS.
- R. D. McCullough and R. D. Lowe, J. Chem. Soc., Chem. Commun., 1992, 70–72 RSC.
- R. D. McCullough, R. D. Lowe, M. Jayaraman and D. L. Anderson, J. Org. Chem., 1993, 58, 904–912 CrossRef CAS.
- R. S. Loewe, P. C. Ewbank, J. S. Liu, L. Zhai and R. D. McCullough, Macromolecules, 2001, 34, 4324–4333 CrossRef CAS.
- T. Yamamoto, D. Komarudin, M. Arai, B. L. Lee, H. Suganuma, N. Asakawa, Y. Inoue, K. Kubota, S. Sasaki, T. Fukuda and H. Matsuda, J. Am. Chem. Soc., 1998, 120, 2047–2058 CrossRef CAS.
- J. K. Politis, J. C. Nemes and M. D. Curtis, J. Am. Chem. Soc., 2001, 123, 2537–2547 CrossRef CAS.
- D. Sainova, S. Janietz, U. Asawapirom, L. Romaner, E. Zojer, N. Koch and A. Vollmer, Chem. Mater., 2007, 19, 1472–1481 CrossRef CAS.
- Z. Bao, A. Dodabalapur and A. J. Lovinger, Appl. Phys. Lett., 1996, 69, 4108–4110 CrossRef CAS.
- T. A. Chen, X. M. Wu and R. D. Rieke, J. Am. Chem. Soc., 1995, 117, 233–244 CrossRef CAS.
- D. Ofer, T. M. Swager and M. S. Wrighton, Chem. Mater., 1995, 7, 418–425 CrossRef CAS.
-
T. M. Swager, in Organized Molecular Assemblies in the Solid State, ed. J. K. Whitesell, John Wiley and Sons Ltd., New York, 1999 Search PubMed.
- A. L. Ayzner, C. J. Tassone, S. H. Tolbert and B. J. Schwartz, J. Phys. Chem. C, 2009, 113, 20050–20060 CrossRef CAS.
- G. Li, Y. Yao, H. Yang, V. Shrotriya, G. Yang and Y. Yang, Adv. Funct. Mater., 2007, 17, 1636–1644 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c0py00222d |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.