Network structure–property relationship in UV-cured organic/inorganic hybrid nanocomposites†
JungHo
Jin
,
SeungCheol
Yang
and
Byeong-Soo
Bae
*
Lab. Optical Materials & Coating, Dept. Materials Science and Engineering, KAIST, 373-1 Guseong-dong, Yuseong-gu, Daejeon 305-701, Korea. E-mail: bsbae@kaist.ac.kr; osirus@kaist.ac.kr; pure0620@kaist.ac.kr
First published on 25th September 2010
Abstract
The UV-induced cross-linked network structures of methacryl-oligosiloxane nanoclusters (MONCs) were analyzed with a hydrofluoric acid dissolution derived gel permeation chromatography technique (HF-GPC) to study their network structure–property relationships. With this method the oligosiloxane cores were dissolved in HF and the remaining organic tethers underwent GPC analysis to observe the tether species and characterize the original cross-linked network structures. It was found that the MONC nanocomposites have a highly cross-linked structure, resulting in oligosiloxane cores which are completely isolated by short methacryl chain segments (two- or three-armed methacryl tethers). This structural analysis was found to be consistent with the macroscale properties of MONC nanocomposites when characterized with thermal (TGA) and thermo-mechanical (DMA and TMA) analyses.
Introduction
Over the last few decades organic/inorganic hybrid nanocomposites have attracted considerable attention and are currently under intense study given some novel properties not found with their non-hybrid counterparts. Thermo-mechanical,1–4 electrical5–8 and optical properties8–10 are included in a range of studies currently being undertaken in industrial and academic settings.11 To date, there have been a significant number of works studying different organic/inorganic hybrid nanocomposites, including those based on polyhedral oligosilsesquioxanes (POSS),12–19 nano-building blocks (NBBs) of metal oxo-clusters4,20–25 and organo-oligosiloxane nanoclusters (ONCs).26–28Compared to pure organic polymers, the desirable properties of organic/inorganic hybrid nanocomposites primarily originate from the natural properties of individual organic/inorganic phases which are synergistically combined at the molecular level, and the manner in which the two phases combine to build a hybrid network. Hence, in order to control the macroscopic properties of each nanocomposite, it is crucial to understand the interfacial interactions between constituent organic/inorganic components and the hybrid network structure. This becomes more critical when the properties of interest are related to thermal, mechanical and viscoelastic properties as these often limit the practical applications of materials.1,29–31 However, for the vast majority of typical hybrid nanocomposites, inorganic components effectively form rigid building blocks which can be usually set as a constant parameter when controlling such properties. In this case, the macroscopic properties of the nanocomposites will be mostly dependent on the cross-linking (or curing) characteristics of the organic components of each network.
Generally speaking, several methods can be used to analyze the structural networks of polymeric materials, including mechanical measurements for the characterization of physical properties and analysis of chemical conversion using spectroscopic methods such as FT-IR, 1H- and 13C-NMR.32 Conventional techniques such as these can offer useful information about the network structures and can be used for the quantitative analysis of chemical cross-links. However, it is difficult to produce even qualitative interpretations of the network structure using these methods due to the complexity of these cross-linked networks. This is because the reacted organic groups form not only chemical cross-links, but also viscoelastically ineffective chains or network defects, such as dangling chains and chain loops.32,33 Furthermore, it is impossible to use conventional spectroscopic techniques to investigate the cross-linking behavior of hybrid nanocomposites since they usually become insoluble after curing. A new analysis technique using HF dissolution-derived GPC (HF-GPC) has been introduced as a useful tool for characterizing organic component network structures and has already been successfully applied to typical siloxane-based hybrid nanocomposites, such as POSS.32,34 In a recent series of papers, Choi et al. have examined the organic network structure of a multi-functional epoxy-POSS/amine (organic spacer) system and demonstrated that its hybrid network is completely discontinuous. In this system both the organic and inorganic components are perfectly isolated from each other, meaning that there must be direct linkages only between epoxy groups from adjacent POSS cages and the amine spacers. This result seems reasonable since the reaction between epoxides is relatively slow compared to epoxy–amine reactions, leading to the absence of epoxy–epoxy links in the final network. In UV-cured (meth)acrylate systems, on the other hand, there is no such reaction preference between reacting groups during free radical polymerization.33 This difference in reaction kinetics between organic groups can result in markedly different nanocomposite network structures when (meth)acrylate is UV-cured.
In this study, our primary goal is to characterize the network structure of a UV-cured (meth)acrylate-based hybrid nanocomposite using the HF-GPC technique, allowing us to investigate structure–property relationships. Here we used methacryl-oligosiloxane nanoclusters (MONCs) as the building blocks of these nanocomposites. MONC is a typical organic/inorganic hybrid material which can be synthesized by a sol–gel reaction of 3-(trimethoxysilyl) propyl methacrylate (MPTS) and diphenylsilanediol (DPSD) (Scheme 1a).26 When UV-cured (Scheme 1b), these MONC nanocomposites exhibit excellent optical properties, including high transparency, low optical loss and low birefringence.28,35MONC-based nanocomposites also have a versatile range of other beneficial properties offering new options for practical applications related to micro-optical devices,9,36,37 soft lithographic technologies,38,39 biosensors,40 and organic electronics.41
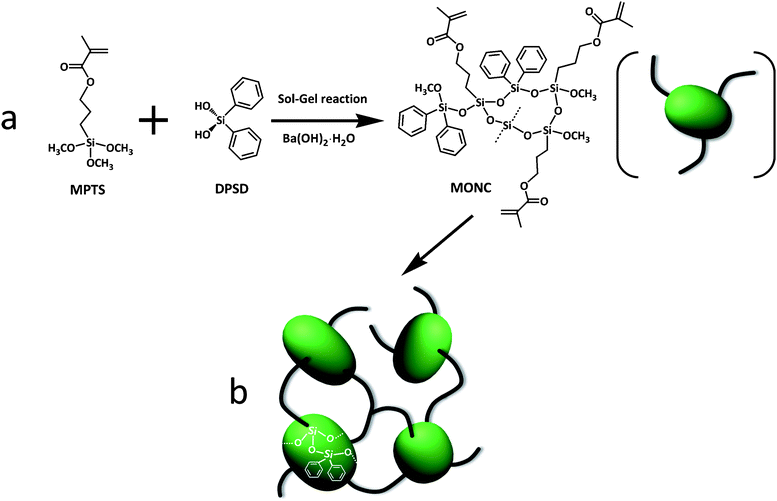 | ||
| Scheme 1 (a) Synthesis of methacryl-oligosiloxane nanoclusters (MONC) and (b) UV-curing of MONC. | ||
Experimental
Materials
3-(Trimethoxysilyl) propyl methacrylate (MPTS), barium hydroxide monohydrate [Ba(OH)2·H2O] and 2,2-dimethoxy-2-phenylacetophenone were purchased from Aldrich (Milwaukee, WI) and used without further purification. Diphenylsilanediol (DPSD) was purchased from Gelest (Morrisville, PA) and used without purification. Ethylene glycol diacrylate (EGDA) was purchased from Aldrich (St Louis, MO) and used as received.Synthesis of MONC resin
Synthesis of MONC resin can be found in the ESI†.Preparation of UV-cured MONC nanocomposites
MONC nanocomposites were prepared by exposing to UV light (Mercury lamp/365 nm, 80 mW cm−2) for 5 minutes under N2 purging and heat-treated at 150 °C for 2 hours in ambient atmosphere. The resultant bulk samples were transparent and hard solids. For MONC/EGDA, EGDA was homogeneously mixed into MONC resin. On blending MONC/EGDA, a useful variable N was defined as a ratio of the number of acryl groups in EGDA/the number of methacryl groups in MONC. Thus, a nanocomposite which is solely composed of MONC is in accordance with N = 0. When N = 0.5, the number of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds in EGDA is half of those in MONC.
C double bonds in EGDA is half of those in MONC.
Characterization
UV-cured nanocomposites were ground into fine powders and 50 mg of each powder was suspended in 4 mL of THF in a polyethylene bottle. Then, 4 mL of HF (50%) were added to the solution. The mixture was slowly stirred and kept at room temperature for 2 days. THF and HF were then evaporated. The residue was mixed with HPLC-grade THF to extract the organic tethers. Remaining coarse powders were removed by filtration using a 0.45 µm Teflon filter before GPC analysis. In previous studies, it was already reported that HF does not deteriorate the organic tethers.34
Results and discussion
The UV cross-linked network structure of MONC nanocomposites
Direct analysis of the organic network structure can be performed with an HF dissolution of the MONC cores, followed by extraction and GPC analysis of the remaining organic fragments. In related studies using this technique, it was reported that organic fragments are retained without damage while Si–O bonds are cleaved by the HF.31 By identifying peaks in the tether GPC the overall network structure can be understood, providing direct insight into the organic network structures of the typical siloxane-based nanocomposites being studied.
Fig. 1 displays the HF-GPC chromatogram of the extracted organic tethers (RI detector) from two materials, with four peaks observable from the MONC (N = 0) sample. Represented in Scheme 2 are four different possible tether species that can be produced during UV-curing of MONC. It is known that fragments of DPSD (D) can be generated during HF-dissolution given that MONC cores are produced by a sol–gel reaction which includes both MPTS and DPSD. Also, due to bulky MONC core-related steric restriction, not all methacryl tethers can participate in cross-linking.46 This means that unreacted methacryl tethers (I) are also expected to be present in the final solution, as evidenced by the reduced peak I in the MONC/EGDA blend. A reference GPC chromatogram was used to identify peaks corresponding to D and I. Fig. 2 shows the GPC chromatogram of a MONC resin containing small traces of unreacted MPTS and DPSD which remain after the sol–gel reaction. Two peaks related to MPTS and DPSD can be observed in the lower molecular weight region. The GPC determined molecular weight of MPTS was 250 g mol−1, almost identical to its formula weight. In contrast, the molecular weight of DPSD appears to be 180 g mol−1, smaller than its formula weight (Table 1). This result suggests that the hydrodynamic volume of DPSD is smaller than that of MPTS.26 Thus it can be expected that the DPSD fragment should be eluted at a longer elution time in the tether GPC. On this basis, peaks appearing in Fig. 1, at 27.7 min and 28.7 min, are thought to be I and D, respectively. The UV-detected HF-GPC chromatogram (Fig. 3) reveals the same tendency and supports the previous result. In this figure the peak appearing at 30 min also shows a stronger UV-response than the peak at 29 min. This can be ascribed to the higher UV-absorbance of benzene derivatives (phenyl groups of D) than the carbonyl compounds of methacryl groups (I).43 After identifying D and I, the remaining peaks appearing at 26.5 min and 27.1 min (Fig. 1) are thought to be three-armed (III) and two-armed tethers (II), respectively. It is worth comparing this result with a completely discontinuous network of epoxy-POSS/amine nanocomposites, which consists of only linear (two-armed) tethers.34 The existence of three-armed tethers (III) in our sample can be attributed to the nature of free radical polymerization in (meth)acrylates, where radical-initiated CC double bonds can propagate to react with other double bonds. Nevertheless, an important feature to note here is the absence of multi-armed tethers of higher molecular weight in the tether GPC. This suggests that these MONC nanocomposites are highly cross-linked, such that their cores are isolated by only a few methacryl chain segments.
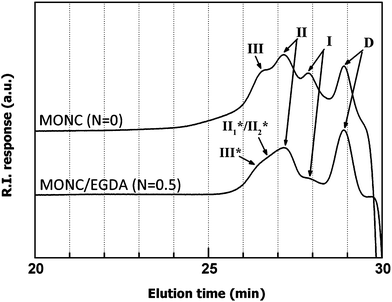 | ||
| Fig. 1 HF-GPC chromatogram of extracted organic tethers from N = 0 (RI detector). For MONC/EGDA (N = 0.5), additionally possible tether species (III* and II1*/II2*) are illustrated in Scheme 3. | ||
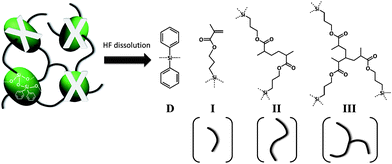 | ||
| Scheme 2 Possible organic tether structures of MONC. Key: (D) fragment of DPSD; (I) unreacted methacryl tether; (II) two-armed tether; and (III) three-armed tether. The dashed bonds represent cleaved state of siloxane bonds due to HF dissolution of MONC cores. | ||
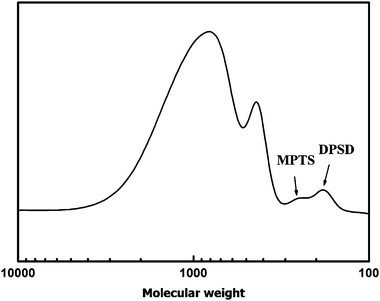 | ||
| Fig. 2 GPC chromatogram of MONC resin. (The molecular weights of MPTS and DPSD determined by GPC analysis were 250 g mol−1 and 180 g mol−1, respectively.) In a GPC test, molecular weights are determined by size exclusion effect. Thus, despite the similar formula weights, the lower molecular weight of DPSD, empirically determined by GPC, is due to the smaller hydrodynamic volume of DPSD than that of MPTS. | ||
 | ||
| Fig. 3 HF-GPC chromatogram of organic tethers (UV detector). | ||
In the MONC/EGDA blend (N = 0.5), several changes are observed in the tether GPC chromatogram (Fig. 1). Firstly, the peak corresponding to I is significantly reduced. This is because the remaining unreacted tethers (I) have reacted with the acryl groups from the EGDA. Secondly, the peak corresponding to III is slightly shifted toward the higher molecular weight region (lower elution time of 26.3 min) and has a profile which overlaps with II. Unlike epoxy–amine reactions, the nature of radical-initiated CC chain reactions can lead to more complicated tether structures.29 Theoretically, however, five additional tether structures can be generated, on the basis that the EGDA can extend from mono- to tetra-functionality (Scheme 3). A shoulder at 26.3 min is thought to be tether III*, however, no clear peak for IV is observed in the tether GPC. Despite the theoretical possibility, it seems, therefore, that the chances of producing tether IV seem to be significantly limited. This may due not only to steric hindrance, but also to the decreased mobility of EGDA after the formation of III*.29 The broad region at around 26.5 min is thought to be II1* and/or II2*; however, the tether structures in this region cannot be differentiated exactly. This heavy overlapping of peaks arises from the similar hydrodynamic volumes of II1* and II2*; these can be estimated properly by a systematic characterization of the macroscopic physical properties.
 | ||
| Scheme 3 (a) Cross-linked network structure of MONC/EGDA blend. (b) Additional possible organic tether structures of MONC/EGDA blend. Functionality of an EGDA monomer can extend from mono- to tetra-, cross-linking with MPTS tether (I) can produce various types of conjugated tethers. | ||
Thermal gravimetric analysis (TGA)
Fig. 4 shows the TGA profile of MONC and MONC/EGDA blends from which the 5% mass loss temperatures (T5%) of both systems can be compared (Table 2). For MONC (N = 0), initial mass loss begins at ∼270 °C, arising from the decomposition of unreacted methacryl tethers (I). The second abrupt mass loss occurs at ∼400 °C and is thought to be due to degradation of cross-links (III and II). For MONC/EGDA (N = 0.5), on the other hand, a similar initial mass loss is not observed. This is because I tethers I have reacted with EGDA, producing additional cross-links, clearly demonstrated by the higher T5% of MONC/EGDA (338 °C vs. 283 °C for MONC). This result shows that the thermal stability of MONC nanocomposites can be strongly enhanced by the reduction of network defects and creation of additional chemical cross-links. In correlation with the above GPC study, it is clear that these cross-links are thermally effective. They are also thought to consist of only III* and II1*, since tethers such as II2* and I* are network defects with dangling chains in the cross-linked structure, which are thermally weak and would be decomposed first (resulting in a lower T5%). This result supports the hypothesis that the macroscopic thermal stabilities of nanocomposites are considerably affected by the organic network structure, provided that the inorganic cores remain unvaried.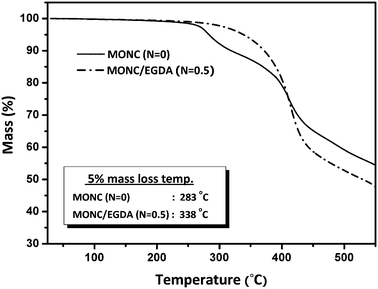 | ||
| Fig. 4 TGA profile of MONC and MONC/EGDA nanocomposites. All tests were carried out with a 5 °C min−1 ramp rate under N2 atmosphere. | ||
| T 5%/°C | T g/°C | E′g/GPa | E′r/MPa | CTE (ppm per °C) | ||
|---|---|---|---|---|---|---|
| Before Tg | After Tg | |||||
| a E′g: Glassy state storage modulus, E′r: rubbery state storage modulus, and CTE: coefficient of thermal expansion. | ||||||
| PET | — | 105 | 4.07 | — | 11 | 64 |
| MONC (N = 0) | 283 | 60 | 3.74 | 281 | 176 | 256 |
| MONC/EGDA (N = 0.5) | 338 | 86 | 3.69 | 521 | 139 | 227 |
Dynamic and thermo-mechanical analysis (DMA and TMA)
Fig. 5 compares the DMA of MONC and MONC/EGDA nanocomposites, and uses polyethylene terephthalate (PET) as a pure organic polymer reference. | ||
| Fig. 5 DMA tests of nanocomposites. PET is used as a reference. All tests were performed with a 5 °C min−1 ramp rate: (a) storage modulus and (b) tan δ. | ||
An important feature can be immediately noticed when comparing the nanocomposites with PET. The storage moduli of the nanocomposites slowly decrease over a wide temperature range, while the PET modulus decreases abruptly (Fig. 5a). This is more clearly observed in the tan δ curves (Fig. 5b). For PET, the maximum tan δ value is as high as ∼0.3 and appears sharply at 105 °C, which is the glass transition temperature (Tg) of PET. For nanocomposites, on the other hand, such a transition is barely noticeable and the maximum tan δ values are only ∼0.1 or less. These values are accompanied by a decrease in the storage modulus of only one order of magnitude (Table 2). A similar trend has also been reported in the early works of Choi et al.34 This contrast can be clearly demonstrated by considering the different network structures of nanocomposites compared to PET.
On the basis of viscoelasticity, the glass transition (α-transition) of an organic polymer is caused by the relaxation motion of chain segments. Tg is where these motions become cooperative, even considerably for main backbone chains.44 These relaxation motions can be slowed through chain entanglements, small crystallinity and rigid side chains, but often most effectively with cross-links.32,45 However, these motions can still become sufficiently cooperative, even through the cross-links of a highly cross-linked polymer. In this context, the lack of pronounced α-transitions in MONC nanocomposites can be interpreted as being due to the absence of main chains (Scheme 4) and the macroscale relaxation of the nanocomposites is the sum of relaxation of each organic tether. This pronounced difference in network structures is illustrated in Scheme 4. Since they are highly cross-linked only by the tethers such as III and II (for N = 0), these short oligomeric chain segments are isolated by inorganic cores and are thus expected to behave differently compared to the continuous chains of a polymer. The relaxation motions of localized tethers are expected to be non-cooperative and even to some extent hindered by the rigid inorganic cores. This accounts for the broad and weak α-transitions in typical highly cross-linked organic/inorganic hybrid nanocomposites, which make them useful in applications where thermo-mechanical properties play an important role.47
 | ||
| Scheme 4 Comparison of network structures: (a) cross-linked polymer and (b) MONC nanocomposites (N = 0). | ||
Another noticeable feature can be observed when comparing MONC and the MONC/EGDA blend. While the glassy state moduli of both systems are similar (∼3.7 GPa), the rubbery state modulus of MONC/EGDA is much higher (521 MPa) than that of MONC (281 MPa). In a cross-linked structure, a rubbery state modulus is directly related to the network cross-link density, often accompanied by a higher Tg. Thus, it is suggested that MONC/EGDA is more highly cross-linked and the relaxation motion of its organic tethers is restricted much more significantly than that of MONC. This result is also consistent with the TGA result that tethers II2* and I* are not likely to exist in MONC/EGDA, since these thermally ineffective tethers are also viscoelastically ineffective. That is, because these chain branches are not bound but are dangling, they would be relaxed easily and their motion is likely to be detectable with DMA, resulting in a decrease of the storage modulus and a lower Tg. tan δ curves also support this result (Fig. 5b), where the maximum tan δ value for MONC/EGDA is lower than that for MONC. It is important to note that the Tg of MONC/EGDA (86 °C) is higher than that of MONC (60 °C), which represents further concrete evidence of the higher cross-link density of MONC/EGDA.
The specific volume of materials changes abruptly at Tg and can usually be measured with the coefficient of thermal expansion (CTE). Fig. 6 shows the TMA of nanocomposites from which the CTEs before and after Tgs of all samples can be compared (Table 2). For PET, CTE changes from 11 to 64 ppm °C−1 around Tg, which is about a six-fold increase. This dramatic change in specific volume provides further evidence of the cooperative thermal relaxation motions of main chains that occur at Tg in PET. In contrast, CTEs of nanocomposites experience a less than two-fold change and the transition zone is much broader. This difference clearly demonstrates the highly cross-linked network structure of the nanocomposites.45 Comparing the two nanocomposites it can be seen that the CTE of MONC/EGDA is 30–40 ppm °C−1 less than MONC both before and after Tg. This result also demonstrates that the network cross-link density of MONC/EGDA is higher than MONC, consistent with the DMA result.
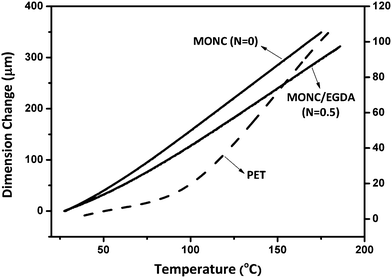 | ||
| Fig. 6 TMA tests of nanocomposites. PET is used as a reference. All tests were performed with a 5 °C min−1 ramp rate. | ||
Conclusion
The hybrid network structure of MONC nanocomposites was investigated with the HF-GPC technique. HF-GPC analyses of organic tethers provide useful information on the network topology of typical siloxane-based nanocomposites. In this study, the results of HF-GPC analyses are also consistently supported by characterizations of the macroscopic physical properties of each material. Systematically relating chemical and physical properties allows for clear interpretations on the hybrid network structures involved. The overall results provide several conclusions for the UV-cured (meth)acrylate/siloxane core-based hybrid nanocomposites and their network structure–property relationships.First, an important point to note is that a typical UV-cured (meth)acrylate–siloxane hybrid network can be discontinuous even in the absence of the core/spacer relation, as seen in the epoxy–amine system. The network can instead be organized by only a few direct linkages between (meth)acryl tethers. This was monitored using DMA and TMA analyses. The results showed that α-transition behavior of each nanocomposite was imperceptible and significantly slowed compared to the pure organic polymer. The highly cross-linked network characteristic and the rigidity of inorganic cores cause the organic tethers to viscoelastically relax slowly and non-cooperatively. However, due to the nature of the radical-initiated polymerization of the (meth)acrylates, three-armed tethers can also be generated.
Second, it was found that nanoscale organic network structures can significantly affect the macroscopic properties of hybrid nanocompsites. For example, nanocomposites' thermal stabilities and thermomechanical properties can be strongly enhanced by introducing additional cross-links, which are thermally and/or viscoelastically effective.
Acknowledgements
This work was supported by the National Research Foundation (NRF) of Korea funded by the Ministry of Education, Science and Technology (MEST) (No. R01-2007-000-20815-0 and CAFDC-20100009898).References
- E. Kopesky, T. Haddad, R. Cohen and G. McKinley, Macromolecules, 2004, 37, 8992 CrossRef CAS.
- C. Sanchez, G. J. de A. A. Soler-Illa, T. Lalot, C. R. Maayer and V. Cabuil, Chem. Mater., 2001, 13, 3061 CrossRef CAS.
- S. Seraji, Y. Wu, M. Forbess, S. J. Limmer, T. Chou and G. Cao, Adv. Mater., 2000, 12, 1695 CrossRef CAS.
- S. Bocchini, G. Fornasieri, L. Rozes, S. Trabelsi, J. Galy, N. E. Zafeiropoulos, M. Stamm, J.-F. Gerard and C. Sanchez, Chem. Commun., 2005, 2600–2602 RSC.
- S. Gross, V. D. Noto and U. Schubert, J. Non-Cryst. Solids, 2003, 322, 154–159 CrossRef CAS.
- B. R. Harkness, K. Takeuchi and M. Tachikawa, Macromolecules, 1998, 31, 4798 CrossRef CAS.
- M. Tsai and W.-T. Whang, Polymer, 2001, 42, 4197 CrossRef CAS.
- J. MacKenzie and E. Bescher, Acc. Chem. Res., 2007, 40, 810 CrossRef CAS.
- D. J. Kang and B. S. Bae, Acc. Chem. Res., 2007, 40, 903 CrossRef CAS.
- C. Rottman, G. Grader, Y. DeHazan, S. Melchior and D. Avnir, J. Am. Chem. Soc., 1999, 121, 8533 CrossRef CAS.
- C. Sanchez, B. Lebeau, F. Chaput and J.-P. Boilot, Adv. Mater., 2003, 15, 1969 CrossRef CAS.
- R. M. Laine, J. Choi and I. Lee, Adv. Mater., 2001, 13, 800 CrossRef CAS.
- O. Toepfer, D. Neumann, N. R. Choudhury, A. Whittaker and J. Matisons, Chem. Mater., 2005, 17, 1027–1035 CrossRef CAS.
- C.-M. Leu, G. M. Reddy, K.-H. Wei and C.-F. Shu, Chem. Mater., 2003, 15, 2261–2265 CrossRef CAS.
- G. Z. Li, L. Wang, H. Toghiani, T. L. Daulton, K. Koyama and C. U. Pittman, Jr, Macromolecules, 2001, 34, 8686–8693 CrossRef CAS.
- L. Matejka, A. Strachota, J. Plestil, P. Whelan, M. Steinhart and M. Slouf, Macromolecules, 2004, 37, 9449–9456 CrossRef CAS.
- H. Xu, B. Yang, X. Gao, C. Li and S. Guang, J. Appl. Polym. Sci., 2006, 101, 3730–3735 CrossRef CAS.
- S. Sulaiman, C. M. Brick, C. M. De Sana, J. M. Katzenstein, R. M. Laine and R. A. Basheer, Macromolecules, 2006, 39, 5167–5169 CrossRef CAS.
- Y. R. Liu, Y. D. Huang and L. Liu, Compos. Sci. Technol., 2007, 67, 2864–2876 CrossRef CAS.
- C. Sanchez, G. J. de A. A. Soler-Illia, F. Ribot, T. Lalot, C. R. Mayer and V. Cabuil, Chem. Mater., 2001, 13, 3061–3083 CrossRef CAS.
- S. Trabelsi, A. Janke, R. Hassler, N. E. Zafeiropoulos, G. Fornasieri, S. Bocchini, L. Rozes, M. Stamm, J.-F. Gerard and C. Sanchez, Macromolecules, 2005, 38, 6068–6078 CrossRef CAS.
- Y. Gao, N. R. Choudhury, J. Matison, U. Schubert and B. Moraru, Chem. Mater., 2002, 14, 4522–4529 CrossRef CAS.
- B. Moraru, G. Kickelbick and U. Schubert, Eur. J. Inorg. Chem., 2001, 1295–1301.
- P. Piszczek, M. Richert, A. Grodzicki, T. Glowiak and A. Wojtczak, Polyhedron, 2005, 24, 663–670 CrossRef CAS.
- G. J. de A. A. Soler-Illia, L. Rozes, M. K. Boggiano, C. Sanchez, C.-O. Turrin, A.-M. Caminade and J.-P. Majoral, Angew. Chem., Int. Ed., 2000, 39(23), 4249–4254 CrossRef CAS.
- Y.-J. Eo, T. H. Lee, S. Y. Kim, J. K. Kang, Y. S. Han and B. S. Bae, J. Polym. Sci., Part B: Polym. Phys., 2005, 43, 827–836 CrossRef CAS.
- J. K. Kim, D. J. Kang and B. S. Bae, Adv. Funct. Mater., 2005, 15, 1870–1876 CrossRef CAS.
- T. H. Lee, J. H. Kim and B. S. Bae, J. Mater. Chem., 2006, 16, 1657–1664 RSC.
- S. Bizet, J. Galy and J.-F. Gerard, Macromolecules, 2006, 39, 2574–2583 CrossRef CAS.
- F. Mammeri, E. L. Bourhis, L. Rozes and C. Sanchez, J. Mater. Chem., 2005, 15, 3787–3811 RSC.
- J. Choi, A. F. Yee and R. M. Laine, Macromolecules, 2003, 36, 5666–5682 CrossRef CAS.
- V. M. Litvinov and A. A. Dias, Macromolecules, 2001, 34, 4051–4060 CrossRef CAS.
- J. E. Elliot and C. N. Bowman, Macromolecules, 2001, 34, 4642–4649 CrossRef.
- J. Choi, J. Harcup, A. F. Yee, Q. Zhu and R. M. Laine, J. Am. Chem. Soc., 2001, 123(46), 11420–11430 CrossRef CAS.
- B. S. Bae, W. S. Kim and K. B. Yoon, Integrated Photonics Research and Application (IPRA) Proceedings, ITuA4, April 11-15, San Diego, USA, 2005.
- J. I. Jung, O. H. Park and B. S. Bae, J. Sol-Gel Sci. Technol., 2003, 26, 897–901 CrossRef CAS.
- K. B. Yoon, B. S. Bae and M. Popall, J. Nonlinear Opt. Phys. Mater., 2005, 14, 399–407 CrossRef CAS.
- W. S. Kim, J. H. Jin and B. S. Bae, Nanotechnology, 2006, 17, 1212–1216 CrossRef CAS.
- D. G. Choi, J. H. Jeong, Y. S. Sim, E. S. Lee, W. S. Kim and B. S. Bae, Langmuir, 2005, 21, 9390–9392 CrossRef CAS.
- W. S. Kim, M. G. Kim, J. H. Ahn and B. S. Bae, Langmuir, 2007, 23, 4732–4736 CrossRef CAS.
- C. G. Choi, S.-Y. Kwak and B. S. Bae, SID Int. Symp. Dig. Tech. Pap., 2008, 39(1), 1239–1242 Search PubMed.
- B. M. Mitsyuk, Theor. Exp. Chem., 1984, 19, 554–559 CrossRef.
- L. D. Field, S. Sternhell and J. R. Kalman, Organic Structures from Spectra, John Wiley & Sons. Ltd, 1986 Search PubMed.
- J. J. Aklonis and W. J. MacKnight, Introduction to Polymer Viscoelasticity, John Wiley & Sons, Inc., 2nd edn, 1983 Search PubMed.
- Y. Rao and T. N. Blanton, Macromolecules, 2008, 41, 935–941 CrossRef CAS.
- J.-S. Kim, S. C. Yang and B. S. Bae, J. Sol-Gel Sci. Technol., 2010, 53, 434–440 CrossRef CAS.
- J. Jin, J.-H. Ko, S. Yang and B. S. Bae, Adv. Mater., 2010 DOI:10.1002/adma.201002198.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of MONC resin. See DOI: 10.1039/c0py00148a |
| This journal is © The Royal Society of Chemistry 2011 |