Sulfonated poly(arylene ether sulfone) ionomers containing di- and tetrasulfonated arylene sulfone segments†
Elin Persson
Jutemar
,
Shogo
Takamuku
and
Patric
Jannasch
*
Polymer & Materials Chemistry, Department of Chemistry, Lund University, P.O. Box 124, SE-221 00, Lund, Sweden. E-mail: patric.jannasch@polymat.lth.se; Fax: +46 462224012; Tel: +46 462229860
First published on 2nd November 2010
Abstract
Poly(arylene ether sulfone) (PSU) ionomers containing disulfonated aryl-SO2-aryl and tetrasulfonated aryl-SO2-aryl-aryl-SO2-aryl segments, respectively, were synthesized and studied to establish their structure–property relationships as proton-exchange membranes. High molecular weight PSUs with different distributions of sulfone bridges in the backbone were prepared by nucleophilic aromatic substitution reactions involving 4,4′-dichlorodiphenyl sulfone (DCDPS), 4,4′-bis[(4-chlorophenyl)sulfonyl]-1,1′-biphenyl (BCPSB), 4,4′-isopropylidenediphenol (bisphenol A), and 4,4′-(1,4-phenylenediisopropylidene)bisphenol (bisphenol P). The polymers were sulfonated viametallation and reaction with sulfur dioxide, followed by oxidation of the resulting sulfinates. This procedure allowed the introduction of two sulfonic acid units on electron-deficient aryl rings in ortho positions to each sulfone bridge of the PSUs. Analysis by small angle X-ray scattering of solvent cast membranes showed that ionic clustering was promoted in ionomers containing sulfonated BCPSB residues and flexible bisphenol P residues. The fully sulfonated PSUs had ion-exchange capacities (IECs) of 3.3–4.1 meq g−1 and were water soluble. However, partly sulfonated polymers with IECs of approx. 1.7 meq g−1 showed high proton conductivity at moderate water uptake and decomposed only above 240 °C during heating 1 °C min−1 under air. This work demonstrated that BCPSB residues can be conveniently and fully tetrasulfonated, which opens possibilities to prepare various aromatic copolymers and membranes with locally very high densities of hydrolytically stable sulfonic acid groups.
Introduction
Highly sulfonated polymers with high thermoxidative and hydrolytic stability are currently investigated and developed for a range of industrial applications. These durable ionomer materials may for example be applied as solid acid catalysts,1 transducers,2 as well as membranes for water purification.3,4 However, perhaps the most important application area of sulfonated polymers is currently as proton-exchange membranes for polymer electrolyte fuel cells, which are highly energy efficient and potentially environmentally benign power sources.5–7 Today, perfluorosulfonic acid polymers such as Nafion® are considered as state-of-the-art membranes due to their high physical and chemical stabilities and high proton conductivity under a wide range of relative humidity at moderate operating temperatures.8 Still, Nafion® ultimately suffers from disadvantages such as high cost, high fuel permeability, low modulus, and low proton conductivity at temperatures above 100 °C at reduced relative humidity (RH).9There is currently an extensive worldwide search and development of alternative sulfonated polymers for proton-exchange membranes.10–13 Over the last few years, a wide range of different sulfonated aromatic hydrocarbon polymers have been evaluated and demonstrated as fuel cell membranes, including sulfonated poly(arylene ether sulfone)s, poly(arylene ether ketone)s and polyphenylenes.11–13 These materials generally have high thermal and chemical stabilities, combined with good mechanical properties. In some cases, membranes based on sulfonated aromatic polymers have shown higher proton conductivity than Nafion® at high temperature under reduced RH.14–16 A number of investigations have demonstrated that it is possible to reach high proton conductivities at low RHs provided that a percolating phase domain is maintained in the membrane where the local concentration of acid is kept high through the presence of densely sulfonated segments in the polymer structure.16–18 In addition, the densely sulfonated segments should preferentially have a high chain stiffness (high glass transition temperature [Tg]) to depress water uptake and solubility of the membrane under immersed or high RH conditions.19
Several different sulfonated polymer chain architectures have been reported to enhance the properties of aromatic hydrocarbon ionomer membranes. In general, the acid units of these ionomers have been concentrated to specific side chains or to segments in the backbone polymer in order to improve the nanophase separation between ionic and non-ionic domains.20 Thus, improved properties have been reported for ionomers with highly sulfonated units in the polymer structure, including end groups,21,22 segments incorporated into the backbone polymer,23 blocks in multiblock copolymers,24–27 and side chains.20,28–30 In addition, various ionomers have been investigated where well-defined highly sulfonated segments are evenly spaced along the polymer backbone by hydrophobic segments.17,31,32 Although advanced copolymers, such as sulfonated hydrophilic–hydrophobic multiblock copolymers, have shown higher conductivities under reduced RH in comparison with homopolymers and statistical copolymers, properly designed polymers of the latter types are still highly interesting for fuel cell applications because of their relative ease of preparation. One strategy to enhance the performance of membranes based on homopolymers and statistical copolymers is to facilitate the formation of large ionic clusters by concentrating the acid units along the backbone polymer.
Sulfonic acid groups in aromatic polymers have most often been introduced via post-modification by using, for example, fuming sulfuric acid, trimethylsilylchlorosulfonate or chlorosulfonic acid.33 These electrophilic substitutions typically lead to sulfonations of the most activated positions in the aromatic polymer structure, often in positions close to electron donating ether bridges. This means that the polymers may be activated for desulfonation during fuel cell operation, especially at temperatures exceeding 100 °C.33 Alternatively, sulfonic acid groups may also be introduced in deactivated aromatic positions close to electron withdrawing bridges in the polymer structure. Kreuer and coworkers have for example prepared a highly sulfonated poly(phenylene sulfone) with an ion-exchange capacity (IEC) of 4.5 meq g−1.14 Each highly electron poor aryl ring in this polymer was monosulfonated, leading to a high hydrolytic stability to prevent desulfonation reactions.14 Although the polymer was water soluble, the proton conductivity at low RHs (15–50%) exceeded that of Nafion®.
In the present work, the prospects of using lithiation chemistry to prepare poly(arylene ether sulfones) (PSUs) having tetrasulfonated segments with the sulfonic acid groups placed in deactivated positions were investigated. In the first step, PSUs were prepared viapolycondensations of 4,4′-bis[(4-chlorophenyl)sulfonyl]-1,1′-biphenyl (BCPSB) with either bisphenol P or bisphenol A. This gave PSUs with evenly spaced aryl-SO2-aryl-aryl-SO2-aryl segments along the polymer backbone. The introduction of four sulfonic acid groups in ortho-sulfone positions of these segments was subsequently achieved via a metallation–sulfination–oxidation route to produce polymers with IECs of 3.6 and 4.1 meq g−1, respectively. Because the fully sulfonated polymers were found to be water soluble, polymers with IECs of approx. 1.7 meq g−1 were also prepared. In addition, a regular PSU of “Udel”-type, containing aryl-SO2-aryl segments, was sulfonated using the same procedure. Sulfonated PSUs having various distributions of densely sulfonated segments along the backbone copolymer (Scheme 1) were characterized with regard to their ability to form ionic clusters in the membranes, as well as their thermal stability, water uptake and proton conductivity.
 | ||
| Scheme 1 Graphical illustration of the fully sulfonated PSU ionomers with different lengths of the bisphenol (filled rectangles) and aryl sulfone segments (unfilled rectangles) with the sulfonic acid groups (filled circles) placed on the latter. The ionomers are designated as sPSUa,b, where a and b are the number of aryl rings in the repeating units which are linked by –C(CH3)2– and –SO2– bridges, respectively. | ||
Experimental
Materials
Poly[oxy-1,4-phenylenesulfonyl-1,4-phenyleneoxy-1,4-phenylene(dimethylmethylene)-1,4-phenylene] (PSU of “Udel-type”, BASF, Ultrason S6010 Natural) was dried under vacuum at 100 °C for 72 h before use. Tetrahydrofuran (THF, Fisher Scientific, HPLC grade) was dried with molecular sieves as described by Burfield et al.34 Potassium carbonate (Acrôs, 99+%) was dried at 120 °C overnight before use. 4,4′-Isopropylidenediphenol (bisphenol A, Acrôs, 97%) and 4,4′-(1,4-phenylenediisopropylidene)bisphenol (bisphenol P, Sigma-Aldrich, 99%) were recrystallized from toluene, while 4,4′-bis[(4-chlorophenyl)sulfonyl]-1,1′-biphenyl (BCPSB, Sigma-Aldrich, 98%) was recrystallized from N,N-dimethylformamide (DMF) before drying in vacuo at 80 °C. N,N-Dimethylacetamide (DMAc, Acrôs, 99%), toluene (Fisher Scientific, HPLC grade), methanol (Fisher Scientific, Analytical grade), n-butyllithium (n-BuLi, Acrôs, 2.5 M in hexanes), chlorotrimethylsilane (Aldrich, redistilled, 99+%), sulfur dioxide gas (Fluka, 99.9%), 2-propanol (IPA, Fisher Scientific, HPLC grade), hydrogen peroxide (Acrôs, 35 wt% in water) and fuming hydrochloric acid (Merck, 37%, pro analysis) were all used as received. Dowex® 50W-X8 (H) ion-exchange resin (Merck) was loaded with Pb2+ using a water solution of lead acetate. Dialysis tubes (Spectrum, Spectra/Pore® Membrane, MWCO: 3500) were placed in distilled water 1 h before use.Polycondensation
Three different PSUs were used in the sulfonations of the present work (Table 1). They were designated as PSUa,b, where a and b denoted the number of aromatic rings linked by isopropylidene and sulfone bridges, respectively, in the repeating units. Thus, the commercially available regular PSU (“Udel-type”) was designated as PSU2,2. PSU2,4 was prepared via a polycondensation of bisphenol A and BCPSB, and PSU3,4 was prepared from bisphenol P and BCPSB, as seen in Scheme 2.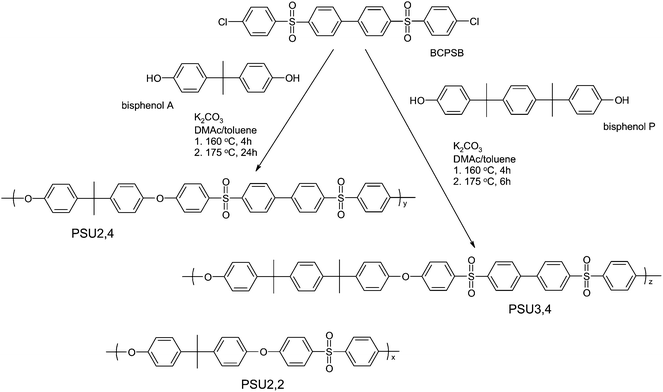 | ||
| Scheme 2 Molecular structures of the PSUs and synthetic pathways to PSU2,4 and PSU3,4via nucleophilic aromatic substitution reactions of BCPSB and bisphenol A and P, respectively. | ||
PSU2,4 was prepared as follows: bisphenol A (5.1979 g, 22.7688 mmol), BCPSB (11.4625 g, 22.7688 mmol) and K2CO3 (3.78 g, 27.35 mmol) were added to a mixture of DMAc (70 mL) and toluene (35 mL) in a two-necked flask equipped with a magnetic stirrer, a nitrogen inlet, and a Dean–Stark trap with both a condenser and an outlet fitted with a calcium chloride filter. The reaction mixture was first heated at 160 °C for 4 h. After dehydration and removal of the toluene, the reaction temperature was increased to 175 °C and kept at this temperature for 24 h to complete the polycondensation. The viscous mixture was then cooled to room temperature before precipitation of the polymer in an excess of distilled water. The water was replaced by fresh hot distilled water and the polymer was washed overnight. The precipitate was filtered and washed with methanol, and filtered again before redissolving in cold THF. Non-soluble components were filtered off before precipitation in methanol. The product was washed repeatedly in methanol and hot water, filtered, and finally rinsed with methanol before being dried in vacuo at 80 °C overnight (overall yield: 87%).
PSU3,4 was prepared by essentially the same procedure with only minor modifications: bisphenol P (6.6888 g, 19.3056 mmol), BCPSB (9.7190 g, 19.3056 mmol) and K2CO3 (3.20 g, 23.15 mmol) were added to a mixture of DMAc (64 mL) and toluene (32 mL) in the two-necked flask described above. As with the PSU2,4, the reaction mixture was first heated at 160 °C for 4 h to dehydrate and remove the toluene. This reaction was kept at 175 °C for 6 h to complete the polycondensation. The product was purified using the same procedure as described above for PSU2,4 (overall yield: 75%).
Polymer silylation
In an initial study to investigate the maximum extent of lithiation and sulfonation possible, both PSU2,4 and PSU3,4 were lithiated and then silylated before characterization by 1H NMR spectroscopy. In a typical procedure, 0.50 g (0.76 mmol) of PSU2,4 polymer was dissolved in 50 mL of THF in a reactor fitted with an argon inlet/outlet, a thermometer, and a septum. The solution was cooled to −40 °C, carefully degassed several times, and left under a blanket of argon. It was then cooled to −70 °C and 10 equivalents (3.0 mL, 7.6 mmol) of n-BuLi were added dropwise. After 60 min chlorotrimethylsilane (2.5 mL, 19 mmol) was added all at once, and the reaction mixture was slowly heated to room temperature, after which the polymer was precipitated in 200 mL of IPA. The trimethylsilyl-modified polymer was filtered and dried in vacuo at 80 °C overnight before analysis.Polymer sulfonation
PSU2,4, PSU3,4 and PSU2,2 were sulfonated via sulfination by lithiation and reaction with sulfur dioxide gas, followed by the oxidation of the –SO2Li groups to –SO3Li. In a typical procedure the polymer was dissolved in THF to reach a concentration of 0.01 g mL−1 in a reactor fitted with an argon inlet/outlet, a thermometer, a sulfur dioxide gas inlet, and a septum. The solution was cooled to −40 °C, carefully degassed, then cooled to −70 °C and left under a blanket of argon. Subsequently, n-BuLi was added dropwise until the color of the solution changed from colorless to light green. At this point, a volume of n-BuLi corresponding to the desired level of sulfination was added (see Table 2), which gave a dark green solution/dispersion. After 30 min the argon was evacuated and gaseous sulfur dioxide was added from the top of the reactor by carefully opening the gas inlet valve under extensive stirring of the reaction solution, resulting in the immediate formation of a white precipitate. Degassing and addition of sulfur dioxide gas were repeated until the supernatant became colorless, indicating that all the lithiated sites had been quenched. During slow heating to room temperature, the reaction mixture was degassed several times. For polymers with a high degree of sulfination (target IEC of 2.0 or higher), the THF was evaporated from the mixture before drying the samples in vacuo at room temperature overnight. For polymers with a low degree of sulfination, the mixture was poured into IPA to isolate the precipitate which was filtered off and dried in vacuo at room temperature overnight.| Ionomer | m polymer a/g | V BuLi b/mL | n BuLi (mmol)/nrepeating unitc (mmol) | IECtarget (meq g−1)/DStargetd | IECcalc (meq g−1)/DScalce | IECexpf (meq g−1) | w water g (wt%) | λ |
|---|---|---|---|---|---|---|---|---|
| a Mass of the polymer charged. b Volume of n-BuLi solution charged. c Mole of n-BuLi per mole of repeating unit of the polymer charged. d Targeted IEC/targeted average number of sulfonic acid groups per repeating unit. e Calculated IEC/calculated average number of sulfonic acid groups per repeating unit, as determined from NMR. f Calculated from titration data of the membrane. g Water uptake after immersion at room temperature. h w/s—water soluble. | ||||||||
| sPSU2,2-1.11 | 9.8 | 7.5 | 0.85 | 1.33/0.66 | 1.11/0.54 | 1.03 | 24 | 13 |
| sPSU2,2-1.80 | 2.0 | 2.3 | 1.25 | 2.00/1.05 | 1.80/0.93 | 1.76 | 56 | 18 |
| sPSU2,2-3.31 | 2.0 | 4.5 | 2.5 | 3.31/2.00 | 3.31/2.00 | — | w/sh | — |
| sPSU2,4-1.09 | 2.0 | 1.2 | 0.98 | 1.33/0.98 | 1.09/0.79 | 0.72 | 17 | 13 |
| sPSU2,4-2.00 | 2.0 | 1.9 | 1.57 | 2.00/1.57 | 2.00/1.57 | 1.74 | 49 | 16 |
| sPSU2,4-4.09 | 0.5 | 1.5 | 5 | 4.09/4.00 | 4.09/4.00 | — | w/sh | — |
| sPSU3,4-1.00 | 1.1 | 0.7 | 1.16 | 1.33/1.16 | 1.00/0.84 | 1.01 | 25 | 14 |
| sPSU3,4-1.91 | 2.0 | 1.9 | 1.85 | 2.00/1.85 | 1.91/1.75 | 1.69 | 46 | 15 |
| sPSU3,4-3.65 | 0.5 | 1.4 | 5 | 3.65/4.00 | 3.65/4.00 | — | w/sh | — |
The sulfinated polymers were subsequently oxidized with H2O2 to produce the sulfonated derivatives, which were designated as sPSUa,b-c where c denoted the IEC value calculated from 1H NMR data. In a typical procedure, 0.75 g of the sulfinated polymer was dispersed or dissolved (in the case of water-insoluble and water-soluble sulfinates, respectively) in 20 mL of distilled water under nitrogen in a reactor. The dispersion/solution was heated to 40 °C and 2 equivalents of H2O2 were added per –SO2Li. After 2 h of reaction, additional 8 equivalents of H2O2 were added and the reaction was allowed to proceed overnight. Water-insoluble polymers were filtered off and washed with distilled water before being dried in vacuo at 80 °C overnight, while water-soluble polymers were dialyzed in tubes with a cut-off molecular weight of 3500 g mol−1. The water was evaporated from the dialyzed polymer solutions before drying the samples in vacuo at 80 °C overnight.
Polymer characterization
Size exclusion chromatography (SEC) was used to determine the molecular weight of the non-sulfonated polymers. The setup included three Shodex columns in series (KF-805, -804, and -802.5) and a refractive index detector. All samples were run at room temperature in chloroform at an elution rate of 1 mL min−1. Calibration was done by using polystyrene standards with molecular weights of 30.3, 96.0, 650 and 1800 kg mol−1 from Polymer Laboratories, Ltd.1H NMR data were collected using a Bruker DRX400 spectrometer. Spectra were recorded at 400.13 MHz and the chemical shifts were reported relative to DMSO-d6 (δ 2.50 ppm). For the silylated polymers, the degree of functionalization (and hence the degree of lithiation) was determined by comparing the integrals of the methyl protons at δ = 0.3–0.7 ppm, originating from the trimethylsilyl groups, the isopropylidene protons at δ = 1.6–1.8 ppm, and the aromatic protons at δ = 6.8–8.1 ppm. For the sulfinated and sulfonated polymers, the degree of functionalization was determined by comparing the integrals of the isopropylidene protons at δ = 1.6–1.8 ppm and the aromatic protons at δ = 6.8–8.4 ppm.
FTIR analysis was carried out on a Bruker IFS 66 spectrometer. The polymers were ground together with KBr before tablets were pressed. Spectra of the samples were recorded between 400 and 4000 cm−1 at a resolution of 4 cm−1 using 128 cumulative scans.
Membrane preparation
Membranes were cast in Petri dishes from 8 wt% solutions of the sulfonated polymers in their lithium salt form dissolved in DMAc. All the membranes were prepared under N2 flow at 65 °C for 48 h, followed by drying under vacuum at 80 °C for 24 h. The membranes had a thickness in the range 100–150 µm and were ion-exchanged to the protonated form by immersion in 1 M HCl for 24 h, followed by leaching with distilled water for two days during which time the water was repeatedly exchanged.X-Ray scattering
SAXS measurements were carried out on membranes in the Pb2+ form to enhance the scattering intensity. The ion-exchange was performed by immersing water non-soluble ionomers in a 1 wt% solution of lead acetate and by passing the water-soluble ionomers through a cation-exchange resin loaded with Pb2+. The scattering experiments were performed on a Kratky compact small angle system equipped with a position sensitive wire detector with 1024 channels having a width of 53.6 µm. A Seifert ID 3000 X-ray generator operating at 55 kV and 40 mA provided CuKα radiation with a wavelength of λ = 1.524 Å. Dry samples were placed between mica sheets in a sealed solid sample cell and the measurements were performed for 2 h at 25 °C. The wave vector (q) was calculated according to:| q = 4π/λ × sin θ | (1) |
| d = 2π/q | (2) |
Thermal characterizations
The thermal stability was evaluated by thermogravimetric analysis (TGA) using a Q500 analyzer from TA Instruments. The thermal stability of samples, both in the protonated form and the salt form, was analyzed under N2 during heating from 50 to 600 °C at 10 °C min−1, and under air during heating from 50 to 600 °C at 1 °C min−1. Prior to the heating scan, all samples were pre-dried under N2 at 150 °C for 10 min. The temperature at which the sample had lost 5% of its original weight during heating was taken as the degradation temperature (Td).A Q1000 calorimeter from TA Instruments was used to carry out the differential scanning calorimetry (DSC) investigations. During the DSC measurements, the polymers were first heated to 400 °C, or alternatively to 10 °C below Td if this temperature was below 410 °C. The samples were then cooled to 50 °C, followed by a heating scan to 400 °C. All heating and cooling rates were kept at 10 °C min−1. Glass transition temperatures (Tgs) were taken as the midpoint of the transition recorded during the second heating scan.
Water uptake and ion-exchange capacity measurements
Membranes were equilibrated in distilled water for at least 48 h to determine the water uptake (wwater) under immersed conditions. To obtain the wet weight (Wwet), the excess water was gently removed with tissue paper before weighing the swollen membranes. The dry weight (Wdry) was obtained after drying in vacuo at 80 °C overnight. The water uptake was then calculated as:| wwater = (Wwet − Wdry)/Wdry × 100% | (3) |
The IEC was measured by titration of acidic membranes. Protonated membranes were soaked in an aqueous 2 M NaCl solution for at least 72 hours. The solutions were then titrated with a 0.01 M KOH solution using phenolphthalein as indicator.
Conductivity measurements
Proton conductivity was evaluated by electrochemical impedance spectroscopy (EIS) using a Novocontrol high-resolution dielectric analyzer V 1.01S equipped with a Novocontrol temperature system. Impedance data were measured using a 2-probe method with the membranes fully immersed in a sealed cell during heating from −20 to 100 °C, then during cooling to −20 °C, and finally during heating to 100 °C. The reported data were collected during the second heating scan. Measurements were performed over a frequency range of 107 to 10−1 Hz at a voltage amplitude of 50 mV, and the data were subsequently analyzed using the software WinDeta® from Novocontrol. The conductivity was evaluated by plotting the imaginary part of the conductivity versus the real part, and the conductivity was then taken as the real value that corresponded to the minimum imaginary response.Results and discussion
Organolithium chemistry allows the introduction of sulfonic acid groups specifically on stable deactivated positions on certain aromatic polymers without the occurrence of side reactions.35–37 In the present study, the possibility of preparing highly sulfonated polymersvia lithiation and sulfonation of PSUs containing a high density of sulfone bridges was investigated (Scheme 1). For this purpose polymers with different concentrations and distributions of sulfone links along the backbone polymer were synthesized.Polymer preparation and characterization
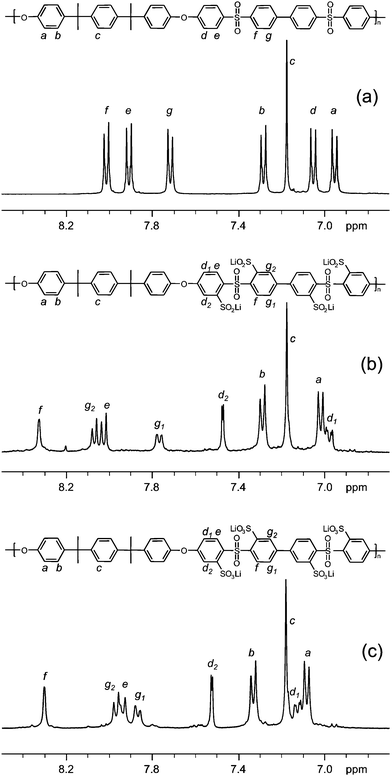
Two PSU samples, PSU2,4 and PSU3,4, were synthesized viapolycondensation of bisphenol A and bisphenol P, respectively, together with BCPSB (Scheme 2). In the present work, bisphenols were chosen as comonomers in order to vary the chain length in-between the sulfonated segments while maintaining the same structural units in the hydrophobic parts of the polymers. Later on, suitable monomers may be chosen to prepare fully aromatic ionomers. BCPSB was charged in equimolar amounts to the respective bisphenol together with a 25% excess of potassium carbonate. After a 4 h dehydration step, the temperature was increased and the polycondensation was allowed to proceed until a high viscosity of the polymerization mixture was reached, which occurred after 24 h for PSU2,4 and after 6 h for PSU3,4. Fig. 1a and 2a show the aromatic region of the 1H NMR spectra of the purified polymers. In the case of PSU2,4, all aromatic shifts were found between δ = 6.9 and 8.1 ppm and the signal arising from the aliphatic isopropylidene links was observed at δ = 1.7 ppm (not shown). The aromatic region of the 1H NMR spectrum of PSU3,4 was identical except for an additional aromatic shift at δ = 7.2 ppm arising from the phenylene rings flanked by the two isopropylidene links in the bisphenol P residue. Additionally, in relation to the spectrum of PSU2,4, an increased signal from the additional aliphatic isopropylidene link was observed at δ = 1.7 ppm. | ||
| Fig. 1 1H NMR spectra of (a) PSU2,4, (b) fully sulfinated PSU2,4, and (c) fully sulfonated sPSU2,4-4.09. The data were collected using DMSO-d6 solutions of the samples. | ||
 | ||
| Fig. 2 1H NMR spectra of (a) PSU3,4, (b) fully sulfinated PSU3,4, and (c) fully sulfonated sPSU3,4-3.65. The data were collected using DMSO-d6 solutions of the samples. | ||
The average molecular weight and the polydispersity of the PSUs were determined by SEC using polystyrene standards. As seen in Table 1, PSU2,2 and PSU2,4 had similar molecular weights, 38 kg mol−1 and 33 kg mol−1, respectively, while PSU3,4 had a molecular weight of 79 kg mol−1 as anticipated by the high viscosity reached during the polymerization.
Polymer silylation
The PSUs were modified by lithiation using n-BuLi, which generally provides a highly specific and efficient route to chemically functionalize this class of polymers. Lithiation of PSUs has the advantage that it is exclusively directed to aromatic positions situated ortho to the sulfone linkages of the polymer backbone.35 In accordance, PSU2,4 and PSU3,4 have four positions per repeating unit that may potentially be lithiated because of the presence of the two sulfone bridges. In order to investigate the extent of modification possible, the two polymers were lithiated using a large excess of n-BuLi followed by reaction with chlorotrimethylsilane in THF at −70 °C (see ESI, Scheme S1†). Chlorotrimethylsilane was chosen as a reactant because of its quantitative reaction with lithiated sites38 and because the shifts of the methyl protons of the resulting trimethylsilyl groups did not overlap with any of the protons of the backbone polymers in the 1H NMR spectra. This enabled a straightforward quantification of the lithiation efficiency.In order to reach the maximum of four lithiated positions per repeating unit, 10 equivalents of n-BuLi per repeating unit of the respective PSU were charged in the lithiations. During the addition, the color of the solution quickly turned deep green. In contrast to PSU3,4, sample PSU2,4 precipitated after the addition of n-BuLi. However, the solubility was regained after the addition of an excess of chlorotrimethylsilane, whereafter the solution turned from light green to colorless at room temperature. The lithiation of four positions per repeating unit was confirmed by 1H NMR for both of the polymers (see ESI, Fig. S1†). The ratio of the integral of the aromatic protons to that of the methyl protons of the attached trimethylsilyl groups was found to be 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :36 for PSU2,4 and 24:36 for PSU3,4, which corresponded to the fully silylated polymers. Thus, PSU2,4 apparently became fully lithiated despite the precipitation of the polymer during the reaction.
:36 for PSU2,4 and 24:36 for PSU3,4, which corresponded to the fully silylated polymers. Thus, PSU2,4 apparently became fully lithiated despite the precipitation of the polymer during the reaction.
Polymer sulfonation
After demonstrating the possibility to lithiate up to four positions per repeating unit of PSU2,4 and PSU3,4, the prospects of synthesizing fully sulfonated PSUs having four sulfonic acid groups per repeating unit were investigated (see Scheme 3). At this point, PSU2,2 was also added to the study with the possibility to introduce two sulfonic acid groups per repeating unit.37 Fully sulfinated PSUs were achieved by reacting the polymers with a 25% molar excess of n-BuLi, followed by the addition of sulfur dioxide as depicted in Scheme 3. The synthesis was conveniently performed as a one-pot procedure in THF at −70 °C. The 1H NMR spectra of the purified polymers, seen in Fig. 1b, 2b and 3b, confirmed the full sulfination, and the peak integrals had all the expected ratios. | ||
| Scheme 3 Sulfonation of PSU2,4via lithiation and sulfination, followed by oxidation to obtain the fully tetrasulfonated derivative (sPSU2,4-4.09). PSU2,2 and PSU3,4 were sulfonated using the same method. | ||
 | ||
| Fig. 3 1H NMR spectra of (a) PSU2,2, (b) fully sulfinated PSU2,2, and (c) fully sulfonated sPSU2,2-3.31. The data were collected using DMSO-d6 solutions of the samples. | ||
The sulfinated polymers were efficiently oxidized to the fully sulfonated polymers sPSU2,2-3.31, sPSU2,4-4.09, and sPSU3,4-3.65 by using H2O2. The change in the position of the shifts and the unaltered integrals in the 1H NMR spectra, seen in Fig. 1c, 2c and 3c, confirmed the complete oxidation of all the sulfinates. The full oxidation was also confirmed by FTIR, as shown in Fig. 4. The absorption band at 976 cm−1, originating from the symmetrical S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching of the sulfinate groups, was replaced by an absorption band at 1012 cm−1 originating from the symmetrical SO stretching of the sulfonate groups. As expected from their high theoretical IEC values, all the three fully sulfonated polymers were readily water soluble.
O stretching of the sulfinate groups, was replaced by an absorption band at 1012 cm−1 originating from the symmetrical SO stretching of the sulfonate groups. As expected from their high theoretical IEC values, all the three fully sulfonated polymers were readily water soluble.
 | ||
| Fig. 4 FTIR spectra of (a) PSU3,4, (b) fully sulfinated PSU3,4 and (c) fully sulfonated PSU3,4 (sPSU3,4-3.65). | ||
Obviously, the water solubility prevented the preparation of membranes of the fully sulfonated PSUs. Consequently, the three different PSUs were also partly sulfonated, aiming at IECs of 1.33 and 2.00 meq g−1, in order to study the influence of the PSU structure on the water uptake characteristics and proton conductivity of the ionomer membranes. Partly sulfinated polymers were prepared using a similar procedure as previously described for the fully sulfinated polymers. However, a lower molar equivalent of n-BuLi in relation to the available sites ortho-to-sulfone links was used. A few droplets of n-BuLi were first added until a light green color appeared, and then a specific volume of n-BuLi was added. Table 2 shows the number of moles of n-BuLi added per repeating unit, corresponding to the target degree of sulfonation (DStarget). For PSU2,2, a slightly higher amount of n-BuLi had to be used in order to reach an IEC in level with that of the corresponding ionomers based on PSU2,4 and PSU3,4.
The resulting degree of sulfination of the PSU3,4 ionomers was determined by 1H NMR data using the sum of the integrals of protonsd2 and f, as shown in Fig. 5. For the PSU2,4 ionomers, the degree of sulfination was determined in a similar way by using the sum of the c2 and eprotons. In all cases, the degree of sulfination was found to be higher on the phenyl rings flanked by an ether and sulfone bridge in comparison to the biphenyl rings, with between 60 and 70% of the sulfinate groups being placed on the former rings. This indicated a higher reactivity of these rings during the lithiation step. The degree of sulfination of the PSU2,2 was determined by comparing the signals arising from the di-, mono-, and non-sulfonated repeating units, respectively, as previously described by Kerres et al.37 All the partly sulfonated PSUs were fully oxidized by employing the same method as for the fully sulfonated polysulfones. The complete oxidation was confirmed by both NMR and FTIR data. Table 2 shows the DS obtained for the different sulfonations. As seen, the DScalc was in the range of 0.54 to 2.00 and 1.00 to 4.00 for the sPSU2,2 ionomers and sPSU2,4 and sPSU3,4 ionomers, respectively.
 | ||
| Fig. 5 Aromatic region of the 1H NMR spectrum of partly sulfinated PSU3,4 showing signals which indicate the fractions of mono- and disubstituted aromatic regions of the BCPSB residue, respectively. The degree of sulfination was quantified by taking into account the integrals of the d2′, d2″, f′, and f″ signals. Z denotes –SO2Li or –H. | ||
X-Ray scattering
The ability of the sulfonated polymers to form ionic clusters was studied by SAXS, a method which allows an identification of the characteristic separation length between the ionic domains in terms of the position (q-value) and width of the so-called ionomer peak.39SAXS has been widely employed to study the ionic clusters of ionomers, including sulfonated poly(ether ether ketone),40–42 poly(ether sulfone)30,41 and poly(1,4-phenylene sulfide),43 as well as Nafion®.39,41,42The SAXS profiles originating from the six PSU ionomer membranes with the highest IEC are shown in Fig. 6 together with the corresponding profile of Nafion®. Prior to the scattering experiments the membranes were ion-exchanged to the Pb2+ form to increase the contrast between the ionic clusters and the hydrophobic polymer-rich matrix phase. As seen in Fig. 6, Nafion® gave rise to a rather sharp ionomer peak at q = 0.18 Å−1, indicating distinct ionic clusters which were rather regularly separated with a characteristic separation length of d = 34 Å.

The profiles of the sulfonated PSU membranes showed much broader ionomer peaks and were shifted to higher q-values, as compared to the profile of Nafion®. This difference indicated a smaller cluster separation, d = 22–30 Å, with a significantly wider distribution of the characteristic separation lengths in comparison with Nafion®. The PSU main chain is far less hydrophobic and less flexible, and the sulfonic acid groups less acidic, than in Nafion®. This led to less regularly spaced phase domains in the sulfonated PSU membranes. These findings are consistent with previously published SAXS data on dry main chain sulfonated aromatic polymers.30,40,42 By comparing the profiles within the two series of fully and partly sulfonated PSUs shown in Fig. 6, it is clearly seen that the ionomer peak position shifted to lower q-values for the polymers containing bisphenol P residues, in comparison to those containing bisphenol A residues. This indicated that long flexible hydrophobic segments gave larger separation lengths between the ionic clusters, and thus promoted the ionic clustering process. In addition, PSUs containing the larger sulfonated BCPSB residues showed lower q-values in comparison with corresponding PSUs with the DCDPS residues. Similar observations have previously been reported by Miyatake et al. after studies of sulfonated poly(arylene ether sulfone) copolymers containing various hydrophobic components.44 As seen in Fig. 6, the ionomer peaks of the sPSU3,4 ionomers were narrower than the peaks of the sPSU2,2 and sPSU2,4 ionomers at comparable IEC values, which indicated a more narrow distribution of the separation lengths in the former membranes. Also seen was a tendency for the ionomers having the same PSU backbone to shift to higher q-values as the IEC increased. This finding was consistent with the expected decrease in distances between the ionic domains for membranes with increasing sulfonic acid concentrations and IEC values. In conclusion, a clear phase separation on the nano scale was found between ion-rich domains and the hydrophobic polymer-rich matrix in all the studied ionomers.
Thermal properties
The Tgs of the non-modified PSUs were determined by DSC and the values are found in Table 1. As seen, PSU2,4 and PSU3,4 with the long and stiff aryl-SO2-aryl-aryl-SO2-aryl segments showed higher Tgs than PSU2,2 with the comparatively short aryl-SO2-aryl segments. The Tgs of PSU2,4 and PSU3,4 were very similar, despite the presence of the longer flexible bisphenol residues in the structure of the latter. This is likely explained by the higher molecular weight of PSU3,4 in relation to PSU2,4, which raised the Tg of the former.DSC analysis of the membranes based on the partly sulfonated ionomers in the salt form showed clear glass transitions. However, no glass transitions were detected for the membranes in the acid form. In addition, no glass transitions were detected for the fully sulfonated polymers in the temperature range up to 400 °C. As expected, the IEC was found to have a profound impact on the Tg of the ionomers (Table 3). For example, an increase in IEC from 0.72 to 1.74 meq g−1 for the ionomers derived from PSU2,4 led to an increase of 110 °C, from Tg = 260 to 371 °C. This increase is consistent with previously reported Tgs of PSUs sulfonated by using mixtures of chlorosulfonic acid and chlorotrimethylsilane,45 or a sulfur trioxide–triethyl phosphate complex.46 In these studies, Tgs up to 300 °C were observed for the highest sulfonated PSUs.
| Membrane | Sodium salt form | Acid form | |||
|---|---|---|---|---|---|
| T g/°C | T d under N2a/°C | T d under airb/°C | T d under N2a/°C | T d under airb/°C | |
| a Measured at 10 °C min−1. b Measured at 1 °C min−1. c n/d—not detected. d n/m—not measured. | |||||
| sPSU2,2-1.11 | 195 | 420 | 376 | 305 | 259 |
| sPSU2,2-1.80 | 273 | 415 | 373 | 281 | 242 |
| sPSU2,2-3.31 | n/dc | 355 | 345 | n/md | n/md |
| sPSU2,4-1.09 | 260 | 435 | 371 | 349 | 308 |
| sPSU2,4-2.00 | 371 | 423 | 393 | 311 | 270 |
| sPSU2,4-4.09 | n/dc | 409 | 364 | n/md | n/md |
| sPSU3,4-1.00 | 279 | 427 | 383 | 323 | 279 |
| sPSU3,4-1.91 | 326 | 421 | 386 | 294 | 254 |
| sPSU3,4-3.65 | n/dc | 384 | 363 | n/md | n/md |
The thermal stability of the polymers and ionomers was investigated by TGA analysis. The Td values of PSU2,2 and PSU3,4 were comparable, 492 and 483 °C, respectively, under nitrogen (Table 1). PSU2,4 had a Td at 455 °C which was 28 and 37 °C lower than that of PSU2,2 and PSU3,4, respectively. The ionomers were analyzed in both the sodium salt and the acid form. Measurements under air at 1 °C min−1 were undertaken to study the stability under oxidative conditions, and measurements under nitrogen at 10 °C min−1 were performed to study the degradation under less drastic conditions. The ionomers sPSU2,4 and sPSU3,4, containing the BCPSB monomer residue, were shown to have higher thermal stability than the sPSU2,2. As mentioned previously, PSU2,4 showed the lowest Td of the PSUs. However, after sulfonation the sPSU2,4 ionomers had the highest thermal stability of the three series of ionomers. As expected, the ionomers in the sodium salt form showed higher Tds than the corresponding ionomers in the acid form, and the stability was higher under nitrogen than under air. In the acid form, the Td decreased strongly with increasing IEC values within all three series of ionomers. It is important to note that the Td of all the ionomers in the present study was higher, by up to 70 °C under nitrogen, than PSUs post-sulfonated by using trimethylsilylchlorosulfonate,47 or chlorosulfonic acid/chlorotrimethylsilane.45 This indicated the advantage of using the lithiation–sulfonation reactions to introduce sulfonic acid on deactivated positions.
Water uptake characteristics and proton conductivity
The level of hydration of proton-exchange membranes is highly dependent on the IEC. However, at high levels of hydration the mechanical properties are typically compromised because of the high degree of swelling. Consequently, the membrane properties should be tuned so that the water uptake is controlled and kept at a moderate level. PSUs with sulfonic acid groups placed directly onto the main chain typically show excessive water uptake at elevated temperatures or sufficiently high IECs.9Tough transparent membranes of the partly sulfonated ionomers were cast from DMAc solutions with the ionomers in the lithium salt form. The membranes were subsequently acidified and titrated to determine the IEC. Table 2 shows the resulting IEC values which, with one exception, were equal to or slightly lower than the IEC calculated from NMR data. However, sPSU2,4-1.09 had a markedly lower titrated IECexp of 0.72 meq g−1 compared to the IEC determined by NMR, IECcalc = 1.09 meq g−1. FTIR results showed the complete disappearance of the absorption band at 976 cm−1 originating from the sulfinate groups. A possible explanation of the discrepancy between the IEC values might be a loss of labile sulfinate groups during the oxidation process.37
The water uptake and λ-value (i.e. the number of water molecules per sulfonic acid group) of each ionomer membrane at room temperature are shown in Table 2. As previously mentioned, all the fully sulfonated ionomers were completely water soluble. For the partly sulfonated polymers, an increase in IEC gave the expected increase in the water uptake within each series of ionomer with low to moderate water uptakes ranging from 17 to 56%, corresponding to λ-values between 13 and 18. These values were similar to or lower than previously published values for sulfonated poly(ether sulfones).45,48 As anticipated from its low IEC, the water uptake of membrane sPSU2,4-1.09 was quite low, 17%. Comparing the three membranes in the narrow IEC range between 1.69 and 1.76 meq g−1, the water uptake of sPSU2,4-2.00 and sPSU3,4-1.91 was lower than that of sPSU2,2-1.80, which may be explained by the higher Tgs of the former ionomers.
The proton conductivity was measured under immersed conditions as a function of temperature during heating from −20 °C and 100 °C. As anticipated, the proton conductivity in all the three series of ionomer membranes increased with increasing IEC (Fig. 7). The larger rate of conductivity increase observed during heating between −20 and 20 °C was due to the melting of water. The proton conductivity of all the membranes then increased rather monotonously between 20 and 80 °C. For two of the ionomers the proton conductivity decreased during heating from 80 to 100 °C. This may be caused by an excessive water uptake, leading to a dilution of the sulfonic acid groups. Under these conditions, the Tg is highly depressed, which might lead to poor dimensional stability. Consistent with the relatively low IEC value and water uptake of the sPSU2,4-1.09 membrane, the proton conductivity was quite low. Two of the samples, sPSU2,2-1.80 and sPSU2,4-2.00, reached conductivities higher than that of Nafion® over the entire temperature range studied.
 | ||
| Fig. 7 Proton conductivity of the PSU ionomer membranes measured by EIS with the membranes fully immersed in water. Corresponding data for Nafion® 117 have been included for comparison. | ||
Conclusions
The possibility to introduce segments with locally very high densities of hydrolytically stable sulfonic acid groups into aromatic polymers was shown by first preparing PSUs with different concentrations and distributions of sulfone links, and then introducing sulfonic acid units in ortho-positions to the sulfone links. The PSUs were prepared by polycondensations using BCPSB and bisphenols, and the aryl-SO2-aryl-aryl-SO2-aryl segments of these polymers were subsequently fully sulfonated viametallation–sulfination–oxidation which resulted in regular sequences of four sulfonated aryl rings in the backbone. The fully sulfonated PSUs reached IECs of 3.3–4.1 meq g−1 and were water soluble. However, also partly sulfonated ionomers with IECs of approx. 1.7 meq g−1 were prepared with good precision. Membranes based on these ionomers showed high proton conductivity at moderate water uptake and decomposed only above 240 °C during heating 1 °C min−1 under air. SAXS analysis revealed that the ionic clustering in the membranes was promoted by ionomers containing sulfonated BCPSB residues and flexible bisphenol P residues. The work demonstrated that BCPSB residues can be conveniently and fully tetrasulfonated. This offers possibilities to prepare different aromatic copolymers and membranes with locally very high densities of hydrolytically stable sulfonic acid groups, which is beneficial for fuel cell operation under low RH conditions.Acknowledgements
We thank the Swedish Foundation for Strategic Environmental Research, MISTRA, for financial support through the MISTRA Fuel Cell programme.References
- S. Iimura, K. Manabe and S. Kobayashi, Org. Lett., 2003, 5, 101 CrossRef CAS.
- A. J. Duncan, D. J. Leo and T. E. Long, Macromolecules, 2008, 41, 7765 CrossRef CAS.
- R. K. Nagarale, G. S. Gohil and V. K. Shahi, Adv. Colloid Interface Sci., 2006, 119, 97 CrossRef CAS.
- G. M. Geise, H.-S. Lee, D. J. Miller, B. D. Freeman, J. E. McGrath and D. R. Paul, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 1685 CrossRef CAS.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS.
- J. Zhang, Z. Xie, J. Zhang, Y. Tang, C. Song, T. Navessin, Z. Shi, D. Song, H. Wang, D. P. Wilkinson, Z.-S. Liu and S. Holdcroft, J. Power Sources, 2006, 160, 872 CrossRef CAS.
- F. de Bruijn, Green Chem., 2005, 7, 132 RSC.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535 CrossRef CAS.
- J. A. Kerres, J. Membr. Sci., 2001, 185, 3 CrossRef CAS.
- A. L. Rusanov, P. V. Kostoglodov, M. J. M. Abadie, V. Y. Voytekunas and D. Y. Likhachev, Adv. Polym. Sci., 2008, 216, 125 CAS.
- J. Rozière and D. J. Jones, Annu. Rev. Mater. Res., 2003, 33, 503 CrossRef CAS.
- G. Maier and J. Meier-Haack, Adv. Polym. Sci., 2008, 216, 1 CAS.
- T. Higashihara, K. Matsumoto and M. Ueda, Polymer, 2009, 50, 5341 CrossRef CAS.
- M. Schuster, C. C. de Araujo, V. Atanasov, H. T. Andersen, K.-D. Kreuer and J. Maier, Macromolecules, 2009, 42, 3129 CrossRef CAS.
- B. Bae, T. Yoda, K. Miyatake, H. Uchida and M. Watanabe, Angew. Chem., Int. Ed., 2010, 49, 317 CAS.
- A. Roy, X. Yu, S. Dunn and J. E. McGrath, J. Membr. Sci., 2009, 327, 118 CrossRef CAS.
- M. L. Einsla, Y. S. Kim, M. Hawley, H.-S. Lee, J. E. McGrath, B. Liu, M. D. Guiver and B. S. Pivovar, Chem. Mater., 2008, 20, 5636 CrossRef CAS.
- B. Bae, T. Yoda, K. Miyatake, M. Uchida, H. Uchida and M. Watanabe, J. Phys. Chem. B, 2010, 114, 10481 CrossRef CAS.
- K. Goto, I. Rozhanskii, Y. Yamakawa, T. Otsuki and Y. Naito, Polym. J. (Tokyo, Jpn.), 2009, 41, 95 CrossRef CAS.
- Y. Yang and S. Holdcroft, Fuel Cells, 2005, 5, 171 CrossRef CAS.
- S. Matsumura, A. R. Hlil, N. Du, C. Lepiller, J. Gaudet, D. Guay, Z. Shi, S. Holdcroft and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 3860 CrossRef CAS.
- S. Matsumura, A. R. Hlil, C. Lepiller, J. Gaudet, D. Guay and A. S. Hay, Macromolecules, 2008, 41, 277 CrossRef CAS.
- K. Matsumoto, T. Higashihara and M. Ueda, Macromolecules, 2009, 42, 1161 CrossRef CAS.
- C. K. Shin, G. Maier, B. Andreaus and G. G. Scherer, J. Membr. Sci., 2004, 245, 147 CrossRef CAS.
- H. Ghassemi, J. E. McGrath and T. A. Zawodzinski, Jr, Polymer, 2006, 47, 4132 CrossRef CAS.
- K. Nakabayashi, K. Matsumoto, T. Higashihara and M. Ueda, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 7332 CrossRef CAS.
- X. Yu, A. Roy, S. Dunn, A. S. Badami, J. Yang, A. S. Good and J. E. McGrath, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 1038 CrossRef CAS.
- D. J. Jones and J. Rozière, J. Membr. Sci., 2001, 185, 41 CrossRef CAS.
- J. H. Pang, H. B. Zhang, X.-F. Li, L. F. Wang, B. J. Liu and Z. H. Jiang, J. Membr. Sci., 2008, 318, 271 CrossRef CAS.
- E. P. Jutemar and P. Jannasch, J. Membr. Sci., 2010, 351, 87 CrossRef CAS.
- Z. Zhu, N. M. Walsby, H. M. Colquhoun, D. Thompsett and E. Petrucco, Fuel Cells, 2009, 9, 305 CrossRef CAS.
- A. Roy, M. A. Hickner, X. Yu, Y. Li, T. E. Glass and J. E. McGrath, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 2226 CrossRef CAS.
- C. Iojoiu, M. Maréchal, F. Chabert and J.-Y. Sanchez, Fuel Cells, 2005, 5, 344 CrossRef CAS.
- D. R. Burfield, G. H. Gan and R. H. Smithers, J. Appl. Chem. Biotechnol., 1978, 28, 23 CAS.
- M. D. Guiver, G. P. Robertson, M. Yoshikawa and C. M. Tam, ACS Symp. Ser., 2000, 744, 137 CAS.
- P. Jannasch, Fuel Cells, 2005, 5, 248 CrossRef CAS.
- J. Kerres, W. Cui and S. Reichle, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 2421 CrossRef CAS.
- M. D. Guiver, J. W. ApSimon, O. Kutowy, US Pat., 4833219, 1989.
- G. Gebel and O. Diat, Fuel Cells, 2005, 5, 261 CrossRef CAS.
- D. X. Luu, E.-B. Cho, O. H. Han and D. Kim, J. Phys. Chem. B, 2009, 113, 10072 CrossRef CAS.
- N. Takimoto, L. Wu, A. Ohira, Y. Takeoka and M. Rikukawa, Polymer, 2009, 50, 534 CrossRef CAS.
- B. Yang and A. J. Manthiram, J. Power Sources, 2006, 153, 29 CrossRef CAS.
- M. A. Barique, L. Wu, N. Takimoto, K. Kidena and A. Ohira, J. Phys. Chem. B, 2009, 113, 15921 CrossRef CAS.
- B. Bae, K. Miyatake and M. Watanabe, Macromolecules, 2009, 42, 1873 CrossRef CAS.
- H. B. Park, H.-S. Shin, Y. M. Lee and J.-W. Rhim, J. Membr. Sci., 2005, 247, 103 CrossRef CAS.
- A. Noshay and L. M. Robeson, J. Appl. Polym. Sci., 1976, 20, 1885 CrossRef CAS.
- S. Guhathakurta and K. Min, J. Appl. Polym. Sci., 2010, 115, 2514 CrossRef CAS.
- F. Wang, M. Hickner, Y. S. Kim, T. A. Zawodzinski and J. E. McGrath, J. Membr. Sci., 2002, 197, 231 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reaction scheme and 1H NMR data for silylated polysulfones. See DOI: 10.1039/c0py00290a |
| This journal is © The Royal Society of Chemistry 2011 |