
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced cellular uptake and photochemotherapeutic potential of a lipophilic strained Ru(II) polypyridyl complex†
Stephanie Mehannaab,
Najwa Mansourab,
Hassib Audia,
Kikki Bodman-Smithb,
Mohamad A. Mrouehc,
Robin I. Taleb a,
Costantine F. Dahera and
Rony S. Khnayzer*a
a,
Costantine F. Dahera and
Rony S. Khnayzer*a
aDepartment of Natural Sciences, Lebanese American University, Chouran, Beirut 1102-2801, Lebanon. E-mail: rony.khnayzer@lau.edu.lb
bFaculty of Health and Medical Sciences, Department of Microbial and Cellular Sciences, University of Surrey, UK
cSchool of Pharmacy, Department of Pharmaceutical Sciences, Lebanese American University, Lebanon
First published on 3rd June 2019
Abstract
The use of ruthenium complexes as chemotherapeutic agents has been recently explored as one of the alternatives to conventional treatments. In the present study, two Ru(II) polypyridyl complexes were synthesized and characterized: a strained [Ru(bipy)2(BC)]Cl2 (complex 1) where [bipy = 2,2′-bipyridine and BC = bathocuproine] along with the unstrained control [Ru(bipy)2(phen)]Cl2 (complex 2) where [phen = 1,10-phenanthroline]. The photophysical and photochemical analyses proved that unlike the photostable complex 2, complex 1 ejected both bipy and BC ligands at a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 respectively. Results showed that the activity of complex 1 was significantly enhanced upon photoactivation. The response was however particularly significant in B16-F10 melanoma cells where phototoxicity index (PI = IC50 dark/IC50 light) was >900. When compared to cisplatin, the photoproducts were more potent against all tested cell lines, implying that the complex acquired significant chemotherapeutic potential upon irradiation. Cellular uptake of complex 1 and the free BC ligand were found to be significantly facilitated as evidenced by 400–600 fold increase in concentration of the compounds inside the cells relative to the extracellular culture medium. Complex 2 exhibited 35 times lower cellular concentration relative to complex 1. Flow cytometry and plasmid DNA gel electrophoresis measurements showed that complex 1 interacts with DNA inducing apoptosis in the dark and either late-apoptosis or necrosis upon irradiation. These findings corroborate the importance of lipophilic ligands such as BC to enhance uptake and subsequently improve the photochemotherapy potential of Ru(II) polypyridyl complexes.
:1 respectively. Results showed that the activity of complex 1 was significantly enhanced upon photoactivation. The response was however particularly significant in B16-F10 melanoma cells where phototoxicity index (PI = IC50 dark/IC50 light) was >900. When compared to cisplatin, the photoproducts were more potent against all tested cell lines, implying that the complex acquired significant chemotherapeutic potential upon irradiation. Cellular uptake of complex 1 and the free BC ligand were found to be significantly facilitated as evidenced by 400–600 fold increase in concentration of the compounds inside the cells relative to the extracellular culture medium. Complex 2 exhibited 35 times lower cellular concentration relative to complex 1. Flow cytometry and plasmid DNA gel electrophoresis measurements showed that complex 1 interacts with DNA inducing apoptosis in the dark and either late-apoptosis or necrosis upon irradiation. These findings corroborate the importance of lipophilic ligands such as BC to enhance uptake and subsequently improve the photochemotherapy potential of Ru(II) polypyridyl complexes.
Introduction
The accidental discovery of cisplatin as a chemotherapeutic drug paved the way to the extensive research on metal-based anticancer agents. Metal complexes have distinct properties and diverse mechanisms of action that might differ from other chemotherapies.1 Cisplatin, oxaliplatin and carboplatin are platinum complexes used clinically for the treatment of many types of cancer such as head, neck, lung, ovarian and bladder cancers.2 However, their clinical application has been limited by adverse side effects, toxicity and development of resistance by cancer cells.3 To overcome these constraints, research has been directed towards platinum derivatives as well as other metal-based chemotherapeutic agents.4More recently, ruthenium has gained a lot of interest mainly due to distinct features that could potentially convey selectivity towards cancer cells.5 The advantage of these drugs could mostly be attributed to their potential selectivity to cancer cells through intrinsic or extrinsic mechanisms, such as photochemotherapy. Advantageous properties of ruthenium-based drugs include (i) the ability of some complexes to achieve different oxidation states in biological media,5 (ii) the possibility to design Ru(III) prodrugs that can be selectively reduced to the active Ru(II) drugs by the hypoxic environment of cancer cells,6 (iii) the possible exchange between O- and N- ligands which can allow binding to biological targets like nucleic acids in a way similar to cisplatin,5 and (iv) intracellular transport by transferring receptors that are overexpressed in cancer cells.7
Selectivity of Ru(II) complexes has been further exploited by the use of light-mediated activation of Ru(II) prodrugs through mechanisms including photodynamic therapy (PDT) and photo-activated chemotherapy (PACT).8 Unlike PDT, the mechanism of PACT is oxygen-independent which is an advantage since the environment of cancer cells is known to be hypoxic.9 Ru(II) polypyridyl complexes are particularly attractive for PACT because of their tunable photophysical and photochemical properties.10 While most of these complexes are photostable, some can undergo ligand photosubstitution reactions that result from distortions in the octahedral geometry.11 Photodissociation typically occurs following the population of the 3d–d state which is thermal accessed from the 3MLCT excited state.12 PACT exploits several mechanisms to induce cytotoxicity depending on the photoactivation strategy employed.8 Ejection of the straining ligand upon irradiation often leads to the formation of aqua complexes that can interact with DNA similarly to cisplatin.8 We have recently reported the quantitative ejection of the non-straining ligand bipy in [Ru(bipy)2dpphen]2+ [where bipy = 2,2′-bipyridine and dpphen = 2,9-diphenyl-1,10-phenanthroline] and the formation of aquated photoproduct.13 Other groups evaluated the photochemistry and photobiology of similar Ru(II) bipyridine complexes in water where aqua complexes like [Ru(bipy)2(H2O)2]2+ and [Ru(bipy)2(H2O)(OH)]+ were produced upon dissociation of the straining ligand. Photoproducts were proved to damage DNA by variable mechanisms including DNA binding, intercalation, or cleavage.12,14–16 Other ligand frameworks with similar binding potential to DNA have also been investigated.17–19 The cytotoxic species in Ru-based PACT is not necessarily the aqua complex formed. In this respect, our group demonstrated that the dissociating ligand 2,9-dimethyl-1,10-phenanthroline (dmphen) in [Ru(bipy)2(dmphen)]Cl2 exhibited higher toxicity on ML-2 acute myeloid leukemia cells as compared to [Ru(bipy)2(H2O)2]2+ and cisplatin.20 Likewise, Bonnet and coworkers confirmed that the phototoxicity of [Ru(bipy)2(dmbipy)]2+ was primarily attributed to the ejected ligand 6,6′-dimethyl-2,2′-bipyridine (dmbipy) rather than the ruthenium bis-aqua complex which was proved to be less potent.21 Turro et al. also exploited the ability of a Ru(II) complex to act as a caging molecule for the active ligand 5-cyanouracil (5CNU) which inhibited pyrimidine catabolism when photoejected from the Ru(II) center.22 Importantly, we have recently demonstrated that 4,7-diphenyl-1,10-phenanthroline ligands significantly enhance the uptake and biological activity of Ru(II) complexes.23
In the current study, a sterically strained Ru(II) bipyridyl complex [Ru(bipy)2(BC)]Cl2 (complex 1) [where BC = bathocuproine] has been synthesized along with the unstrained control [Ru(bipy)2(phen)]Cl2 (complex 2), Fig. 1. The aim of introducing the BC ligand was to create steric strain around the ruthenium center due to the methyl groups at the 2,9-positions and increase lipophilicity of the drug by introducing phenyl groups at the 4,7-positions of the 1,10-phenanthroline ligand. Here, the photochemistry of complex 1 was studied and photolysis products were analyzed.
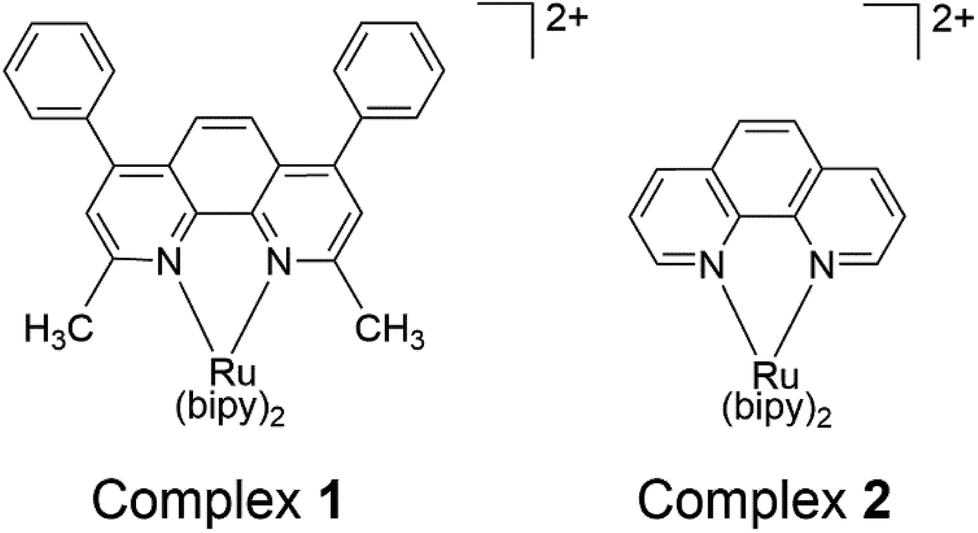 | ||
| Fig. 1 Structures of complexes 1 and 2 (bipy = 2,2′-bipyridine). | ||
The in vitro cytotoxicity in the dark and upon light-activation was then evaluated on a panel of cell lines and compared to the control (complex 2). The cellular uptake of complexes 1, 2 and the BC ligand were assessed using LC-MS/MS and rationalized based on their lipophilicity. Finally, the apoptotic potential of complex 1 was investigated using Annexin V/PI staining flow cytometry and plasmid DNA gel electrophoresis experiments.
Experimental
Instrumentation
The instrumental parameters used for 1H NMR, 13C NMR, MALDI-TOF MS, LC-MS/MS and irradiation were previously reported.13 Elemental analysis and HR-ESI MS were performed at Atlantic Microlab and Michigan State University respectively. Cell death was assessed by Annexin V/PI staining, using a C6 flow cytometer (BD Accuri, Ann Arbor, MI) and data were analyzed on FL1-H versus FL2-H scatter plot.Materials
cis-Bis(2,2′-bipyridine)dichlororuthenium(II) hydrate [Ru(bipy)2Cl2·2H2O], bathocuproine (BC), 1,10-phenanthroline (phen), dowex 22 chloride, stationary phase material (Sephadex LH 20), DMEM medium, penicillin G sodium salt, FBS, and all other chemicals and solvents were purchased from Aldrich. Formic acid and LC-MS grade water were from Fisher Chemical. WST-1 reagent was from Roche©. Annexin V-Fluorescein Isothiocyanate (Annexin V-FITC) and Propidium Iodide (PI) apoptosis/necrosis detection kit was from Abcam (Cambridge, MA) and all cell lines were received from ATCC.Synthesis of [Ru(bipy)2(BC)](PF6)2 (complex 1)
Synthesis was performed following a slightly modified published method.24 Briefly, Ru(bipy)2Cl2·2H2O (100 mg, 0.19 mmol) and BC (74.25 mg, 0.21 mmol, MW 360.45 g mol−1) were mixed in 8 mL ethylene glycol and the solution was degassed for 1 h under argon and refluxed in a pressure vessel (260 °C for 6 h). The product was then cooled at room temperature and filtered through micropores (PVDF sterile syringe filters, 33 mm, 0.45 μm, Millipore® Millex®). The hexafluorophosphate (PF6) salt was precipitated by adding a saturated aqueous solution of KPF6 dropwise and purification was achieved by column chromatography on sephadex LH 20 column (methanol). Yield: 179.7 mg, 89%. 1H NMR (CD3CN, 500 MHz) δ = 8.54 (d, J = 10 Hz, 2H), 8.47 (d, J = 10 Hz, 2H), 8.07 (dd, J = 12.5 Hz, 10 Hz, 2H), 8.04 (s, 2H), 7.99 (dd, J = 12.5 Hz, 10 Hz, 2H), 7.76 (dd, J = 5 Hz, J = 1 Hz, 2H), 7.73 (dd, J = 5 Hz, J = 1 Hz, 2H), 7.63–7.58 (m, 12H), 7.36 (dd, J = 10 Hz, J = 5 Hz, 2H), 7.28 (dd, J = 10 Hz, J = 5 Hz, 2H), 1.97 (s, 6H) (Fig. S1†). 13C NMR (CD3CN, 500 MHz) δ = 167.36, 158.62, 158.34, 153.86, 152.76, 150.40, 150.23, 138.77, 138.60, 136.73, 130.66, 130.55, 130.03, 128.78, 128.42, 128.32, 128.24, 125.78, 125.46, 125.37, 26.39 (Fig. S2†). Elemental anal calcd for C46H36F12N6P2Ru: C, 51.94; H, 3.41; N, 7.9. Found: C, 51.69; H, 3.57; N, 7.73. HRMS (ESI/QTOF) for C46H34N6Ru [M]2+: m/z calcd 387.1029; found: 387.1034 (Fig. S3†). MS (MALDI/TOF) for C46H36F12N6P2Ru [M]+: 773.1 (Fig. S4†). UV/vis (ACN): λmax(ε M−1 cm−1) 454 nm (22000). Conversion of the complex to chloride salt was performed as previously reported, using dowex beads.13
Synthesis of [Ru(bipy)2(phen)]Cl2 (complex 2)
Synthesis was performed according to a published procedure.25Study of the photochemistry of complexes 1 and 2 by 1H NMR and LC-MS/MS
The photochemistry of complex 1 (PF6 salt) was evaluated by 1H NMR in deuterated acetonitrile (CD3CN) and LC-MS/MS was performed on the chloride form of the complex to establish whether the photochemical transformation is similar in water. Complex 2 was assessed by LC-MS/MS and all analyses were conducted with and without irradiation. For 1H NMR, photoactivation was performed at an output of 100 mW cm−2 for 6 h (high concentration) as compared to 35 min for LC-MS/MS (low concentration). BC and bipy ligands were also independently inserted into the MS to enable their identification in the spectra. The dissociation ratio of BC and bipy was determined by 1H NMR analysis of the photolyzed complex as well as LC-MS/MS using a 1:1 bipy:BC solution as a standard.
Comparison of the stability of complex 1 between dark and light conditions by UV/vis spectroscopy
The chloride salt of complex 1, [Ru(bipy)2(BC)]Cl2] was dissolved in water and irradiated with blue LED light (460 nm, 100 mW cm−2) or incubated in the dark for different durations. UV/vis absorption spectra were then recorded at each time point using the same parameters for all conditions tested.Lipophilicity
logP values were measured for complexes 1 and 2 according to a modified procedure.26 Briefly, 0.5–2 mg of the chloride form of the complex were dissolved in 1 mL water saturated with octanol. 1 mL octanol saturated with water was then added and the mixture was shaken at 1400 rpm for 1 h at room temperature. Complete separation of the aqueous and organic phases was obtained by centrifugation at 4300 rpm for 10 min. The lower aqueous phase was then aspirated using a glass syringe while degassing through the octanol phase to prevent any traces of octanol from contaminating the needle. The two phases were sequentially aspirated, and UV-vis absorption spectra were recorded. logP values were computed using the following formula:| logP = log (Coctanol/Cwater) |
Quantification of cellular uptake by LC-MS/MS
| [Ru(bipy)2(BC)]Cl2 (complex 1) | [Ru(bipy)2(Phen)]Cl2 (complex 2) | BC | |
|---|---|---|---|
| Mobile phase | Isocratic 5% A: H2O (0.1% formic acid), 95% B: MeOH (0.1% formic acid), flow rate (0.5 mL min−1) | ||
| Precursor ion (m/z) | 386.86 | 296.78 | 361.16 |
| Product ions (m/z) | 308.7, 361.16 | 181.1, 218.67 | 345.23, 359.16 |
| Collision energy (V) | 27 | 27 | 52 |
| Retention time (min) | 0.40 | 0.38 | 0.50 |
| Mass mode | ESI positive mode | ESI positive mode | ESI positive mode |
| Scan type | Single reaction monitoring (SRM) | SRM | SRM |
Cellular uptake
Human alveolar adenocarcinoma cells (A549) were seeded in 6-well plates at a final concentration of 1.2 × 105 cells per well and allowed to adhere overnight at 37 °C/5% CO2. 3 μM of complex 1, complex 2, or BC were then added, and the medium was decanted immediately, or else plates were left in the dark for 1, 3, 6, 12, or 24 h at 37 °C/5% CO2. After removing the supernatant, cells were washed with PBS containing 1.5% DMSO (3 times) and harvested by scraping in the presence of 1.5 mL acetonitrile (used as the extraction solvent). Cells were then completely lysed using an ultrasonic homogenizer (model 150VT; continuous mode at medium power for 1 min) and compounds were extracted in the supernatant by centrifugation at 13000 rpm for 10 min. Concentrations were obtained by LC-MS/MS based on the calibration curves designed above and results were reported as mean ± SEM (n = 4).
Isolation of mesenchymal stem cells (MSCs) from rat bone marrow
Cell viability assay
Cell survival assays were performed on human alveolar adenocarcinoma A549, murine melanoma B16-F10, human colon adenocarcinoma Caco-2, human colorectal adenocarcinoma HT-29 and triple negative human breast adenocarcinoma MDA-MB-231 cell lines treated with complexes 1 and 2, cisplatin, bipy, and BC, as previously described.13 Briefly, DMEM containing 10% FBS and 1.5% pen-strep at 37 °C with 5% CO2 was used to maintain cells. The latter were seeded and allowed to adhere at a concentration of 104 cells per well in a 96 well-plate. Drugs were added to the media at different concentrations using 3-fold serial dilution. Cells were incubated with drugs for 12 h followed by 35 min exposure to light (LED Engin, 460 nm peak wavelength and 100 mW cm−2 power output) or else the plates were left in the dark. Cell survival was measured using WST-1 kit (Roche©) after 72 h incubation. Three independent experiments were performed, each comprised of triplicate measurements. To test the activity of complex 1 on normal cells, the isolated MSCs were treated with the complex using the same conditions described for cancer cells.Analysis of cell death by flow cytometry
The type of cell death was determined by flow cytometry using a modified protocol.33,34 Briefly, B16-F10 cells were treated with complex 1 (10 μM) and irradiated as described above or left in dark conditions. After 24 h incubation at 37 °C/5% CO2, cells were harvested and treated for 5 min with PI (5 mg mL−1) and FITC-conjugated annexin V antibody (5 mg mL−1) at 37 °C. Annexin V/PI data were then analyzed by flow cytometry on FL1-H versus FL2-H scatter plot.Assessment of DNA damage by gel electrophoresis
The plasmid (pUC8) was added to 10 mM potassium phosphate buffer (pH 7.4) in a 96-well plate followed by treatment with complex 1 at 10 or 200 μM concentrations. Plates were then irradiated at 460 nm (100 mW cm−2) or left in the dark. After 12 h incubation, DNA suspensions were mixed with loading dye and electrophoresis was performed in the presence of a negative control (untreated pUC8) on a 2% agarose gel containing 0.1 mg mL−1 ethidium bromide (AppliChem, USA) and TAE buffer (Tris base, acetic acid and EDTA). The gel was then visualized using Bio-Rad ChemiDoc™ gel imaging system.Statistical analysis
The results were analyzed for statistical significance using one-way analysis of variance (ANOVA). Values of the different tested parameters within each group are presented as mean ± SEM. All data were analyzed with the statistical package SPSS 18, and differences between groups were considered statistically significant if p-value < 0.05. The IC50 values were calculated using the nonlinear regression curve with the use of Graph Pad Prism version 5.0 software for Windows.Results and discussion
We report here the synthesis, characterization, and photochemotherapeutic activity of [Ru(bipy)2BC]2+. The incorporation of methyl groups on the 2,9-positions of the 1,10-phenanthroline ligand near the Ru(II) coordination sphere imparts a steric strain on the Ru(II) complex. The latter likely distorted the octahedral geometry of the complex, making it photochemically labile and thus prone to photolysis.When the complex absorbed visible light (460 nm), ligand ejection occurred. This can be attributed to the population of the dissociative triplet metal-centered state (3*dd) from the triplet metal-to-ligand charge transfer band (3*MLCT). The MLCT absorption of complex 1 was first assessed by UV/vis. The band was centered around 454 nm whereas the average extinction coefficient was 22000 ± 2460 M−1 cm−1 in acetonitrile ([Ru(bipy)2BC](PF6)2), Fig. 2. The maximum peak in the UV range (287 nm) can be assigned to intra-ligand π → π* transition. An LED system with a peak wavelength of 460 nm (light output ca. 100 mW cm−2) was therefore utilized as the excitation source for all photochemical and photobiological studies.
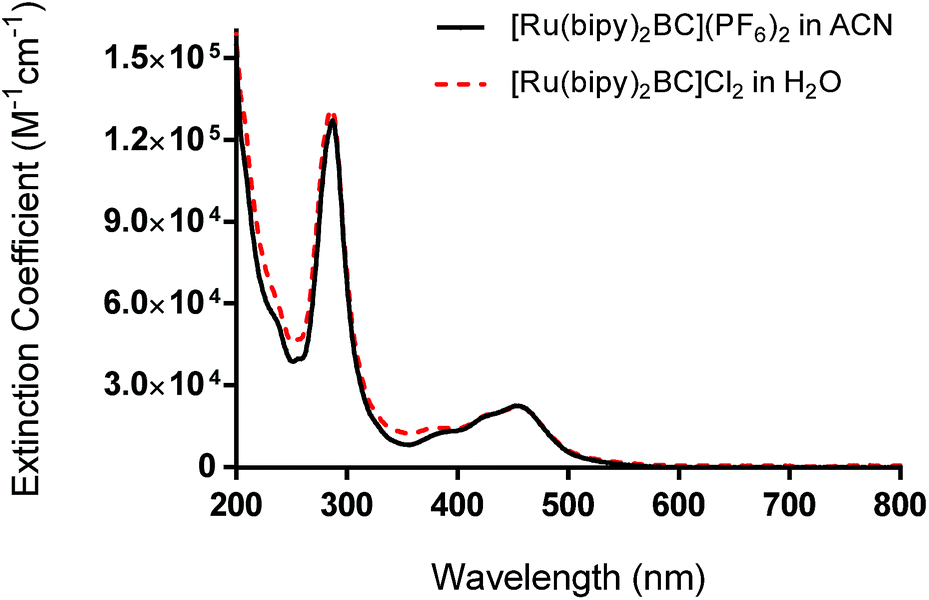 | ||
| Fig. 2 UV-vis absorption spectra of complex 1 as hexafluorophosphate salt (in acetonitrile) and chloride salt (in water). | ||
UV-vis performed on complex 1 in the dark indicated that the compound is thermally stable at room temperature. However, upon exposure to blue light (100 mW cm−2, 460 nm), complex 1 undergoes a photochemical ligand displacement reaction that leads to the depletion of the starting material with MLCT absorbance maximum at 455 nm and the formation of ruthenium aqua complexes with a red-shifted absorption maximum around 490 nm. The half-life of complex 1 under those irradiation conditions was ∼2 minutes, Fig. 3.
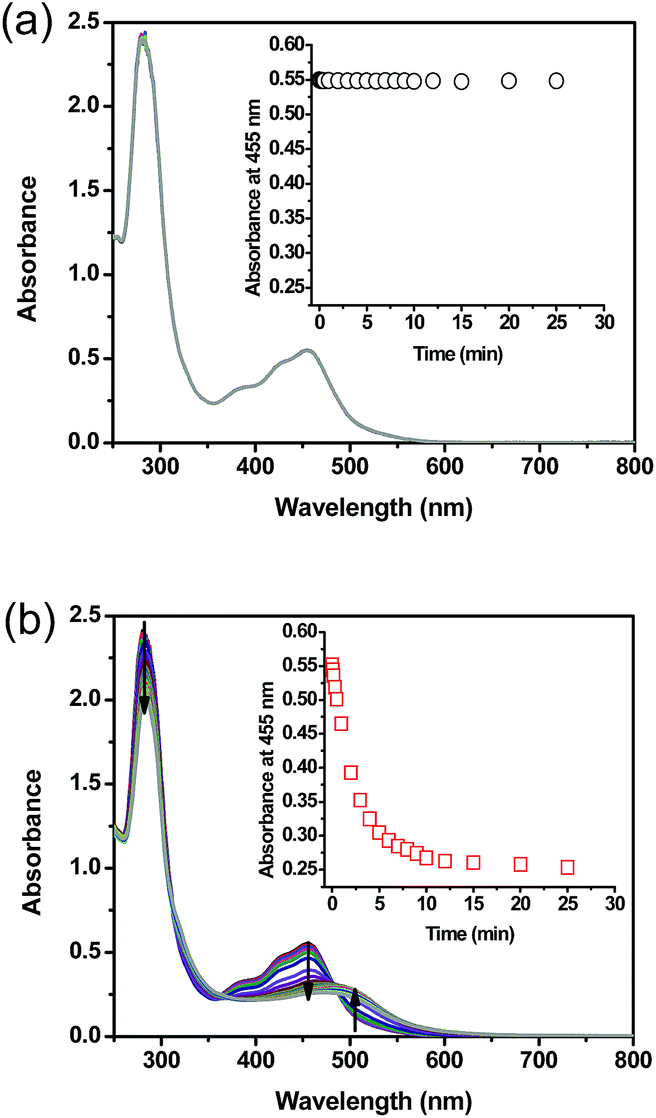 | ||
| Fig. 3 Absorption spectra of complex 1 in water as a function of time (a) in the dark (b) upon exposure to blue light (100 mW cm−2, 460 nm); inset: absorbance values at 455 nm as a function of time. Arrows indicate the spectral changes trend throughout light exposure. | ||
1H NMR spectra were obtained to determine the photolysis products of complex 1 and compare it to the spectra of the free ligands bipy and BC. The peaks assigned to the starting material (before irradiation) were absent in the photolysis products thus proving that [Ru(bipy)2BC](PF6)2 underwent a complete conversion in CD3CN upon irradiation. 1H NMR spectra were examined before and after irradiation to specify which of the ligands dissociate from the metal center, Fig. 4. The spectrum of the photolyzed solution included chemical shifts assigned to the free bipy and BC ligands which indicated that both ligands were prone to ejection from the complex upon light activation. Typically, the straining ligand is more likely to dissociate from the complex due to the weaker ligand field axis.24
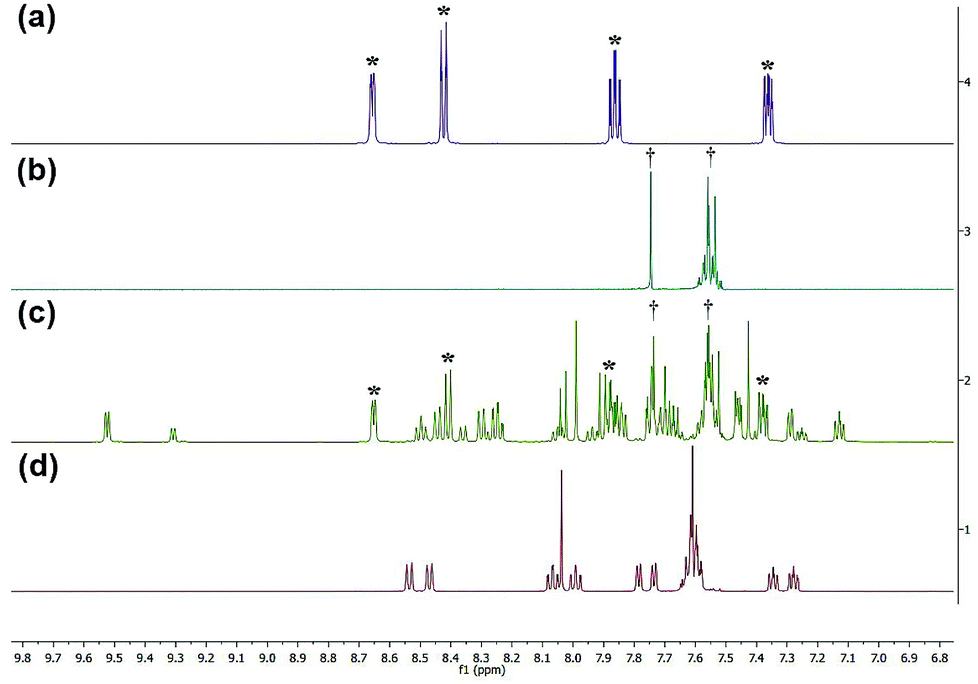 | ||
| Fig. 4 1H NMR spectra of bpy (a), BC (b), complex 1 after photolysis (c) and complex 1 before photolysis (d). *Denotes peaks attributed to bipy, and †represents peaks of BC. | ||
However, previous studies have described a similar dissociative potential of bipy upon irradiation of the strained complex [Ru(bipy)2(dpphen)]2+ [where dpphen = 2,9-diphenyl-1,10-phenanthroline]. Ejection of the bipy ligand could be favored due to the free rotation around C2–C2′ axis in addition to the asymmetrical distortions of the octahedral complex.13,24
Peaks labeled as a and b (Fig. 4) refer to the methyl groups on the [Ru(bipy)BC(CD3CN)x](PF6)2 (where x = 1 or 2) and BC respectively and are generated by different ligand substitution pathways, thus allowing relative signal quantification. Upon ejection of bipy, complex 1 undergoes photosubstitution in d-acetonitrile to form [Ru(bipy)BC(CD3CN)x](PF6)2 (where x = 1 or 2) and bipy. In this case, the two methyl groups of BC are not equivalent and therefore produced signals with equal integration at different chemical shifts (Fig. 5a). The ejection of BC led to the formation [Ru(bipy)2(CD3CN)x](PF6)2 (where x = 1 or 2) and free BC ligand with a characteristic single peak in the aliphatic region attributed to the two equivalent methyl groups (letter b, Fig. 5). The 1H NMR aliphatic peaks pertaining to the methyl substituents of the BC were integrated and relatively-quantified in the photolyzed complex, Fig. 5. The dissociation ratio was found to be ∼3:1 bipy to BC.
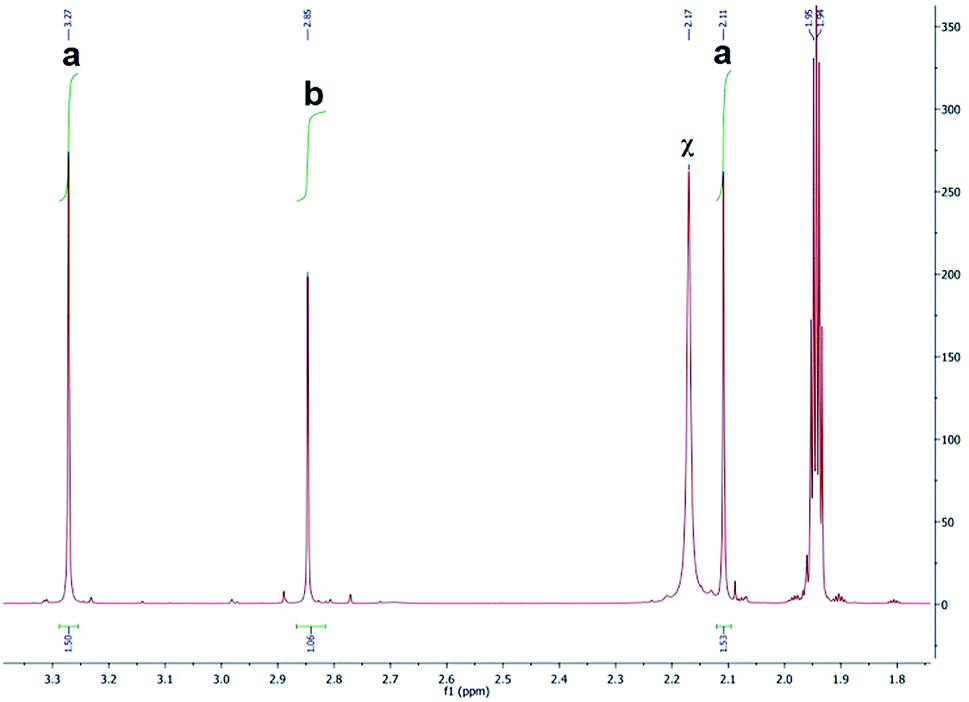 | ||
| Fig. 5 1H NMR spectrum (aliphatic region) of photolyzed complex 1. “a” represent the protons on the methyl groups of BC in [Ru(bpy)BC(CH3CN)x](PF6)2 (where x = 1 or 2), “b” denotes proton signals attributed to the methyl groups of BC and “χ” refers to solvent residual. | ||
LC-MS studies were conducted to probe the photochemical products of [Ru(bipy)2BC]Cl2 in water. The ESI-MS spectrum of the complex before photactivation (Fig. 6a) displayed only one peak at m/z [M − 2Cl]2+ = 387.20 which corresponds to [Ru(bipy)2BC]2+, whereas bipy, BC and Ru aqua photoproducts ([Ru(bipy)2(H2O)2]2+ and [Ru(bipy)BC(H2O)]2+) (Table 2) were detected after irradiation (Fig. 6b). Complex 1 is likely to undergo aquation upon photochemical ligand dissociation in water. However, the number of aqua or hydroxo ligands per Ru center cannot be confirmed by our results because the electrospray ionization used in the LC-MS may alter the aquation process. Upon photoactivation of [Ru(bipy)2(dmbipy)]2+ in water, various products were reported such as [Ru(bipy)2(H2O)2]2+, [Ru(bipy)2(H2O)(OH)]+, and the ligand deficient [Ru(bipy)2]2+.12,14,21 Furthermore, we have detected [Ru(bipy)(dpphen)(OH)]2+ as a photoproduct of [Ru(bipy)2(dpphen)]Cl2 in aqueous medium.13
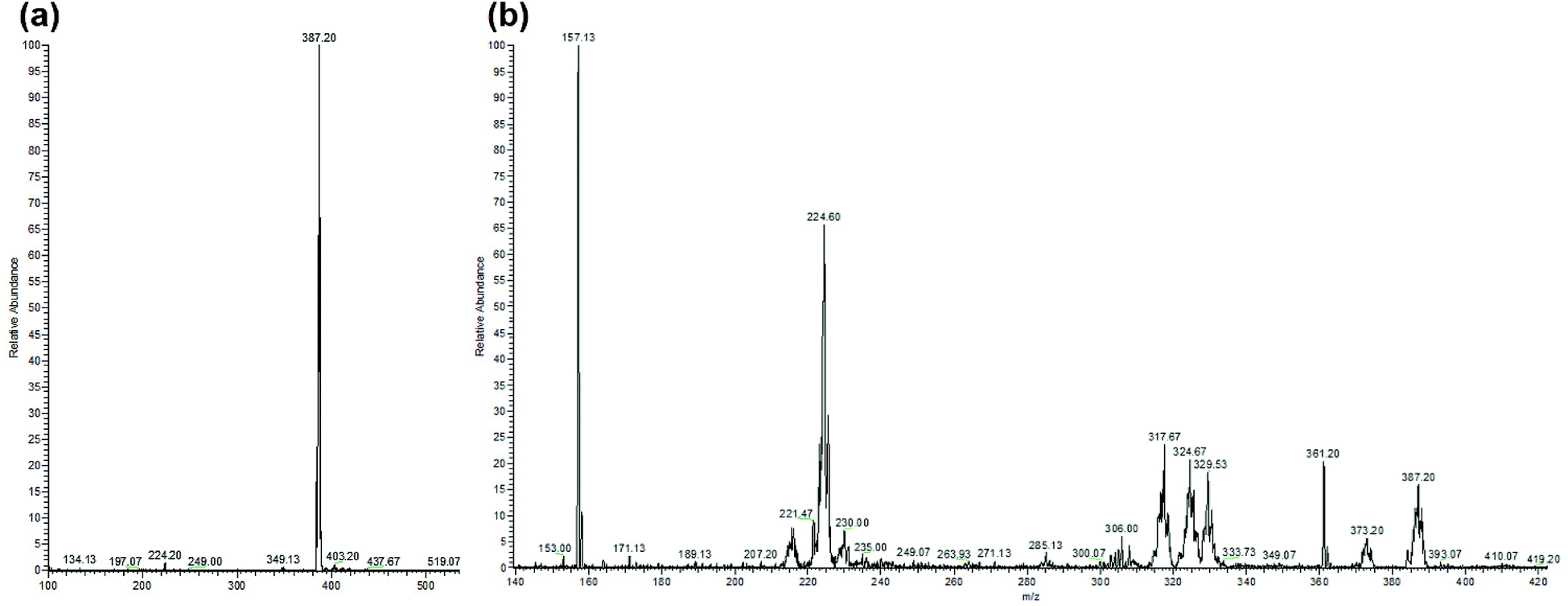 | ||
| Fig. 6 ESI-MS spectra of [Ru(bipy)2BC]Cl2 in water, (a) before photolysis and (b) after 35 min photolysis with blue LED (output: 100 mW cm−2). Peaks are assigned in Table 2. | ||
| Molecule | m/z found | m/z calculated |
|---|---|---|
| Bipy | 157.13 | [M + H+]+ 157.07 |
| [Ru(bipy)2(H2O)(OH)]2+ | 224.60 | [M]2+ 224.53 |
| [Ru(bipy)BC(OH)]2+ | 317.67 | [M]2+ 317.57 |
| BC | 361.20 | [M + H+]+ 361.17 |
| Complex 1: [Ru(bipy)2BC]2+ | 387.20 | [M]2+ 387.10 |
To determine the bipy:BC dissociation ratio in water, a 1:1 mixture of the two ligands was prepared as a standard. The bipy:BC ratio found by LC-MS was ∼3.5:1 as deduced from the results of the averaged relative intensity of the peaks. LC-MS data thus correlated with 1H NMR data and both suggested that dissociation was favoured towards the substitution of bipy by solvent molecules. 1H NMR (Fig. S1†), 13C NMR (Fig. S2†) and LC-MS/MS (Fig. 6a) spectra revealed that complex 1 is kinetically inert in dark conditions and thus can be stored for days without decomposition when protected from light. As previously demonstrated,13 complex 2 did not undergo any detectable ligand substitution upon irradiation with the blue LED setup used here.
The cellular uptake of complex 1 was measured and values were used to determine the optimal incubation period with the drug prior to photoactivation. After 6 to 12 hours of incubation with A549 cells, complex 1 reached a maximum concentration of nearly 2.2 nmol/106 cells (Fig. S9a†). Assuming that cells have an average volume of 1.7 pL35 and that the complex is homogeneously distributed within the cell, the concentration of complex 1 is around 1400 μM per cell (Fig. 7a), which is about 430 times the initial concentration (3 μM). Complex 2 also reached an elevated cellular concentration of around 40 μM per cell after 12 h (Fig. 7b), yet was still approximately 35 times lower than complex 1. Transport of complex 2 inside the cell must similarly be facilitated but to a lesser extent. The ligand BC tested independently reached a maximum concentration of 2000 μM per cell with similar kinetic profile as complex 1, Fig. 7a, inset. Complex 1 is therefore considerably concentrated in the cells suggesting a facilitated mode of transport which is likely mediated through the presence of the lipophilic BC ligand.
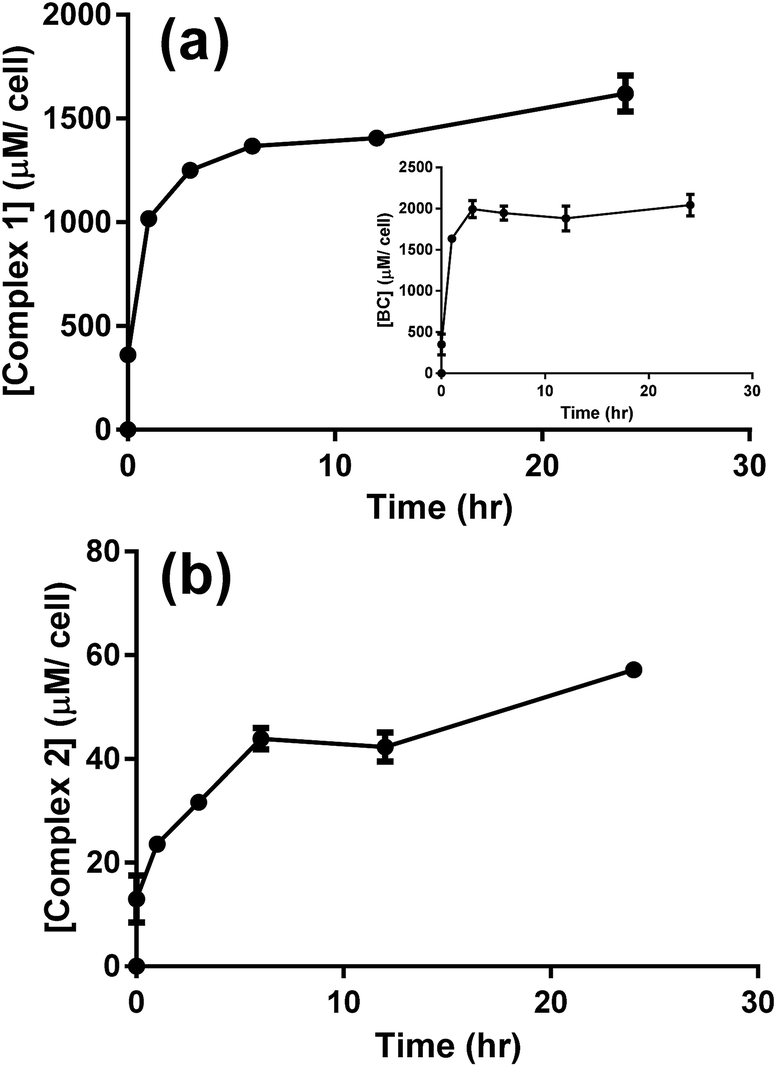 | ||
| Fig. 7 LC-MS/MS analysis for the uptake of 3 μM of complex 1 (a), BC (a, inset), and complex 2 (b) by A549 cells. Results are averages of four different experiments (±SEM) and are expressed in μM per cell, as estimated from the calibration curves (Fig. S5–S7†). | ||
Puckett and Barton demonstrated that cellular uptake of dppz complexes of Ru(II) was significantly enhanced by the presence of the lipophilic ligand 4,7-diphenyl-1,10-phenanthroline, as evidenced by the increased photoluminescence intensity across the cell. Similarly, these reported results imply that cellular transport of Ru(II) complexes was facilitated by the lipophilic BC ligand.36,37
Studies on metal complexes including platinum- and ruthenium-based chemotherapeutics suggested that cellular uptake increased with the lipophilicity of the complexes.36,38 The partition coefficient (logP) between the hydrophobic octanol phase and the hydrophilic water phase was thus measured to determine whether the extent of lipophilicity could explain the results of cellular uptake, Table 3. The dicationic complex 1 (logP = −1.57) was found to be significantly more lipophilic as compared to complex 2 (logP = −2.82). The lipophilic property of complex 1, which is largely imparted by the coordinated BC ligand (calculated logP = 6.96), can explain the fast uptake of complex 1 by the cells with accumulated concentration of 0.6 nmol/106 cells immediately after incubation with the drug, Fig. S9a.† The dicationic complex 2 proved to be rather hydrophilic (logP = −2.82), which correlates well with the lower uptake of the complex through the cell membrane.36 Although cisplatin is only slightly more lipophilic than complex 2 (logP = −2.21), studies have recently emphasized the role of active transport in the cellular uptake of this neutral Pt(II) complex.39
| IC50 valuesa (μM) | logP |
|||||
|---|---|---|---|---|---|---|
| Treatment | A549 | B16-F10 | Caco-2 | HT-29 | MDA-MB-231 | |
| a Values in a column not sharing a common superscript a, b, or c are significantly different (p < 0.05).b IC50 data previously reported by our group.13c logP values reported in the literature.26,47d logP values estimated by ChemDraw Professional (v15.0, CambridgeSoft). |
||||||
| Complex 1 | Dark: 51.8a,c ± 18.8 | Dark: >100a | Dark: 29.8a ± 2.9 | Dark: 91.0a ± 5.90 | Dark: 5.55a ± 1.45 | −1.57 ± 0.01 |
| Light: 4.96a ± 1.16 | Light: 0.11b ± 0.10 | Light: 14.8a ± 0.81 | Light: 12.0b ± 1.9 | Light: 0.63a ± 0.90 | ||
| PI: 11.1 | PI: >900 | PI: 2.0 | PI: 7.6 | PI: 8.9 | ||
| BC | >100b | >100a | >100b | >100c | >100b | 6.96c |
| bipy | >100b | >100 | >100 | >100c | 37.5b ± 0.14 | 1.88d |
| Complex 2b | Dark: >100b,c | Dark: >100a | Dark: >100b | Dark: >100c | Dark: >100b | −2.82 ± 0.10 |
| Light: >100b,c | Light: >100b | Light: >100b | Light: >100c | Light: >100b | ||
| PI: — | PI: — | PI: — | PI: — | PI: — | ||
| Cisplatin | 7.74b,c ± 1.00 | 7.70a ± 0.20 | 21.8b ± 0.54 | >100c | 30.9b ± 0.82 | −2.21c |
Complex 1 is a potential candidate for PACT owing to its significant uptake and photochemical properties. However for efficient PACT, the compounds should be active predominantly upon photolysis, thus inducing selective toxicity while minimizing side effects.40 This is commonly assessed by measuring the difference in cytotoxicity in vitro between dark and light conditions, known as the photoindex or PI (PI = IC50 dark/IC50 light).41 Cellular uptake plateaued after 12 h for complexes 1 and 2 (Fig. 7), thus cell survival assays were performed after a 12 h incubation period with the complexes. In all cell lines tested, the potency of complex 1 improved upon photoactivation. White light, more specifically blue light, was reported to induce cytotoxicity in vitro and in vivo as compared to dark conditions. Bonnet and coworkers demonstrated that the viability of certain types of cancer cells, like human malignant melanoma (A375), A549 and MDA-MB-231 cells, decreased when irradiated with blue light and thus highlighted the necessity of including the adequate controls when studying photoactivable anticancer agents.42 Likewise, Wang and coworkers showed that blue light decreased the viability and mediated the death of human promyelocytic leukemia cells (HL60) in a time-dependent fashion. A 24 hour exposure lead to overproduction of reactive oxygen species (ROS) and mitochondrial depolarization. When tested in vivo, irradiation with blue light caused regression of HL60-xenografted tumors and Western blot analyses proved that the effect resulted from the activation of the mitochondrial apoptosis pathway.43 In our irradiation experiments, blue light (λ = 460 nm, 35 min irradiation at 100 mW cm−2) resulted in an average of 35% loss of cell viability versus the dark control, which was consistent with the results of Bonnet on the same cell lines.42 However cell viability results, under photoexcitation, were assessed against control cells exposed to light on the same plate.13,20,23,26 In that perspective, the influence of light, if any, was subtracted from the data allowing the assessment of the effect of drugs. While IC50 values ranged between 5.55 μM (in MDA-BD-231 cells) and >100 μM (B16-F10 cells) in dark conditions, the values were as low as 0.11 μM (in B16-F10 cells) upon irradiation (Table 1). The response was however particularly significant in B16-F10 cells where PI > 900, as compared to only 2–11 in the other cell lines (Table 3, Fig. 8). Complex 1 exhibited substantial PACT potential on B16-F10 cells in view of the high PI which is comparable to the highest reported in the literature44 and 70-fold lower IC50 than the prototypical cisplatin (where IC50 only reached a minimum of 7.70 ± 0.20 μM in B16-F10 cells). The photoproduct of complex 1 was consistently more potent than cisplatin in all cell lines tested indicating that this complex acquired significant chemotherapeutic properties upon irradiation. When tested on HT-29 cells, cisplatin exhibited a large IC50 value > 100 μM, suggesting that these cells could have developed resistance to cisplatin. This finding is consistent with a published report confirming that colon cancer cells were less responsive to cisplatin treatment possibly due to their potential for DNA repair.45 Control cell viability experiments were done on normal cells (isolated MSCs). The IC50 in the dark was 52 ± 13 μM and decreased significantly down to 0.024 ± 0.002 μM upon exposure to light, Fig. S10.† These results indicate photoactivatable potential of complex 1 on all tested cell lines, suggesting a lack of intrinsic mechanism of discrimination between normal and cancerous cells. However, it is envisioned that a delicate spatial and temporal control of light delivery can infer selectively in PACT as demonstrated by the high PI of complex 1, Table 3.
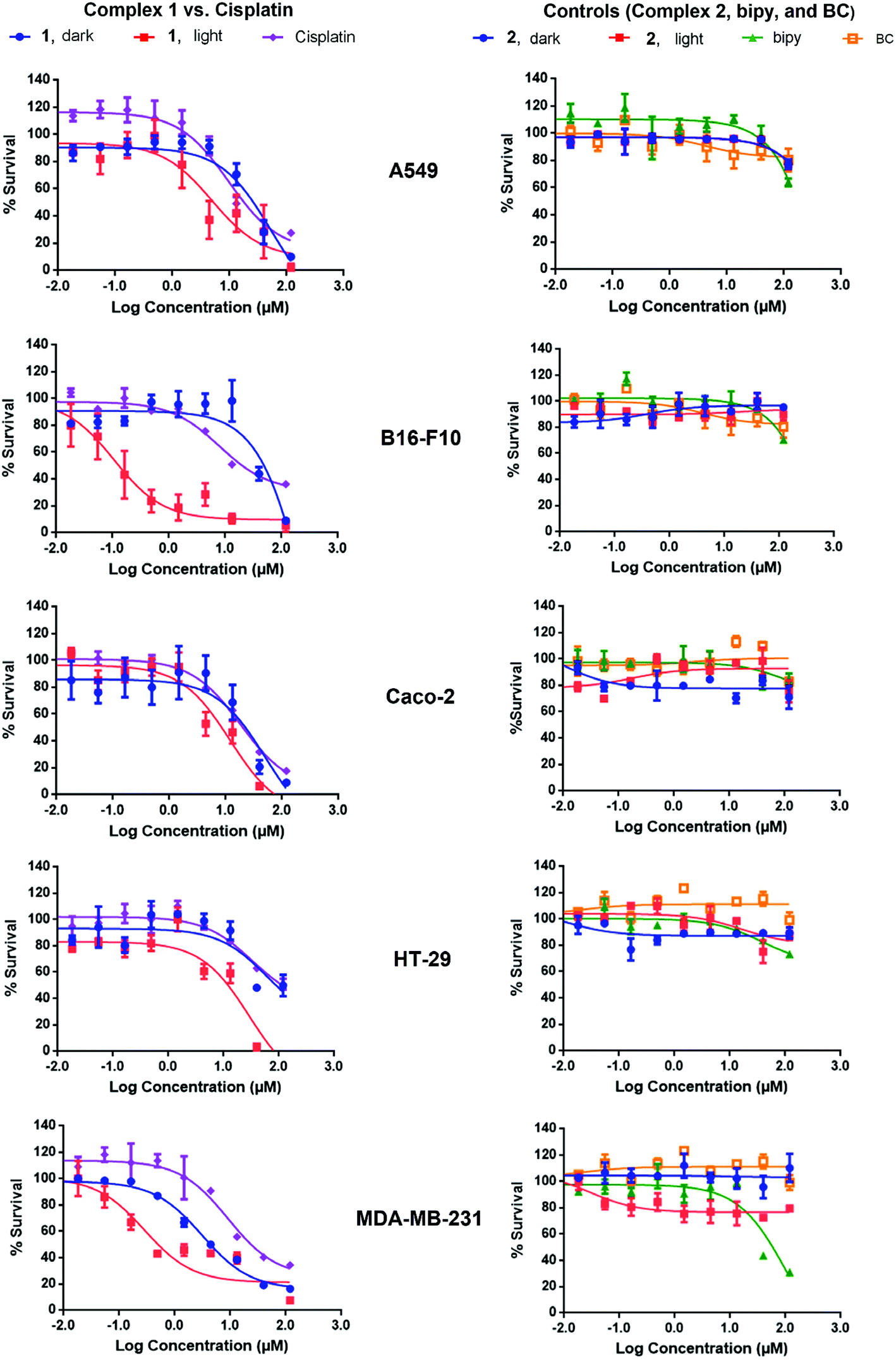 | ||
| Fig. 8 Effect of complexes 1, 2, bipy, BC, and cisplatin on the survival of A549, B16-F10, Caco-2, HT-29, and MDA-MB-231 cells. Treatment was performed with 3-fold dilutions of the complexes and ligands, starting at 120 μM. The cytotoxicity of complexes 1 and 2 was evaluated in both dark and light conditions (blue LED at a power of 100 mW cm−2) and all others were kept in the dark. Each experiment was repeated three times and measurements were performed in triplicates. The graphs shown here correspond to a single representative trial. | ||
The low IC50 in the dark, which was most notable in MDA-MB-231 cells (5.55 ± 1.45 μM), could have resulted from the geometry of the complex. When studying the binding geometry of [Ru(bipy)2phen]2+ with DNA, Lincoln and Nordén found that the phenanthroline ligand was oriented nearly perpendicular to the DNA double helix and was able to achieve a stacking interaction with the nucleobases.46 The phenanthroline derivative, BC, in complex 1 is most likely exposed, which could have led to similar interactions with DNA thus mediating dark cytotoxicity. Control experiments showed no differences between the cytotoxicity of complex 1 when the supernatant containing the compound was replaced with fresh medium or kept prior to the photoactivation step. This emphasized the role of cellular uptake in mediating cytotoxicity. In accordance with reported results, complex 2 was neither potent in the dark nor upon light irradiation on all cell lines tested (IC50 > 100 μM).12,13 Complex 2, with similar characteristic MLCT absorption in the blue as complex 1, is a valid control in PACT since it remains both structurally unchanged and biologically inactive upon irradiation. Previous reports demonstrated that [Ru(bipy)2phen]2+ displayed an ordered binding with DNA.46 However, the poor cellular uptake of complex 2 is likely the limiting factor inducing low cytotoxicity and IC50 > 100 μM.
The photolysis products of complex 1 are consistently cytotoxic on all tested cell lines. Generally, it is expected that the photochemically generated metal aqua complexes are responsible for the biological effect of the complex, similarly to cisplatin.47 However, our group and others have demonstrated that the ejected ligand could be potent.20–22 Both dissociating ligands, bipy and BC, were therefore included in cell survival studies to investigate their potential cytotoxic effect. While only slightly toxic on MDA-MB-231 cells (IC50 = 37.5 ± 0.140 μM, Table 3), bipy was not potent on all other cell lines tested (IC50 > 100 μM, Table 3). These results were expected since bipy is known to form electrostatic interactions rather than intercalation with DNA.25 Likewise, BC was consistently inactive against all cancer cells studied (IC50 > 100 μM), which, however, was not anticipated since BC, unlike bipy, could intercalate DNA and subsequently lead to cell death.48 To experimentally rule out the lack of uptake, the cellular concentration of BC was measured in cultured A549 cells at different time points. When compared to complex 1 (maximum concentration of 1400 μM per cell after 12 h, Fig. 7a), the optimal cellular uptake of BC was achieved after 3 h and it leveled off at a higher concentration of 2000 μM per cell (Fig. 7a, inset). BC is therefore effectively transported across the cell membrane. Owing to the high lipophilicity of bipy and BC, with logP values of 1.88 and 6.96 respectively (Table 3), cellular uptake should not have been compromised for these free ligands. BC is a copper-specific chelator; in the presence of endogenous Cu2+ or in a medium supplemented with Cu2+, it could bind to the metal ion, forming a copper chelate complex.49 Rabinovitz and coworkers previously demonstrated that 2,9-dimethyl-1,10-phenanthroline (neocuproine), a similar copper chelator, was cytotoxic exclusively when bound to Cu2+. Upon incubation of neocuproine with either cells seeded in a medium enriched with Cu2+ in vitro or in vivo, a significant chemotherapeutic effect was observed. To prove that the copper chelate was the active complex, they oversaturated the medium with a copper-specific chelating agent, bathocuproine sulfonate (BCS). This led to normal cell growth and thus confirmed that neocuproine is dependent on the presence of copper for inducing cytotoxicity.50,51 Typically cuprous compounds, which result from the intracellular reduction of cupric precursors, are significantly more potent and often deemed the active species.52,53 McMillin and coworkers have reported the interaction of Cu(BC)2+ with DNA and described a nonclassical mode of binding involving bridging structures that lead to DNA aggregation and elongation without intercalation.54–56 However, we recently tested the cytotoxic effect of [Cu(BC)2]+ in the dark, and the IC50 values obtained (21.8 and 25.5 μM in A549 and A375 cells, respectively) reflected moderate to low toxicity compared to cuprous neocuproine complex (IC50 of 0.9 and 1.8 μM on A549 and A375 cells, respectively). These results indicated that, unlike neocuproine, the activity of BC is not considerably enhanced by the presence of copper.26 Therefore, the potency of complex 1 photoproducts was primarily attributed to the formed ruthenium aqua complexes and to a less extent the dissociating ligands, bipy and BC. The photoproducts may possibly have synergistic effect on the various tested cells, leading to the high potency after light irradiation. Numerous studies have indicated the role of metal aqua complexes in mediating cytotoxicity through DNA interaction and/or modification.15,57
The in vitro mechanism of death induced by complex 1 was determined on B16-F10 cells using Annexin V/PI staining. In dark conditions, ∼26% of the cells stained positive for annexin V and negative for PI whereas 21% of the cells stained positively for both annexin V and PI (Fig. 9b). The precursor appeared to induce apoptosis as cells are distributed between living (annexin V and PI negative), early apoptosis (annexin V positive and PI negative) and late apoptosis/necrosis (annexin V and PI positive).58 Upon photoactivation of the complex, the percentage of cells that were annexin V and PI positive increased to ∼90% (Fig. 9c). Due to the lack of cells undergoing early apoptosis in the lower right quadrant (annexin V positive and PI negative), the mechanism of cell death of the photolysis products could be through either late-apoptosis or necrosis. It is worth mentioning that the photoactivation of the complex considerably enhanced the percentage of cells undergoing cell death, which is in accordance with the results of the cell survival assay.
 | ||
| Fig. 9 Analysis of the mechanism of complex 1-mediated cytotoxicity in B16-F10 cells using Annexin/PI staining on a flow cytometer. (a) Control: no treatment, (b) cells treated with 10 μM of the complex and incubated for 24 h in the dark, (c) cells treated with 10 μM of the complex and irradiated for 35 min using the blue LED system at an output of 100 mW cm−2. FL1-H: Annexin staining, FL2-H: PI staining. | ||
To further assess the effect of complex 1 on DNA, gel electrophoresis was performed on pUC8 plasmid, Fig. 10. At low concentration of the precursor complex (in dark conditions, lane 2), DNA migrated slower than the untreated pUC8 plasmid control (lane 1). These results suggest binding of complex 1 to plasmid DNA and possibly, intercalation, as observed with other compounds like cisplatin.59 Similarly, EtBr intercalation decreased at low concentrations of the photoactivated complex (Fig. 10, lane 3) whereas EtBr signal was completely lost at high concentrations of both the precursor and the photoproducts (lanes 4 and 5 respectively). Such behavior strongly suggest binding to DNA which likely changes in structure, charge, and molecular weight while hindering the intercalation of EtBr.60 These results therefore imply that binding of the complex to DNA is not exclusively photoinduced and that the precursor molecule is capable of interacting with nucleic acids. This is potentially due to the presence of BC ligand in complex 1, which facilitates interaction with DNA nucleobases46 leading to the dark cytotoxicity (Fig. 8, Table 3).
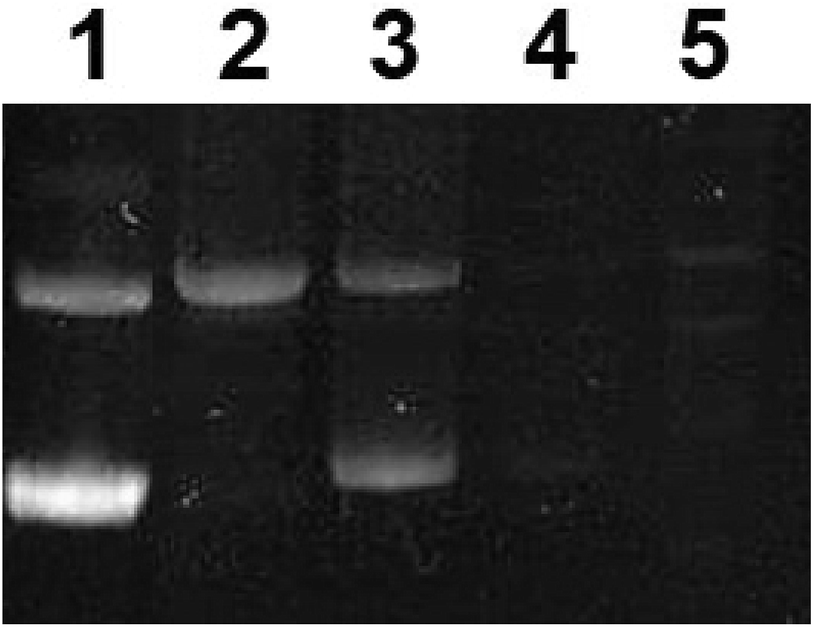 | ||
| Fig. 10 Agarose gel electrophoresis of pUC8 plasmid in 10 mM potassium phosphate buffer (pH 7.4). Lane 1: pUC8 untreated, lanes 2 and 4: complex 1 in dark conditions, 10 μM and 200 μM respectively, lanes 3 and 5: complex 1 in light conditions (blue LED at a power of 100 mW cm−2), 10 μM and 200 μM respectively. | ||
The cell death induced by complex 1 in both dark and light conditions could correlate with the observed DNA interactions. Apoptosis is typically one of the major mechanisms that lead to cell inactivation in response to DNA damage.61 Many signaling pathways are involved, including p53-mediated activation of pro-apoptotic factors as a result of DNA double-stranded breaks, MAP-kinase activation, and the capacity for DNA repair.62–64 It was previously demonstrated that Ru(II) anticancer drugs can trigger DNA damage which in turn activates apoptosis via the intrinsic pathway.65 Zhang and coworkers reported the intercalative potential of Ru(II) polypyridyl complexes which were shown to induce apoptosis in A549 cells, as concluded from flow cytometric studies.66 Likewise, Xu and his colleagues found that Ru(II) bipyridyl complexes were capable of DNA intercalation and photo-induced DNA cleavage and described an apoptosis-mediated death of human hepatocellular carcinoma cells (BEL-7402).67 Many other studies on Ru(II) anticancer drugs have reported a similar intersection between DNA damage and apoptosis.68–70 Further in vitro experiments are currently underway to investigate the cascade of events and molecular mechanisms of action associated with Ru(II) polypyridyl complexes.
Conclusions
Owing to their tuneable photophysical and photochemical properties, Ru(II) polypyridyl complexes and particularly those having a distorted octahedral geometry are attractive for PACT. Upon MLCT photoexcitation of complex 1, both bipy and BC were ejected at a ratio of 3:1, respectively. The formed Ru aqua complexes were likely the origin of the photocytotoxicity. The potential of this complex in PACT appeared to be cell-line dependent, as evidenced by disparate PI values. Cell death mechanism with complex 1 is most likely occurring through apoptosis in the dark and late-apoptosis or necrosis upon irradiation. Both the precursor and the photoproducts of complex 1 are likely to bind DNA leading to cellular damages. Complex 1, on the other hand, proved to be photostable and was relatively less potent in both dark and light conditions, which could be partly attributed to its low uptake into the cells. Finally, the design of Ru(II) polypyridyl prodrugs with lipophilic ligands is of prime importance to ameliorate cellular uptake, a cornerstone towards efficient photochemotherapy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the School Research and Development Council at the Lebanese American University and the Lebanese National Council for Scientific Research (Ref: 05-06-14).Notes and references
- N. Muhammad and Z. Guo, Curr. Opin. Chem. Biol., 2014, 19, 144–153 CrossRef CAS PubMed.
- G. Y. Ho, N. Woodward and J. I. G. Coward, Crit. Rev. Oncol. Hematol., 2016, 102, 37–46 CrossRef PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- N. P. E. Barry and P. J. Sadler, Chem. Commun., 2013, 49, 5106–5131 RSC.
- A. Bergamo, C. Gaiddon, J. H. M. Schellens, J. H. Beijnen and G. Sava, J. Inorg. Biochem., 2012, 106, 90–99 CrossRef CAS PubMed.
- P. Schluga, C. G. Hartinger, A. Egger, E. Reisner, M. Galanski, M. A. Jakupec and B. K. Keppler, Dalton Trans., 2003, 2006, 1796–1802 Search PubMed.
- W. Guo, W. Zheng, Q. Luo, X. Li, Y. Zhao, S. Xiong and F. Wang, Inorg. Chem., 2013, 52, 5328–5338 CrossRef CAS PubMed.
- C. Mari, V. Pierroz, S. Ferrari and G. Gasser, Chem. Sci., 2015, 6, 2660–2686 RSC.
- A. L. Harris, Nat. Rev. Cancer, 2002, 2, 38–47 CrossRef CAS PubMed.
- S. Campagna, F. Puntoriero, F. Nastasi, G. Bergamini and V. Balzani, in Photochemistry and Photophysics of Coordination Compounds I, Springer, Berlin, Heidelberg, 2007, pp. 117–214 Search PubMed.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef CAS.
- B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324–8327 CrossRef CAS PubMed.
- N. Mansour, S. Mehanna, M. A. Mroueh, H. Audi, K. Bodman-Smith, C. F. Daher, R. I. Taleb, M. El-Sibai and R. S. Khnayzer, Eur. J. Inorg. Chem., 2018, 2524–2532 CrossRef CAS.
- T. N. Singh and C. Turro, Inorg. Chem., 2004, 43, 7260–7262 CrossRef CAS PubMed.
- R. N. Garner, L. E. Joyce and C. Turro, Inorg. Chem., 2011, 50, 4384–4391 CrossRef CAS PubMed.
- M. Cristina, P. Vanessa, R. Riccardo, P. Malay, J. Hess, S. Bernhard, O. Luciano, S. Julia, I. Ott, S. Luca, F. Stefano and G. Gilles, Chem.–Eur. J., 2014, 20, 14421–14436 CrossRef PubMed.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649–9651 RSC.
- T. Sainuddin, M. Pinto, H. Yin, M. Hetu, J. Colpitts and S. A. McFarland, J. Inorg. Biochem., 2016, 158, 45–54 CrossRef CAS PubMed.
- V. H. S. van Rixel, B. Siewert, S. L. Hopkins, S. H. C. Askes, A. Busemann, M. A. Siegler and S. Bonnet, Chem. Sci., 2016, 7, 4922–4929 RSC.
- D. F. Azar, H. Audi, S. Farhat, M. El-Sibai, R. J. Abi-Habib and R. S. Khnayzer, Dalton Trans., 2017, 46, 11529–11532 RSC.
- J.-A. Cuello-Garibo, M. S. Meijer and S. Bonnet, Chem. Commun., 2017, 53, 6768–6771 RSC.
- M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274–11282 CrossRef CAS PubMed.
- H. Audi, D. F. Azar, F. Mahjoub, S. Farhat, Z. El Masri, M. El-Sibai, R. J. Abi-Habib and R. S. Khnayzer, J. Photochem. Photobiol., A, 2018, 351, 59–68 CrossRef CAS.
- A.-C. Laemmel, J.-P. Collin and J.-P. Sauvage, Eur. J. Inorg. Chem., 1999, 1999, 383–386 CrossRef.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051–3058 CrossRef CAS.
- C. A. Hageh, M. A. Assaad, Z. E. Masri, N. Samaan, M. El-Sibai, C. Khalil and R. S. Khnayzer, Dalton Trans., 2018, 47, 4959–4967 RSC.
- National Research Council (US), Committee for the Update of the Guide for the Care and Use of Laboratory Animals, Guide for the Care and Use of Laboratory Animals, National Academies Press (US), Washington (DC), 8th edn, 2011 Search PubMed.
- N. Zeeni, C. Daher, G. Fromentin, D. Tome, N. Darcel and C. Chaumontet, Stress, 2013, 16, 211–219 CrossRef CAS PubMed.
- L. E. Hayek, M. Khalifeh, V. Zibara, R. A. Assaad, N. Emmanuel, N. Karnib, R. El-Ghandour, P. Nasrallah, M. Bilen, P. Ibrahim, J. Younes, E. A. Haidar, N. Barmo, V. Jabre, J. S. Stephan and S. F. Sleiman, J. Neurosci., 2019, 39, 2369–2382 Search PubMed.
- N. Karnib, R. El-Ghandour, L. E. Hayek, P. Nasrallah, M. Khalifeh, N. Barmo, V. Jabre, P. Ibrahim, M. Bilen, J. S. Stephan, E. B. Holson, R. R. Ratan and S. F. Sleiman, Neuropsychopharmacology, 2019, 44, 1152 CrossRef PubMed.
- A. Smajilagić, M. Aljičević, A. Redžić, S. Filipović and A. C. Lagumdžija, Bosnian J. Basic Med. Sci., 2013, 13, 27–30 CrossRef PubMed.
- M. Najar, H. Fayyad-Kazan, W. H. Faour, M. Merimi, E. M. Sokal, C. A. Lombard and H. Fahmi, Inflammation Res., 2019, 68, 203–213 CrossRef CAS PubMed.
- G. Nasreddine, M. El-Sibai and R. J. Abi-Habib, Invest. New Drugs, 2019, 37, 1–10 CrossRef PubMed.
- E. Kassab, M. Darwish, Z. Timsah, S. Liu, S. H. Leppla, A. E. Frankel and R. J. Abi-Habib, Transl. Oncol., 2013, 6, 25–32 CrossRef PubMed.
- D. N. Wheatley, M. S. Inglis, M. A. Foster and J. E. Rimington, J. Cell Sci., 1987, 88(pt 1), 13–23 Search PubMed.
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2007, 129, 46–47 CrossRef CAS PubMed.
- C. A. Puckett, R. J. Ernst and J. K. Barton, Dalton Trans., 2010, 39, 1159–1170 RSC.
- A. Ghezzi, M. Aceto, C. Cassino, E. Gabano and D. Osella, J. Inorg. Biochem., 2004, 98, 73–78 CrossRef CAS PubMed.
- N. D. Eljack, H.-Y. M. Ma, J. Drucker, C. Shen, T. W. Hambley, E. J. New, T. Friedrich and R. J. Clarke, Met. Integr. Biometal Sci., 2014, 6, 2126–2133 RSC.
- M. Dickerson, Y. Sun, B. Howerton and E. C. Glazer, Inorg. Chem., 2014, 53, 10370–10377 CrossRef CAS PubMed.
- C. Mari and G. Gasser, Chimia, 2015, 69, 176–181 CrossRef CAS PubMed.
- S. L. Hopkins, B. Siewert, S. H. C. Askes, P. Veldhuizen, R. Zwier, M. Heger and S. Bonnet, Photochem. Photobiol. Sci., 2016, 15, 644–653 RSC.
- J. Zhuang, Y. Liu, Q. Yuan, J. Liu, Y. Liu, H. Li and D. Wang, Oncol. Lett., 2018, 15, 6291–6296 Search PubMed.
- A. N. Hidayatullah, E. Wachter, D. K. Heidary, S. Parkin and E. C. Glazer, Inorg. Chem., 2014, 53, 10030–10032 CrossRef CAS PubMed.
- K. Gharehbaghi, T. Szekeres, J. A. Yalowitz, M. Fritzer-Szekeres, Y. G. Pommier and H. N. Jayaram, Life Sci., 2000, 68, 1–11 CrossRef CAS PubMed.
- P. Lincoln and B. Nordén, J. Phys. Chem. B, 1998, 102, 9583–9594 CrossRef CAS.
- F. Qu, S. Park, K. Martinez, J. L. Gray, F. S. Thowfeik, J. A. Lundeen, A. E. Kuhn, D. J. Charboneau, D. L. Gerlach, M. M. Lockart, J. A. Law, K. L. Jernigan, N. Chambers, M. Zeller, N. A. Piro, W. S. Kassel, R. H. Schmehl, J. J. Paul, E. J. Merino, Y. Kim and E. T. Papish, Inorg. Chem., 2017, 56, 7519–7532 CrossRef CAS PubMed.
- E. Grueso, G. López-Pérez, M. Castellano and R. Prado-Gotor, J. Inorg. Biochem., 2012, 106, 1–9 CrossRef CAS PubMed.
- K. T. Spencer, P. D. Lindower, G. R. Buettner and R. E. Kerber, J. Cardiovasc. Pharmacol., 1998, 32, 343–348 CrossRef CAS PubMed.
- A. Mohindru, J. M. Fisher and M. Rabinovitz, Biochem. Pharmacol., 1983, 32, 3627–3632 CrossRef CAS PubMed.
- A. Mohindru, J. M. Fisher and M. Rabinovitz, Nature, 1983, 303, 64–65 CrossRef CAS PubMed.
- C. R. Kowol, P. Heffeter, W. Miklos, L. Gille, R. Trondl, L. Cappellacci, W. Berger and B. K. Keppler, JBIC, J. Biol. Inorg. Chem., 2012, 17, 409–423 CrossRef CAS PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2013, 114, 815–862 CrossRef PubMed.
- F. Liu, K. A. Meadows and D. R. McMillin, J. Am. Chem. Soc., 1993, 115, 6699–6704 CrossRef CAS.
- D. R. McMillin, F. Liu, K. A. Meadows, T. K. Aldridge and B. P. Hudson, Coord. Chem. Rev., 1994, 132, 105–112 CrossRef CAS.
- D. R. McMillin and K. M. McNett, Chem. Rev., 1998, 98, 1201–1220 CrossRef PubMed.
- V. Brabec and O. Nováková, Drug Resist. Updates, 2006, 9, 111–122 CrossRef CAS PubMed.
- S. Abou Najem, G. Khawaja and S. Rizk, Clin. Lymphoma, Myeloma Leuk., 2018, 18, S207 CrossRef.
- E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649–9651 RSC.
- Y. Chen, X. Luo, L. Bai, X. Hu, J. Zhou, P. Zhang and Y. Yu, New J. Chem., 2017, 41, 10225–10230 RSC.
- S. Nowsheen and E. S. Yang, Exp. Oncol., 2012, 34, 243–254 CAS.
- M. H. Hodroj, A. Jardaly, S. Abi Raad, A. Zouein and S. Rizk, Cancer Manage. Res., 2018, 10, 1079–1088 CrossRef PubMed.
- C. Khalil and W. Shebaby, Toxicol. Rep., 2017, 4, 441–449 CrossRef CAS PubMed.
- W. P. Roos and B. Kaina, Trends Mol. Med., 2006, 12, 440–450 CrossRef CAS PubMed.
- L. Zeng, P. Gupta, Y. Chen, E. Wang, L. Ji, H. Chao and Z.-S. Chen, Chem. Soc. Rev., 2017, 46, 5771–5804 RSC.
- P. Zhang, J. Chen and Y. Liang, Acta Biochim. Biophys. Sin., 2010, 42, 440–449 CrossRef CAS PubMed.
- L. Xu, N.-J. Zhong, Y.-Y. Xie, H.-L. Huang, G.-B. Jiang and Y.-J. Liu, PLoS One, 2014, 9, e96082 CrossRef PubMed.
- W. Li, G.-B. Jiang, J.-H. Yao, X.-Z. Wang, J. Wang, B.-J. Han, Y.-Y. Xie, G.-J. Lin, H.-L. Huang and Y.-J. Liu, J. Photochem. Photobiol., B, 2014, 140, 94–104 CrossRef CAS PubMed.
- A. Srishailam, Y. P. Kumar, P. Venkat Reddy, N. Nambigari, U. Vuruputuri, S. S. Singh and S. Satyanarayana, J. Photochem. Photobiol., B, 2014, 132, 111–123 CrossRef CAS PubMed.
- Y.-Y. Xie, H.-L. Huang, J.-H. Yao, G.-J. Lin, G.-B. Jiang and Y.-J. Liu, Eur. J. Med. Chem., 2013, 63, 603–610 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra02615k |
| This journal is © The Royal Society of Chemistry 2019 |