
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous Suzuki–Miyaura coupling of heteroaryl ester via chemoselective C(acyl)–O bond activation†
Hongpeng Ma,
Chaolumen Bai and
Yong-Sheng Bao *
*
College of Chemistry and Environmental Science, Inner Mongolia Key Laboratory of Green Catalysis, Inner Mongolia Normal University, Hohhot, 010022, China. E-mail: sbbys197812@163.com; Tel: +86-471-4392442
First published on 3rd June 2019
Abstract
A site-selective supported palladium nanoparticle catalyzed Suzuki–Miyaura cross-coupling reaction with heteroaryl esters and arylboronic acids as coupling partners was developed. This methodology provides a heterogeneous catalytic route for aryl ketone formation via C(acyl)–O bond activation of esters by successful suppression of the undesired decarbonylation phenomenon. The catalyst can be reused and shows high activity after eight cycles. The XPS analysis of the catalyst before and after the reaction suggested that the reaction might be performed via a Pd0/PdII catalytic cycle that began with Pd0.
Introduction
Transition metal-catalyzed Suzuki–Miyaura reactions have emerged as a powerful and indispensable tool to forge linkages between carbon atoms because of the inherent advantages of organoboron reagents.1 Conventionally, halocarbon electrophiles are the most applied coupling partners in Suzuki–Miyaura reactions.2 However, the corrosive halide-containing waste production does not meet the modern synthetic chemistry expectations with the growing interest in environmentally friendly protocol development nowadays. Hence, expanding the types of electrophiles that react via this pathway is thus an ongoing goal in organic synthesis.Recently, (hetero)aryl esters, due to their ubiquitous nature, have gained significant interest as new aryl-coupling partners via nickel catalyzed C(aryl)–O cleavage (see Scheme 1a).3 Actually, the bond dissociation energy (BDE) of the C(acyl)–O bond is lower than the C(aryl)–O bond in (hetero)aryl esters.3h But, catalyzed by transition-metal, there are challenges associated with C(acyl)–O bond cleavage because of the decarbonylation phenomenon.4 In 2001, Yamamoto reported the first example of palladium catalyzed coupling reaction of aryl esters with organoboron compounds via C(acyl)–O cleavage with carbonyl retention (see Scheme 1b). However, the reaction was limited to electronically activated esters, such as perfluoroaliphatic carboxylic esters.5 Chatani reported the palladium catalyzed coupling reaction of 2-pyridyl esters with organoboron compounds (see Scheme 1c).6 More recently, Newman developed an NHC-based Pd catalyst which can catalyze Suzuki–Miyaura coupling of phenyl esters (see Scheme 1d).7
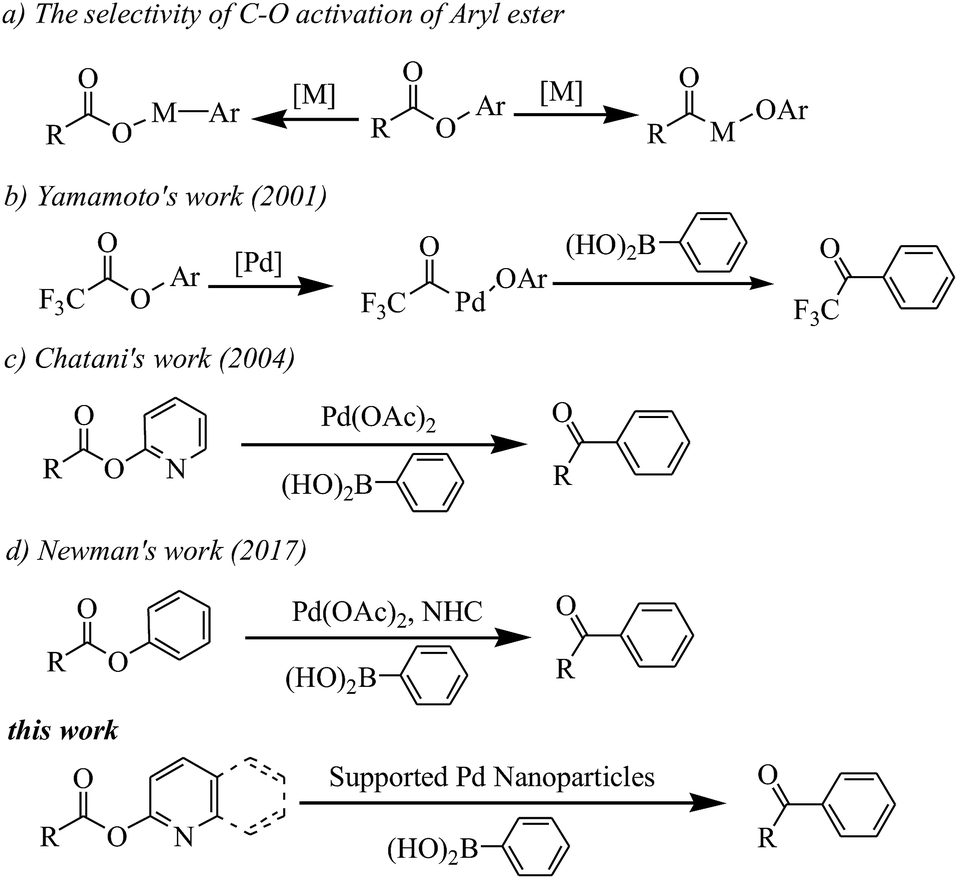 | ||
| Scheme 1 C–O activation of (hetero)aryl ester. | ||
Although these homogeneous palladium catalysts offer high selectivity and yields under relatively mild operating conditions, but their industrial applicability is limited by the inherent problem of catalyst separation from the product and its recycle.8 Moreover, the palladium residues in the product stream could be a serious issue in the pharmaceutical industry.9 With these considerations in mind, a wide variety of alternative methods for heterogeneous Pd-catalyzed Suzuki–Miyaura reactions have been introduced.10 But, to the best of our knowledge, there is no report for heterogeneous Pd-catalyzed Suzuki–Miyaura reactions using ester as coupling partners.
Our previous work has confirmed that (hetero)aryl esters can generate an activated acyl intermediate and perform amidation reactions with tertiary amines or formamides to form amides under supported palladium nanoparticles catalyzed conditions.11 These results prompt us to explore the heterogeneous palladium catalyzed Suzuki–Miyaura reaction of (hetero)aryl esters. But one obstacle to establish this heterogeneous catalysis is that the undesired Pd-catalyzed homocoupling of phenylboronic acid could be easily facilitated under these reaction conditions.12 Another is that in most of the cases, Suzuki–Miyaura reaction need base condition which will give rise to side reactions for ester. Therefore, the choose of catalyst with moderate activity and (hetero)aryl ester with high selectivity is vital.
Herein, we describe a nano-palladium catalyzed Suzuki–Miyaura cross-coupling variant with heteroaryl ester and arylboronic acid components as coupling partners. This newly developed methodology provides a heterogeneous catalysis route for C–C bond formation in a straightforward fashion, which successfully suppresses the undesired decarbonylation phenomenon attendant upon C(acyl)–O bond activation of ester. In a series of supported PdNPs, Pd/γ-Al2O3 catalyst with a PdNPs mean diameter of 3.34 nm exhibited the best catalytic activity and it maintained a relatively high activity after several recover cycles. The XPS analysis of the catalyst before and after reaction suggested that the reaction might be performed via a Pd0/PdII catalytic cycle that began with Pd0.
Experimental
Catalyst preparation
The PdNPs on the γ-Al2O3 and other supports were prepared by a low density lysine solution protected impregnation–reduction method. For example, 3 wt% Pd/γ-Al2O3 was prepared by the following procedure: 0.97 g of γ-Al2O3 powder was dispersed into 50 mL of deionized water, followed by adding 28.2 mL of 0.01 M PdCl2 aqueous solution while magnetically stirring. One milliliter of 0.03 M L-lysine was added with vigorous stirring. Then 0.1 M NaOH aqueous solution was added into the mixture to adjust the pH to 7. To this suspension, 4.5 mL of 0.35 M NaBH4 solution was added dropwise in 10 min. The mixture was aged for 24 h, and then the solid was separated, washed with water (4 times) and ethanol (once), and dried at 80 °C. The dried solid was used directly as the catalyst.Preparation of various esters (1a–s, 4a–h)
A mixture of carboxylic acid (10 mmol), pyridinol or phenol (10 mmol), DMAP (4-(dimethylamino)pyridine, 1 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)carbodiamide hydrochloride (EDC·HCl, 10 mmol) in THF (50 mL) was stirred overnight at 25 °C. The resulting mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl ether/petroleum ether = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 to 1:10 as eluent), affording a corresponding esters 1a–s and 4a–h.
:3 to 1:10 as eluent), affording a corresponding esters 1a–s and 4a–h.
Activity test
The reaction in 25 mL over-dried reaction tube. The Suzuki–Miyaura cross coupling reaction between 2-pyridyl 4-methylbenzoate 1a and phenylboronic acid 2a was used as the model reaction. In a typical reaction catalyst (25 mg), 2-pyridyl 4-methylbenzoate (21.3 mg, 0.10 mmol), phenylboronic acid (24.4 mg, 0.20 mmol), K3PO4 (31.8 mg, 0.15 mmol), H2O (4.5 μL, 0.25 mmol), toluene (2 mL) were added into the reaction tube. The resulting solution was stirred at 120 °C for 48 h in an oil bath. After cooling to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1:10 to 1:15 as an eluent) to afford the desired ketone 3. All products were confirmed by comparison with the previously reported 1H NMR and 13C NMR data.
Catalyst recycle experiment
After each reaction cycle, the solvent, substrate, and the products were removed by centrifugation; the separated catalyst was washed thoroughly with 0.1 M NaOH ethanol solution (twice), distilled water (4 times), and then washed twice with ethanol followed by centrifugal separation and drying at 80 °C for 12 h. The recovered catalyst was used for the next cycle.Hot filtration test
In a testing reaction 3 wt% Pd/γ-Al2O3 (25 mg), 2-pyridyl 4-methylbenzoate 1a (21.3 mg, 0.10 mmol), phenylboronic acid 2a (24.4 mg, 0.20 mmol), K3PO4 (31.8 mg, 0.15 mmol), H2O (4.5 μL, 0.25 mmol), toluene (2 mL) were added into the reaction tube. The hot filtration test is applied by filtering the reaction mixture through a pre-heated Celite pad after the reaction for 2 h. The filtrate was detected by GC to obtain the conversion of 1a. Then the filtered reaction solution continued to react for 2 h under normal conditions, and was detected by GC again.Gram-scale synthesis
In a gram-scale reaction, catalyst (1.17 g), 2-pyridyl 4-methylbenzoate 1a (1.00 g, 4.69 mmol), phenylboronic acid 2a (1.15 g, 9.39 mmol), K3PO4 (1.49 g, 7.04 mmol), H2O (211 μL, 11.73 mmol) and toluene (60 mL) were charged in a 100 mL ovendried round flask. The resulting solution was stirred at 120 °C for 48 h in an oil bath. After cooling to room temperature, the mixture was filtered, and the filtrate was evaporated in vacuo. The residue was purified by flash column chromatography (silica gel, ethyl acetate/petroleum ether = 1:10 as an eluent) to afford the desired product 3aa (0.92 g, 84% yield).
Results and discussion
Our initial attempt was executed toward the supported palladium nanoparticles catalyzed Suzuki–Miyaura cross-coupling reaction using 2-pyridyl 4-methylbenzoate 1a and phenylboronic acid 2a as the coupling partners. Using 3 wt% Pd/γ-Al2O3 as the catalyst, Cs2CO3 as base, and toluene as solvent, after 24 h of refluxing the desired ketone 3aa was obtained in 54% yield (Table 1, entry 1). Meantime, along with the desired product only 9% yield of homocoupling byproduct of phenylboronic acid was isolated. This indicated that the heterogeneous palladium catalyst is showed higher selectivity. Then, various reaction parameters including catalyst, base, solvent, additive and temperature were screened. The PdNPs supported on other oxide powders, including α-Al2O3, CeO2, TiO2 and Fe3O4 were prepared by the impregnation–reduction method and applied to the model reaction (entries 2–5). Among them, the catalyst with 3 wt% Pd/γ-Al2O3 exhibited the best performance. The effect of solvent on the coupling reaction was rather important, toluene proved to the best choice of solvent, whereas other solvents such as cumene and dioxane gave rise to slightly lower yields (entries 6–8). Base was also found to show significant influence on the coupling reaction, and anhydrous K3PO4 showed the best efficiency for this transformation (entries 9–12). In sharp contrast, no reaction was occurred in the absence of the base. It is noteworthy that with the additive of water, higher conversion was achieved and the desired product was obtained in 68% yield (entry 13). A small amount of water might help dissolve the base into the reaction system. But only used water as solvent is inadvisable because water-insoluble property of ester. We also screened the effect of different temperature on the reaction (entries 14–16). We are pleased to find that increasing the temperature to 120 °C led to generate the highest yield of product, and further increasing the temperature of the reaction did not have any improvement on the yield. No product was observed under otherwise identical conditions in the absence of Pd (entry 17).
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Additive | Temp (°C) | Yield (%) |
| a Reaction condition: 1a (0.10 mmol), catalyst (25 mg), Pd (7 mol%), phenylboronic acid 2a (2.0 equiv), base (1.5 equiv), H2O (2.5 equiv), solvent (2 mL), 120 °C, 48 h. | ||||||
| 1 | 3% Pd/γ-Al2O3 | Cs2CO3 | Toluene | — | 100 | 54 |
| 2 | 3% Pd/α-Al2O3 | Cs2CO3 | Toluene | — | 100 | 25 |
| 3 | 3% Pd/CeO2 | Cs2CO3 | Toluene | — | 100 | 38 |
| 4 | 3% Pd/TiO2 | Cs2CO3 | Toluene | — | 100 | 49 |
| 5 | 3% Pd/Fe3O4 | Cs2CO3 | Toluene | — | 100 | 37 |
| 6 | 3% Pd/γ-Al2O3 | Cs2CO3 | Cumene | — | 100 | 35 |
| 7 | 3% Pd/γ-Al2O3 | Cs2CO3 | Dioxane | — | 100 | 20 |
| 8 | 3% Pd/γ-Al2O3 | Cs2CO3 | THF | — | 100 | NP |
| 9 | 3% Pd/γ-Al2O3 | Na2CO3 | Toluene | — | 100 | 30 |
| 10 | 3% Pd/γ-Al2O3 | K2CO3 | Toluene | — | 100 | 47 |
| 11 | 3% Pd/γ-Al2O3 | — | Toluene | — | 100 | NP |
| 12 | 3% Pd/γ-Al2O3 | K3PO4 | Toluene | — | 100 | 58 |
| 13 | 3% Pd/γ-Al2O3 | K3PO4 | Toluene | H2O | 100 | 68 |
| 14 | 3% Pd/γ-Al2O3 | K3PO4 | Toluene | H2O | 110 | 73 |
| 15 | 3% Pd/γ-Al2O3 | K3PO4 | Toluene | H2O | 120 | 88 |
| 16 | 3% Pd/γ-Al2O3 | K3PO4 | Toluene | H2O | 130 | 70 |
| 17 | γ-Al2O3 | K3PO4 | Toluene | H2O | 120 | NP |
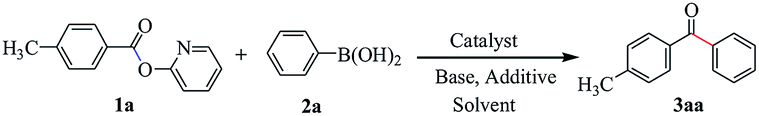
The Suzuki–Miyaura cross-coupling of phenylboronic acid 2a with a set of 2-pyridyl esters was performed under the optimized reaction conditions, and the results are presented in Table 2. Incorporation of electron-donating 3ca–3ga, electron-withdrawing 3ha–3na or naphthyl 3oa, groups on the esters were tolerated. Compared with their para-isomers, 2-pyridyl 2-methylbenzoate 1c, 2-pyridyl 3-methylbenzoate 1d and 2-pyridyl 3-nitrobenzoate 1m delivered a lower yield, which resulted from the steric hindrance effect. Encouragingly, various heterocyclic species could also be utilized 3pa–3ra and (1H-indol-2-yl)(phenyl)methanone 3pa was obtained quantitatively. The ability to obtain high yields of indolyl aromatic ketones is a significant advantage of this methodology. But when pyridin-2-yl propionate 1s react with phenylboronic acid 2a, no product was detected. The result indicated that the acyl C–O bond cleavage with the aid of palladium cannot tolerate aliphatic substituents. The ease of activating this type of bond can be attributed to the stabilizing interaction of the arene π system with palladium.14
|
|---|
| a Reaction conditions: 1 (0.10 mmol), 2a (2.0 equiv), 3 wt% Pd/γ-Al2O3 (25 mg), Pd (7 mol%), K3PO4 (1.5 equiv), H2O (2.5 equiv), toluene (2 mL), 120 °C, 48 h. Isolated yield. |
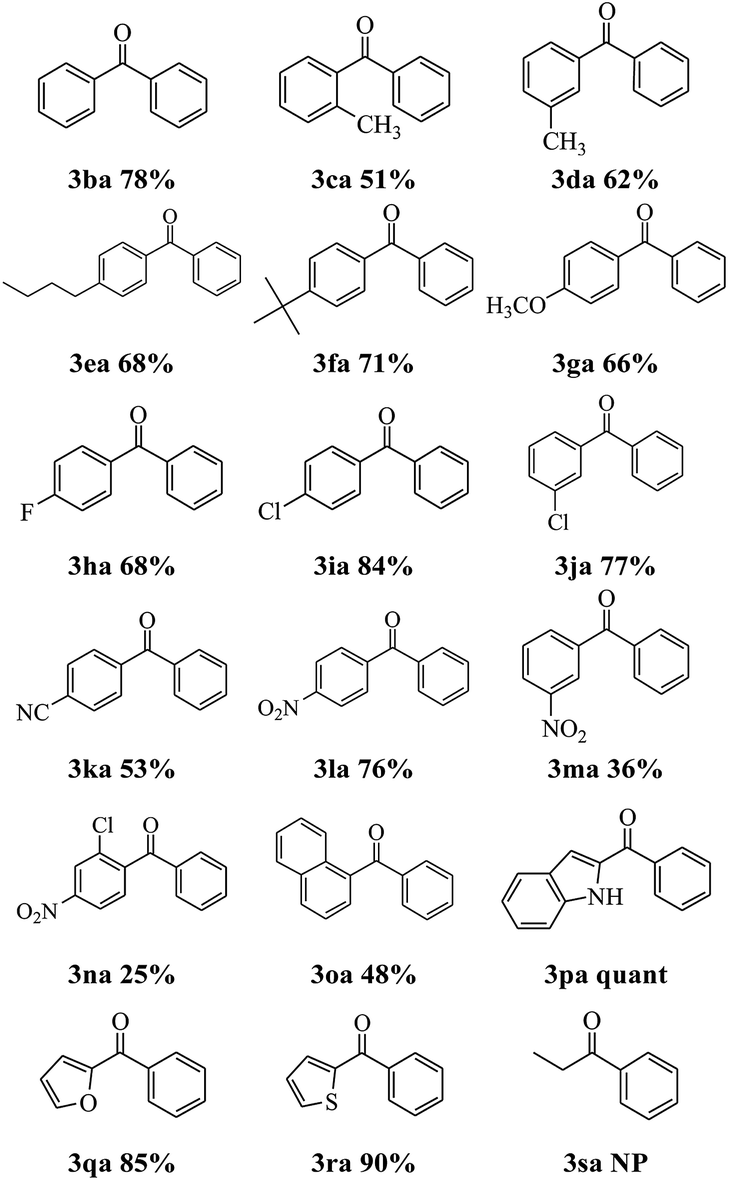 |
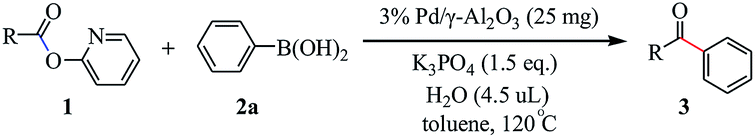
Next, the scope of this reaction between 2-pyridyl 4-methylbenzoate 1a with a variety of boronic acids was investigated and the results are summarized in Table 3. Various functional groups including alkyl, ethoxy, fluoro, bromo, trifluoromethyl, thiomethyl and phenoxyl were compatible and the desired products were achieved in moderate to good yields. But the ortho-ethoxy phenyl boronic acid did not perform the reaction. The result indicated that the steric hindrance effect of boronic acids is a determining factor of the coupling reaction (see 3ag). Interestingly, the Suzuki–Miyaura could be used to synthesize benzodioxole ketone 3an which is an important pharmaceutical intermediate albeit in lower yield.
|
|---|
| a Reaction conditions: 1a (0.10 mmol), 2 (2.0 equiv), 3 wt% Pd/γ-Al2O3 (25 mg), Pd (7 mol%), K3PO4 (1.5 equiv), H2O (2.5 equiv), toluene (2 mL), 120 °C, 48 h. Isolated yield. |
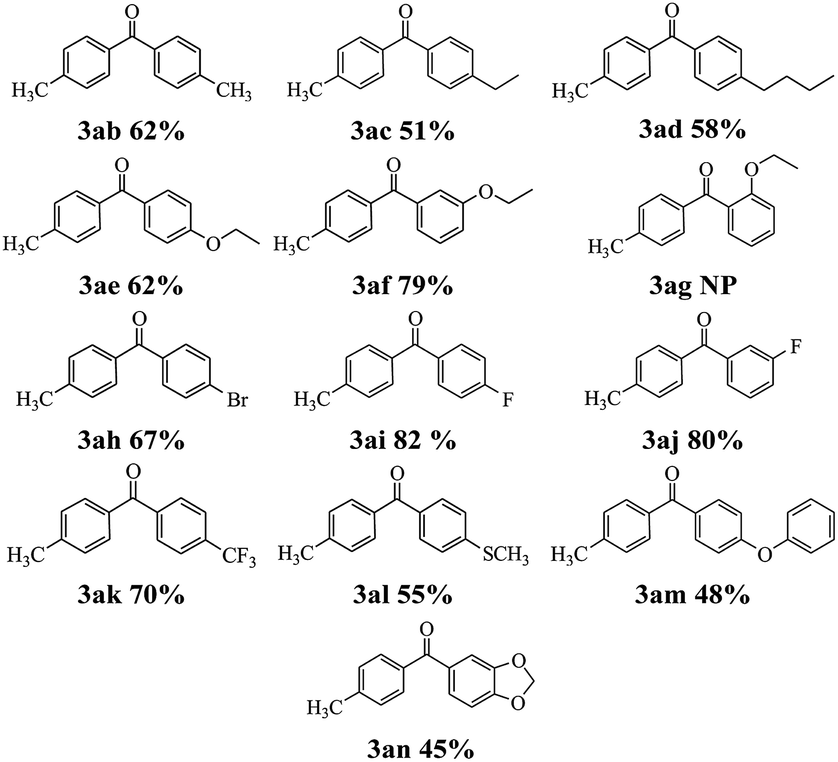 |
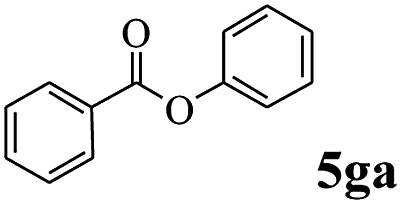
In order to verify that the ortho-directed effect of nitrogen on C(acyl)–O bond cleavage of ester, pyridin-3-yl 2-methylbenzoate 4a and pyridin-4-yl 2-methylbenzoate 4b were used to react with phenylboronic acid 2a (see Table 4, entries 1 and 2). After 24 h reaction, the desired ketone was not obtained, but instead phenyl 4-methyl benzoate 5aa was obtained in 48% and 44% yield, respectively. These results confirmed the ortho-directed effect of nitrogen on C(acyl)–O bond cleavage of ester catalyzed by palladium. The reason for generating phenyl ester is that catalyzed by palladium, phenylboronic acid transferred to phenol11 in the presence of H2O which performed the transesterification reaction with pyridin-2-yl 2-methylbenzoate 1a to generate new ester catalyzed by K3PO4.15 Interestingly, when quinolin-8-yl 4-methylbenzoate 4c containing transannular directing group was used as substrate, no reaction was detected (entry 3). When other heteroaryl benzoate containing ortho-nitrogen, quinolin-2-yl 4-methylbenzoate 4d, 4f and pyrazin-2-yl 4-methylbenzoate 4e were employed instead of 1a, the Suzuki–Miyaura cross-coupling reaction also proceed to afford the same ketone 3aa but with the lower selectivity (entries 4–6). Under the identical reaction conditions, the activated ester perfluorophenyl benzoate 4g only give the transesterification product rather than desired ketone (entry 7). Not only that, when p-tolyl picolinate 4h was used as substrate, no reaction was performed (entry 8). These results clearly show that the pyridyl group serves as a directing group for cleavage of the acyl C–O bond of the ester and that coordination of the nitrogen atom to the catalyst is crucial for the catalyzed reaction. This result is different from Chatani's work.
|
|||
|---|---|---|---|
| Entry | Esters | Product | Yield (%) |
| a Reaction conditions: 4 (0.10 mmol), 2a (2.0 equiv), 3 wt% Pd/γ-Al2O3 (25 mg), Pd (7 mol%), K3PO4 (1.5 equiv), H2O (2.5 equiv), toluene (2 mL), 120 °C, 48 h. Isolated yield. | |||
| 1 | 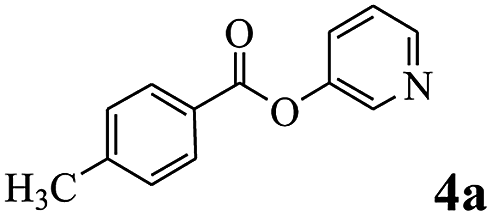 |
 |
48% |
| 2 | 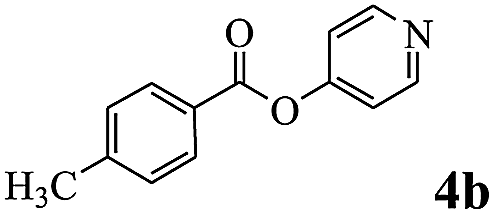 |
5aa | 44% |
| 3 | 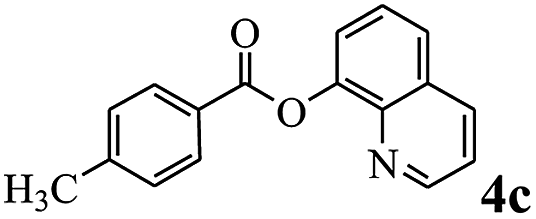 |
— | NP |
| 4 | 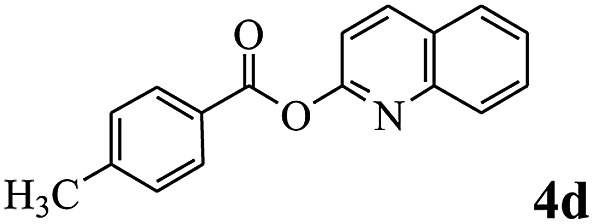 |
3aa | 53% |
| 5aa | 39% | ||
| 5 | 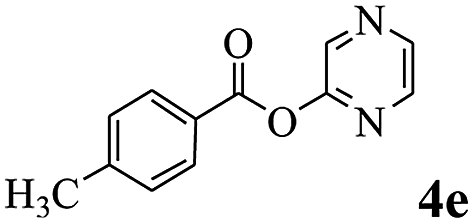 |
3aa | 25% |
| 5aa | 36% | ||
| 6 | 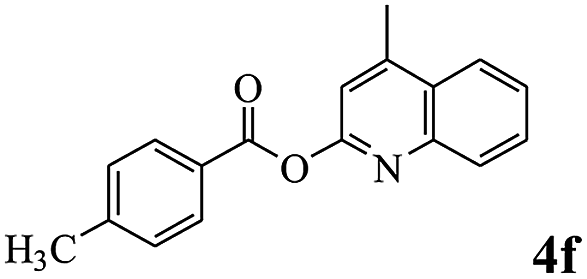 |
3aa | 62% |
| 7 | 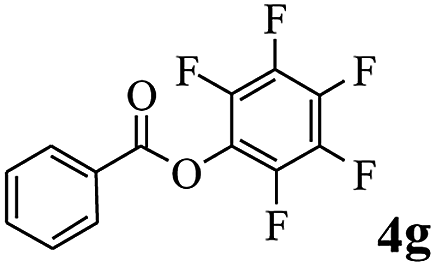 |
 |
32% |
| 8 | 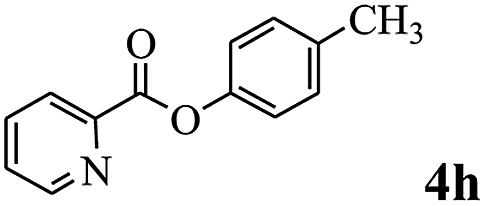 |
— | NP |
To investigate the practical application of this newly developed Suzuki–Miyaura cross-coupling reaction in organic synthesis, we conducted a gram-scale reaction of 1a (1 g) with 2a in the presence of 3 wt% Pd/γ-Al2O3 catalyst, and isolated the desired product 3aa in 84% yield. As we can see, even though the reaction scale was magnified up to 47 times, ideal synthetically yields could be still obtained.
The recyclability of catalyst was examined in the reaction of 2-pyridyl 4-methylbenzoate 1a and phenylboronic acid 2a as provided in the experimental section. As shown in Fig. 1, the catalyst can be reused for eight cycles with only 13% decline of activity after the 8th recycle.
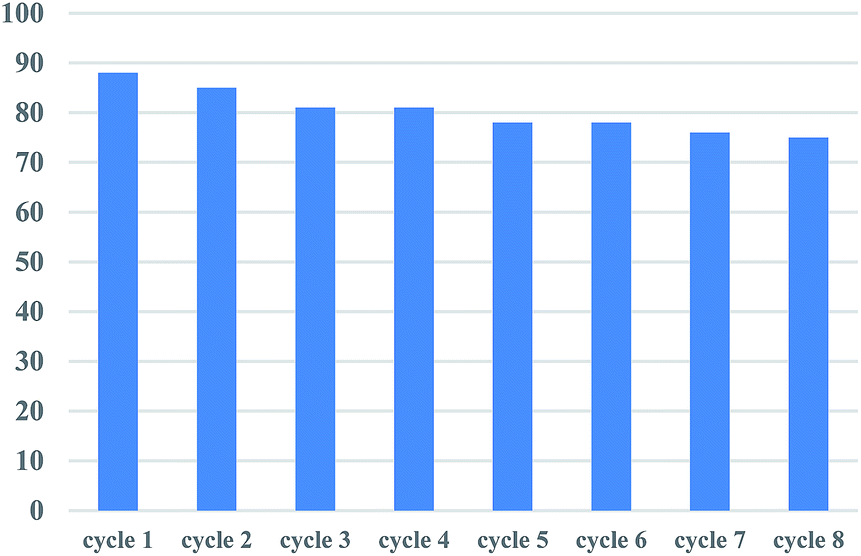 | ||
| Fig. 1 Recyclability of the catalyst. | ||
To have a good knowledge of the information for the catalyst, the fresh and used (recovered after 8th cycle) PdNPs on γ-Al2O3 catalysts were studied by transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS) and so on. The transmission electron microscopic (TEM) analysis of the fresh and used catalysts are represented in Fig. 2. The PdNPs are distributed evenly on the γ-Al2O3 surface, and the mean diameters of the PdNPs are 3.34 nm of fresh catalyst and 3.48 nm of used catalyst, respectively. It does not cause obvious increase in the average size of PdNPs after eight runs.
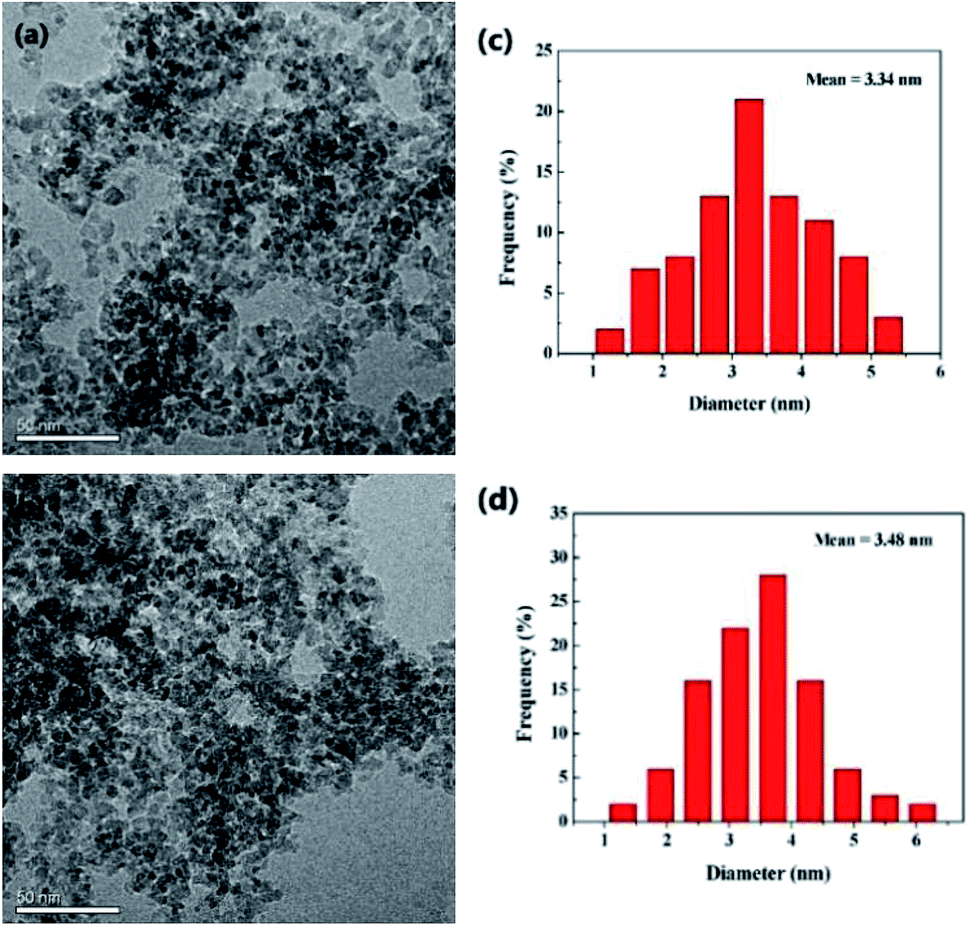 | ||
| Fig. 2 (a and b) TEM images of fresh 3 wt% Pd/γ-Al2O3 and used 3 wt% Pd/γ-Al2O3, respectively; (c and d) PdNPs size distributions of fresh and used catalysts, respectively. | ||
In order to understand the state of PdNPs supported on γ-Al2O3, the catalysts before and after reaction were tested by XPS analysis. The XPS results of the catalysts confirm that palladium exists in the metallic state on γ-Al2O3 supports before and after reaction. As shown in Fig. 3, the binding energies of Pd 3d5/2 and Pd 3d3/2 electrons are 335.69 eV and 340.93 eV of fresh catalyst and 335.80 eV and 341.00 eV of used catalyst respectively. The binding energies of Pd0 in literature are 335.20 eV of Pd 3d5/2 and 340.50 eV of Pd 3d3/2, respectively, whereas the values of PdII are 336.70 eV of Pd 3d5/2 and 342.00 eV of Pd 3d3/2, respectively.13 It is clearly shown that Pd0 as the active center completes the catalytic cycle.
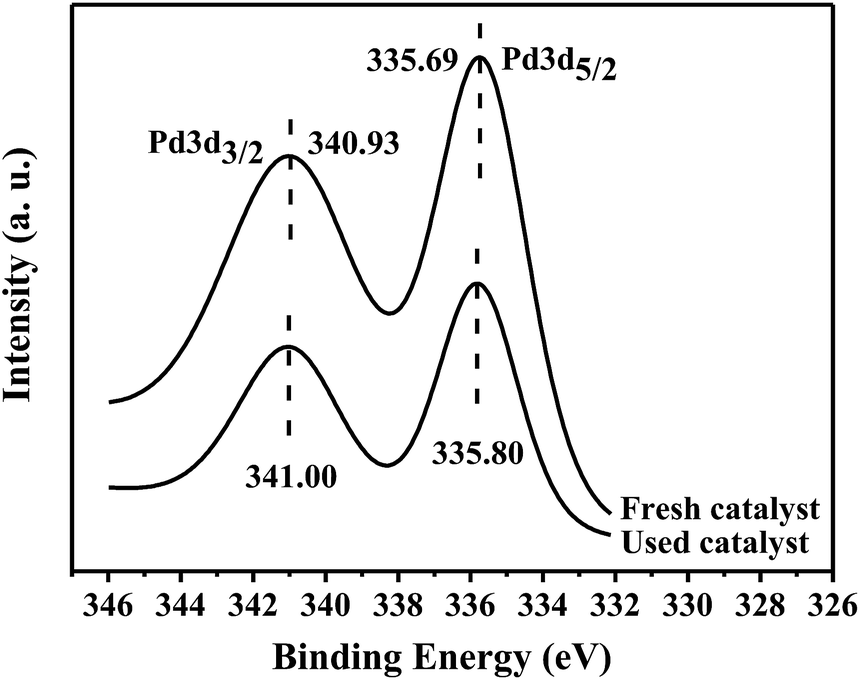 | ||
| Fig. 3 XPS spectra of fresh and used 3 wt% Pd/γ-Al2O3. | ||
The Brunauer–Emmett–Teller (BET) specific surface areas of the catalysts were derived from N2 physical sorption data of the samples using the BET model (Table 5). The γ-Al2O3 is a kind of support which has a large specific surface area, and it does not cause obvious change in the specific surface area of these catalysts after loading a small amount of the PdNPs. The amounts of Pd loading of the catalysts were derived from atomic absorption spectrophotometer (AAS), and the Pd content of fresh catalyst is approximately 3 wt%. We did note a slight decrease in Pd content after being cycled eight times (2.89%, Table 5), which can decrease the catalytic activity on the basis of available Pd on the support surface.
| Samples | SBET (m2 g−1) | Pd loading (wt%) |
|---|---|---|
| γ-Al2O3 | 161 | — |
| 3 wt% Pd/γ-Al2O3 (fresh) | 166 | 3.02 |
| 3 wt% Pd/γ-Al2O3 (used) | 159 | 2.89 |
In order to prove whether the present reaction proceeded with a heterogeneous catalyst or homogeneous catalyst, the hot filtration test of the model reaction mixture was executed and GC data are given in Table 6. The conversion rate of 1a indicated that after filtration the substrate turnover ceases. This is intended to preclude the possibility of homogeneous catalysis. This result was also proved by Hg(0) poisoning test (see ESI†).
To identify a byproduct, which is formed by the cleavage of the C(acyl)–O bond, the model reaction solution was detected with GCMS after the end of the reaction (see the ESI†). The GCMS data indicated that along with desired product 3aa a small amount of biphenyl was obtained and that alkoxyl group of ester eliminated to pyridin-2-ol. Based on the GC-MS analysis results and the commonly accepted mechanism from the literature, the proposed reaction pathway is shown in Scheme 2. Initially, the nitrogen atom in 1a coordinates with Pd0 to form intermediate I, and then Pd attacks the carbonyl carbon to give five-membered palladium ring intermediate II. C–O bond cleavage takes place with re-formation of the carbonyl group and acylpalladium III is generated. Transmetalation between III and organoboron compounds affords intermediate IV.6,16 Finally, reductive elimination from IV yields ketone with regeneration of active Pd0.
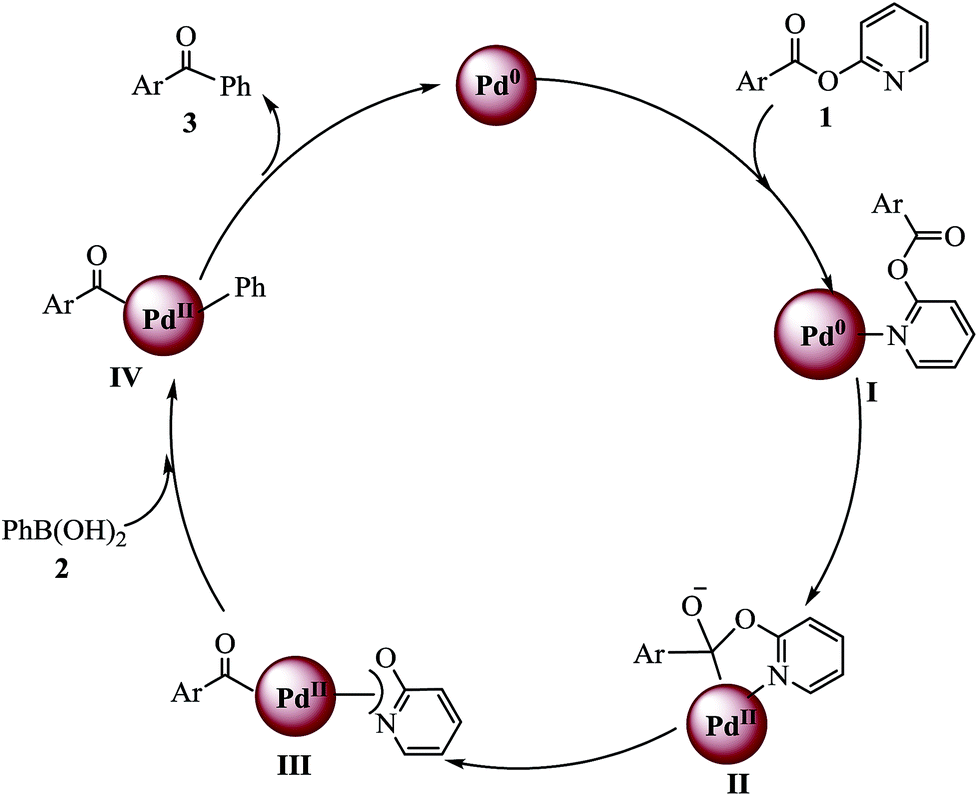 | ||
| Scheme 2 Proposed reaction mechanism. | ||
Conclusions
In summary, the results reported herein represent the first example of heterogeneous and site-selective nano-palladium catalyzed Suzuki–Miyaura cross-coupling reaction with heteroaryl ester and arylboronic acids as coupling partners. The utility of this newly developed protocol has been demonstrated by the broad functional group tolerance and the application in the synthesis of bioactive compounds. It is also shown that the coordination of the nitrogen of the pyridyl group to the catalyst is essential for the efficient reaction. This reaction involves the heterogeneous catalytic generation of acylpalladium intermediates from esters by successful suppression of the undesired decarbonylation phenomenon, and its application to aryl ketone synthesis is achieved. The catalyst can be readily recovered and reused for eight cycles with only 13% decline of activity after the 8th recycle. The XPS analysis of the catalyst before and after reaction suggested that the reaction might be performed via a Pd0/PdII catalytic cycle that began with Pd0. The practicality of this study may inspire further studies on heterogeneous catalyzed C–O activation reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was financially supported by the National Science Foundation of China (21861030, 21462031), the Program for Young Talents of Science and Technology in Universities of Inner Mongolia Autonomous Region (NJYT-17-A22) and Research Innovation Fund of Inner Mongolia Normal University Graduate (CXJJS17090).Notes and references
-
(a) A. D. Meijere and F. E. Diederich, Metal-Catalyzed Cross-coupling Reaction, Wiley-VCH, Weinheim, 2004, vol. 1–2 CrossRef
; (b) I. P. Beletskaya, F. Alonso and V. Tyurin, Coord. Chem. Rev., 2019, 385, 137–173 CrossRef CAS
- A. Chatupheeraphat, H. H. Liao, W. Srimontree, L. Guo, Y. Minenkov, A. Poater and L. Cavallo, J. Am. Chem. Soc., 2018, 140, 3724–3735 CrossRef CAS PubMed
-
(a) H. Xu, K. Muto, J. Yamaguchi, C. Zhao, K. Itami and D. Musaev, J. Am. Chem. Soc., 2014, 136, 14834–14844 CrossRef CAS PubMed
-
(a) L. J. Gooßen and J. Paetzold, Angew. Chem., Int. Ed., 2002, 41, 1237–1241 CrossRef
- R. Kakino, I. Shimizu and A. Yamamoto, Bull. Chem. Soc. Jpn., 2001, 74, 371–376 CrossRef CAS
- H. Tatamidani, F. Kakiuchi and N. Chatani, Org. Lett., 2004, 6, 3597–3599 CrossRef CAS PubMed
- T. B. Halima, W. Zhang, I. Yalaoui, X. Hong, Y. Yang, K. N. Houk and S. G. Newman, J. Am. Chem. Soc., 2017, 139, 1311–1318 CrossRef PubMed
- S. Wittmann, A. Shätz, R. N. Grass, W. J. Stark and O. Reiser, Angew. Chem., Int. Ed., 2010, 49, 1867 CrossRef CAS PubMed
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889 CrossRef CAS
- A. Fihri, M. Bouhrara, B. Nekoueishahraki, J.-M. Basset and V. Polshettiwar, Chem. Soc. Rev., 2011, 40, 5181 RSC
-
(a) Y. S. Bao, M. Baiyin, B. Agula, M. L. Jia and Z. Bao, J. Org. Chem., 2014, 79, 6715 CrossRef CAS PubMed
- L. Wang, W. Zhang, D. S. Su, X. Meng and F. S. Xiao, Chem. Commun., 2012, 48, 5476–5478 RSC
-
(a) T. Pillo, R. Zimmermann, P. Steiner and S. Hüfner, J. Phys.: Condens. Matter, 1997, 9, 3987–3999 CrossRef CAS
- G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 2–20 CrossRef PubMed
- J. Chen, E. Namila, C. Bai, M. Baiyin, B. Agula and Y. S. Bao, RSC Adv., 2018, 8, 25168–25176 RSC
- G. L. Bras and J. Muzart, Eur. J. Org. Chem., 2018, 1176–1203 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: GC-MS analysis of model reaction, characterization data for the products, 1H NMR and 13C NMR spectra of the products. See DOI: 10.1039/c9ra02394a |
| This journal is © The Royal Society of Chemistry 2019 |