
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral biomolecule based dodecanuclear dysprosium(III)–copper(II) clusters: structural analyses and magnetic properties†
Biplab
Joarder
a,
Soumya
Mukherjee
a,
Mahendra
Patil
a,
Shufang
Xue
b,
Jinkui
Tang
*b and
Sujit K.
Ghosh
 *a
*a
aIndian Institute of Science Education and Research (IISER), Pune. Dr Homi Bhabha Road, Pashan, Pune, India 411008. E-mail: sghosh@iiserpune.ac.in; Fax: +91 20 2589 8022; Tel: +91 20 2590 8076
bState Key Laboratory of Rare Earth Resource Utilization, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China. E-mail: tang@ciac.ac.cn
First published on 7th July 2015
Abstract
An isostructural family of three dodecanuclear chiral M4Cu8 (M = Dy, Y) complexes has been synthesized by adopting a mixed-ligand strategy, wherein one of the constituent linkers, pyroglutamic acid, a recognized biomolecule, has been credibly functioning as the chiral precursor imparting the chirality to the aforementioned complex. The highly symmetric new-fangled M4Cu8 coordination-core, comprising of four square-symmetrically coordinated Dy/Y-vertex sharing M2Cu2 cubane units, has been synthesized as an unprecedented discrete coordination complex, which has been analysed by magnetic measurements.
Introduction
Discrete coordination architectures of diverse nuclearities have steadily drawn the attention of chemists and materials scientists for the intriguing functional aspects emanating from the simple design principles based on coordination chemistry driven self-assembly processes involving metal nodes and a vast spectrum of numerous linkers.1–6 Among these, coordination architectures with excellent symmetric features have been the most investigated ones owing to their captivating coordination environments, giving rise to utilitarian attributes as a consequence of their symmetry-aided structure–property correlation.7–13 The inherent symmetry in such complexes makes the structure–property correlation much easier to predict, leading to a strategic rationale based design principle for attaining the target properties and an insight thereinto. Over the decades, employment of a binary or ternary combination of linkers for yielding such complexes has proved quite an efficient protocol. Since the multiple coordination sites involved with their distinct denticities and coordination geometries, in unison with the associated non-covalent interactions, govern the crucial nuclearity aspect for such species; more than one appropriately functionalized linker is frequently employed for obtaining coordination complexes promising from the standpoint of application.14–25 Higher nuclearity-lanthanide discrete complexes or clusters have been the most sought-after ones considering the flurry of such materials evolved in the last decade exhibiting slow magnetic relaxation-triggered single molecule magnet (SMM) characteristics,26–31 ubiquitously imperative behind the development of novel molecular nanomagnets functioning as miniaturized devices for high-density information storage, molecule spintronics and quantum computing.32–40Biomolecules can be expediently employed as the building blocks of functional coordination complexes considering their biocompatibility, structural diversity, intrinsic self-assembly characteristics via their different metal-binding sites, low cost and ample availability coupled with individual chiral signatures of the precursor biomolecule-based synthons.41–45 The chiral features with multidentate facets particularly make such biomolecule-based coordination complexes an exciting domain to investigate, because of their prospective biological applications, including drug delivery or intracellular imaging. Although much effort has been devoted to lanthanide (4f)-based and lanthanide-transition (3d–4f)-based coordination complexes, resulting into the discovery of noteworthy magnetic properties such as slow magnetic relaxation-triggered single molecule magnet behaviour, magnetic refrigeration etc.; biomolecule based coordination complexes46 have never been the focus from the standpoint of magnetism phenomena, in spite of the recently explored spectrum of applications presented by this emerging class of materials.46–52 Despite some recent reports, pyroglutamic acid (PGA) or pidolic acid, a scarcely found proline derivative recently commercialized as a dietary supplement, wherein the free amino group of glutamic acid or glutamine undergoes cyclization to form a lactam, is yet to be comprehensively harnessed for coordination complex reactions.53–57 Importantly, this oxo-derivative of a secondary amino acid and a noteworthy metabolite in the glutathione cycle is a vital biomolecule possessing memory-enhancement effect, hair-follicle growth and dermal penetration enhancing consequences.58–60 Hence, the employment of PGA in the construction of 3d–4f based coordination complexes seems quite a daunting task, considering the unification of the dual features of biocompatibility and magnetism in a single discrete coordination entity. This indeed seems crucial from the standpoint of exploring magnetic materials with the requisite biocompatibility.61,62
Furthermore, to synthesize such desired coordination complexes based on hard Lewis acid (PGA) with high hydrooxophilic LnIII and CuII ions,63–69 we have purposefully chosen pyridine-based auxiliary ligands with flexible alkoxide ends, which might act as chelating and bridging ligands, owing to hard Lewis base O and N binding sites. Herein, we have exploited the oxophilic biomolecule-linker PGA (L and D enantiomers) with ancillary ligand 2-(2-hydroxyethyl)pyridine (HEP) (Fig. S1†) for the synthesis of a series of three isostructural, chiral biomolecule based M4Cu8 coordination complexes [M4Cu8(HEP)8(PGA)8(OH)8(NO3)4] (M = Dy and Y) and have analyzed their magnetism properties with the aid of direct and alternate current susceptibility measurements by SQUID (superconducting quantum interference device). The difference brought about by the presence of the high-anisotropy Dy(III) centres, while coupled with Cu(II) centres in this aforementioned core, was to be compared with the isostructural Y4Cu8 core, highlighting the role of 3d–4f interactions in the Dy-analogue in exhibiting magnetic properties.
Experimental section
Materials and measurements
All the reagents and solvents were commercially available and were used without further purification. The powder X-ray patterns (PXRD) were recorded on Bruker D8 Advanced X-ray diffractometer at room temperature using Cu Kα radiation (λ = 1.5406 Å). FT-IR spectra were measured on NICOLET 6700 FT-IR Spectrophotometer using KBr pellets.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) of MeOH and MeCN was further added to the aforementioned reaction mixture, following which solid Cu(NO3)2·3H2O (12.1 mg, 0.05 mmol) and 3 ml of 0.03 mmol (13.6 mg) methanolic solution of Dy(NO3)3·xH2O is slowly added while sonicating continuously. Slow evaporation of solvent mixture over the next ten days with no mechanical disturbance, yielded excellent quality blue cube-shaped single crystals of compound [Dy4Cu8(HEP)8(L-PGA)8(OH)8(NO3)4] (1L), suitable for single crystal X-ray analysis. Yield ∼60%. IR (KBr, cm−1): 3644(s), 2845(m), 2719(m), 2407(m), 2086(w), 1709(b), 1428(w), 1291(w), 1234(w), 1160(m), 1088(w), 1033(s), 971(w), 879(s), 779(s), 652(s) (Fig. S2†). Anal. Calcd (found) for C96 H112 Cu8 Dy4 N16 O40: C, 35.06 (35.38); H, 3.43 (2.98); N, 6.82 (6.65)%.
:2) of MeOH and MeCN was further added to the aforementioned reaction mixture, following which solid Cu(NO3)2·3H2O (12.1 mg, 0.05 mmol) and 3 ml of 0.03 mmol (13.6 mg) methanolic solution of Dy(NO3)3·xH2O is slowly added while sonicating continuously. Slow evaporation of solvent mixture over the next ten days with no mechanical disturbance, yielded excellent quality blue cube-shaped single crystals of compound [Dy4Cu8(HEP)8(L-PGA)8(OH)8(NO3)4] (1L), suitable for single crystal X-ray analysis. Yield ∼60%. IR (KBr, cm−1): 3644(s), 2845(m), 2719(m), 2407(m), 2086(w), 1709(b), 1428(w), 1291(w), 1234(w), 1160(m), 1088(w), 1033(s), 971(w), 879(s), 779(s), 652(s) (Fig. S2†). Anal. Calcd (found) for C96 H112 Cu8 Dy4 N16 O40: C, 35.06 (35.38); H, 3.43 (2.98); N, 6.82 (6.65)%.
IR (KBr, cm−1) for 1D: 3634(s), 2663(w), 2720(w), 2401(m), 2083(w), 1701(b), 1426(b), 1292(m), 1159(sh), 1088(w), 1034(s), 968(w) (Fig. S2†). IR (KBr, cm−1) for 2L: 3630(s), 2848(m), 2729(m), 2391(m), 2080(w), 1699(b), 1443(w), 1295(w), 1234(w), 1155(m), 1083(w), 1037(s), 967(w), 880(s), 778(s), 653(s) (Fig. S2†). Anal. Found for 1D (%): C, 35.13; H, 3.11; N, 6.90. Anal. Found for 2L (%): C, 35.29; H, 3.57; N, 6.77.
X-ray structural studies
Single-crystal X-ray data of compound 1L was collected at 200 K on a Bruker KAPPA APEX II CCD Duo diffractometer (operated at 1500 W power: 50 kV, 30 mA) using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å). A cube-shaped blue crystal was mounted using nylon CryoLoops (Hampton Research) with Paratone-N (Hampton Research). The data integration and reduction were processed with SAINT70 software. A multi-scan absorption correction was applied to the collected reflections. The structure was solved by the direct method using SHELXTL71 and was refined on F2 by full-matrix least-squares technique using the SHELXL-9772 program package within the WINGX73 programme. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were located in successive difference Fourier maps and they were treated as riding atoms using SHELXL default parameters. The structures were scrutinized by the Adsym subroutine of PLATON74 to ensure that no additional symmetry could be applied to the models. Crystal data and structure refinement details for complex 1L is summarized in Table S1.†Magnetic measurement details
Magnetic susceptibility measurements were carried out on a Quantum Design MPMS-XL7 SQUID magnetometer equipped with a 7 T magnet. The direct current (dc) measurements were collected with an external magnetic field of 1000 Oe in the temperature range 1.9–300 K, and the alternating-current (ac) measurements were carried out in a 3.0 Oe ac field oscillating at 1000 Hz in the temperature range 2–40 K. The experimental magnetic susceptibility data are corrected for the diamagnetism estimated from Pascal's tables and sample holder calibration.75Results and discussion
Compounds 1L and 2L derived from L-PGA, and 1D derived from D-PGA were prepared at room temperature by slow evaporation of the respective reaction mixtures, as described in the Experimental section. Single crystal X-ray analysis reveals that compound 1L crystallized in the tetragonal space group P4222 with Z = 1. The molecular structure of compound 1L, showing the central square-shaped Dy4 core flanked by four Dy2Cu2 cubanes at each of the central Dy-centres is shown in Fig. 1; the precise arrangement of which is shown in sharper detail in Fig. 2a. The 3d–4f heterometallic dodecahedron (Dy4Cu8) is highly symmetric, since all the cubanes and each of the analogous elements therein are found to have crystallographically identical features (Fig. 2a). The four cubanes are interconnected via the Dy(III) vertices in a typical vertex-sharing arrangement. Interestingly, considering the entire bimetallic central core, only one type of Dy(III): Dy1, one kind of Cu(II): Cu1, and just two different types of O atoms (O1 and O5) constitute the entire assemblage of Dy4Cu8O16, providing testimony to the symmetric attributes of this cluster core. While O1 centers are found to connect the two different metals Dy(III) and Cu(II), O5 centers act as bridging oxo-linkers between the 4f-Dy(III) apices of this bimetallic core.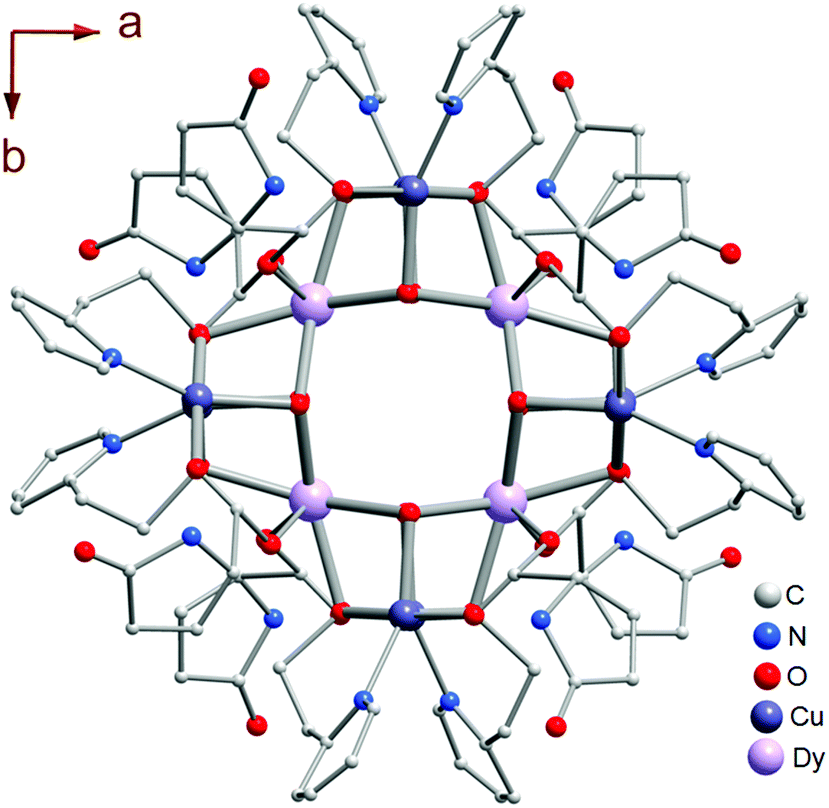 | ||
| Fig. 1 Molecular structure of complex 1L (along crystallographic c-axis), as obtained from SC-XRD analyses presenting a highly symmetric Dy(III)-vertex sharing tetracubane-assemblage based dodecahedral Dy4Cu8 heterometallic cluster core. | ||
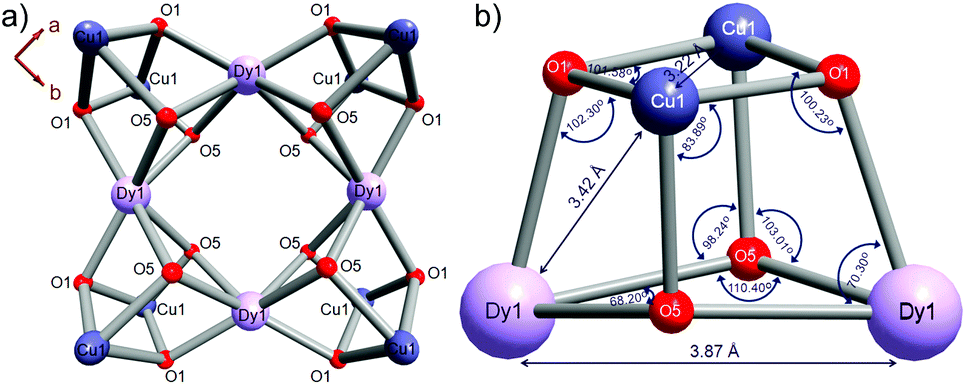 | ||
| Fig. 2 (a) The Dy(III) vertex-sharing arrangement of four Dy2Cu2 cubanes to constitute a Dy4Cu8 dodecahedron architecture, viewed along c-axis; (b) intermetallic distances and bond angles spanned at different vertices of each of the four Dy2Cu2 constituent cubanes of the symmetric heterometallic core. | ||
The bond angles and intermetallic distances spanned across each of the solitary cubane units have been represented in Fig. 2b. Considering the homometallic pairs; while the two larger Dy(III) centers are separated by a distance of 3.87 Å, at the very central core of the motif, the distant Cu(II) centers are in quite close proximity (3.22 Å), resulting in the observed sets of acute and obtuse angles across the metal-vertices, characteristic of a typical cubane. Since the Cu2O2 and Dy2O2 units are apically connected via O-bridges (O1 and O5 respectively), the Cu–Dy closest intermetallic distance (3.42 Å) falls midway in view of the ones between the smaller Cu(II) duo (3.22 Å) and the larger Dy(III) pair (3.87 Å), which is quite in accord with the structural predictions regarding heterometallic cubane species.
Similar PXRD profiles being recorded for two of the analogues unequivocally indicate the isostructural attributes for both the phases 1D and 2L, in comparison to that of 1L (Fig. 3a), while similar IR stretching frequencies and elemental analyses for these merely reinstate the same fact.
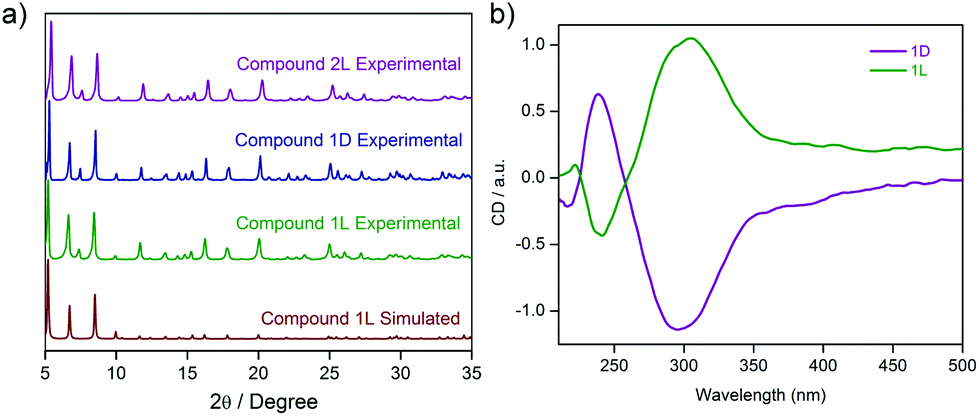 | ||
| Fig. 3 (a) PXRD profiles suggesting a similar nature and phase purity for the three analogous M4Cu8 compounds reported herein, viz. 1L, 1D, and 2L respectively, as compared to the simulated pattern for the L-PGA based cluster 1L; (b) solid-state CD spectra of 1L and 1D; the two chiral clusters synthesized from PGA ligands with different chirality signatures, viz. L-PGA and D-PGA respectively. | ||
Solid state CD spectra for two isostructural complexes (1D and 1L) arising out of different enantiomeric ligands (D and L respectively) are the reverse of each other as predicted (Fig. 3b). Although few in number, some chiral coordination complexes with interesting magnetic properties have been reported in the literature;76–82 however, biomolecule-derived chirality based coordination complexes acting as molecular magnets have not yet been reported.
The direct-current (dc) magnetic susceptibilities of 1L and 2L have been measured in an applied dc magnetic field of 1000 Oe between 300 and 2 K. The plots of χMT versus T, where χM is the molar magnetic susceptibility, are shown in Fig. 4a and 4b. At room temperature, the corresponding χMT values equal to 3.19 and 59.87 cm3 K mol−1 for 2L and 1L respectively (Fig. 4a and 4b) are in agreement with the expected values for eight uncoupled CuII ions (S = 1/2, g = 2, C = 0.375 cm3 K mol−1) for 2L and eight uncoupled CuII ions altogether with four uncoupled DyIII ions (S = 5/2, L = 5, 6H15/2, g = 4/3, C = 14.18 cm3 K mol−1) for 1L in the free-ion approximation.
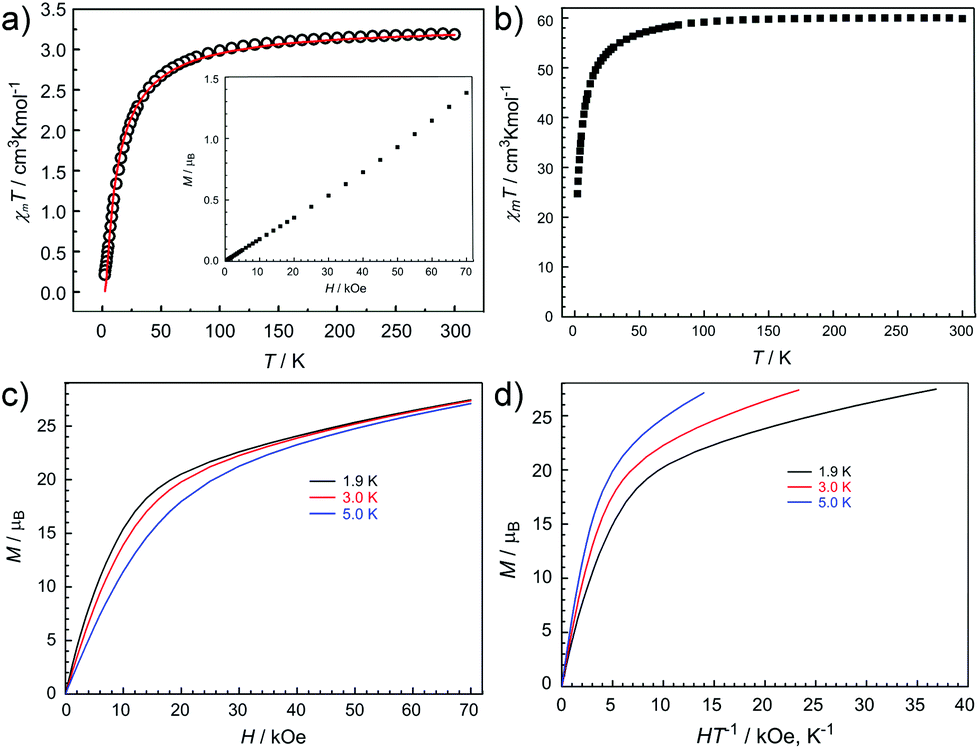 | ||
| Fig. 4 Temperature dependence of χMT in 2L (left; a) and 1L (right; b) under 1000 Oe field, χM being the molar magnetic susceptibility. The red solid line corresponds to the calculated behavior of compound 2L. Inset of a: plot of field dependence of the magnetization (M) of 2L at 2 K; (c) Plots of M vs. H (left) and (d) the reduced magnetization M versus H/T (right) at the indicated temperatures for compound 1L. | ||
For 2L, the χMT value remains unchanged with decreasing temperature until about 100 K, where it sharply decreases to 0.21 cm3 K mol−1 at 2 K. Considering the diamagnetic YIII ion, this decrease means the occurrence of antiferromagnetic interactions among CuII ions. In order to quantify the magnetic coupling, we simulated the magnetic data by using one J coupling parameter. The experimental susceptibility data for 2L was fitted by PHI program83 using the isotropic one-J model and the Hamiltonian Ĥ = −2JŜ1Ŝ2, where J represents the exchange parameter between CuII ions. The fit provides a set of parameters, J = −5.71 cm−1, g = 2.10 and the intermolecular antiferromagnetic interactions zj = −0.5 cm−1. The variable-field magnetization measurement of 2L at 1.8 K is shown in the inset of Fig. 4a. Moreover, the field-dependent magnetization at low temperatures reveals a steady increase approaching the value of 1.4μB for 2L at 70 kOe without saturation (Fig. 4a, inset). The profile of the M vs. H plot confirms the existence of antiferromagnetic interactions within the clusters.
Upon cooling, the χMT value for 1L gradually decreases from 300 to 50 K, subsequently followed by further rapid decline to reach the minimum of 24.70 cm3 K mol−1 at 2 K. This thermal evolution may be ascribed to the depopulation of the Stark sublevels and/or significant magnetic anisotropy present in Dy-containing systems,84,85 and does not preclude very weak antiferromagnetic magnetic interactions between spin carriers.
Magnetization (M) data for 1L were collected in the 0–70 kOe field range below 5 K. The field dependence of the magnetization (M) shows that M increases smoothly with increasing applied dc field without saturation even at 7 T (Fig. 4c), which is ascribed to the anisotropy and the crystal-field effect.86 Furthermore, The non-superimpose M vs. H/T plot (Fig. 4d) also indicates the presence of significant magnetic anisotropy and/or low-lying excited states.
In order to investigate the dynamics of the magnetization, temperature dependence of the in-phase (χ′) and out-of-phase (χ′′) alternating current (ac) susceptibility measurements were carried out at 1000 Hz in zero dc field and 3 Oe ac field for 1L. However, the absence of signals in the out-of-phase magnetic susceptibility operating in a 3.0 Oe ac field and a zero dc field (Fig. 5) indicates the lack of slow magnetic relaxation. This may be ascribed to the fast quantum tunneling phenomena.
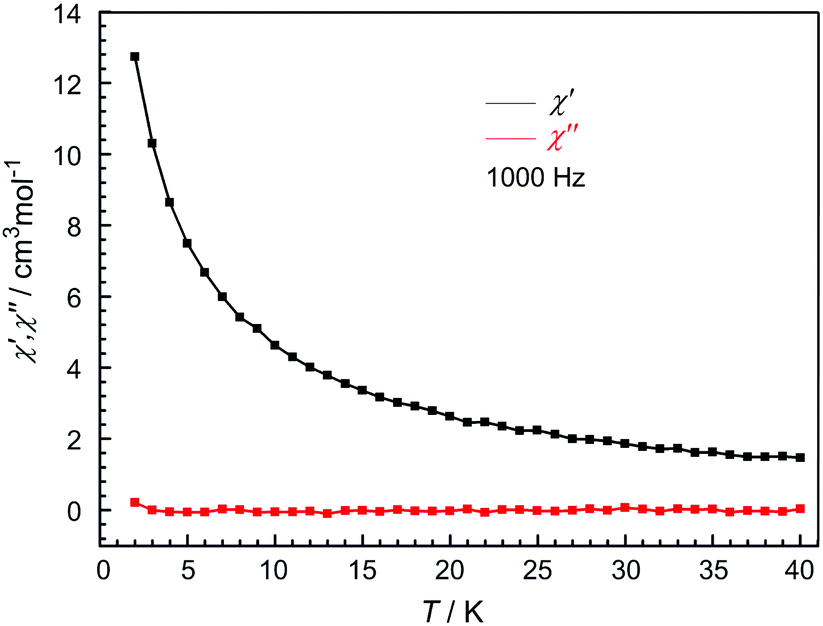 | ||
| Fig. 5 Temperature dependence of in-phase (χ′) (black) and out-of-phase (χ′′) (red) ac susceptibilities of 1L at 1000 Hz in zero dc field and 3 Oe ac field. | ||
Conclusions
In conclusion, magnetic studies on a biomolecule based symmetric chiral M4Cu8 cluster core are reported, which might be further exploited in the future to develop biocompatible magnetic materials from simple inexpensive precursors by coordination chemistry-driven self-assembly guided design principles. Considering the much less-explored domain of biomolecule-based magnetic materials, this report should help to make new inroads for the development of this new class of materials featuring the unique fusion of magnetism and biocompatibility.Acknowledgements
B. J. and S. M. are thankful to CSIR and IISER Pune for senior research fellowships respectively. We are grateful to IISER Pune for research facilities. DST (project no. GAP/DST/CHE-12-0083) is acknowledged for the financial support. DST-FIST (SR/FST/CSII-023/2012) is acknowledged for micro-focus SC-XRD facility.Notes and references
- R. Mukherjee, Coord. Chem. Rev., 2000, 203, 151–218 CrossRef CAS
.
- C. B. Aakeroy, N. Schultheiss, J. Desper and C. Moore, CrystEngComm, 2007, 9, 421–426 RSC
- A. Rajput and R. Mukherjee, Coord. Chem. Rev., 2013, 257, 350–368 CrossRef CAS PubMed
- S.-i. Noro, K. Fukuhara, K. Sugimoto, Y. Hijikata, K. Kubo and T. Nakamura, Dalton Trans., 2013, 42, 11100–11110 RSC
- X.-D. Chen and T. C. W. Mak, Chem. Commun., 2005, 3529–3531, 10.1039/B505919D
- Z. Ma and B. Moulton, Coord. Chem. Rev., 2011, 255, 1623–1641 CrossRef CAS PubMed
- P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502–518 CrossRef CAS
- C.-Y. Su, M. D. Smith and H.-C. zur Loye, Angew. Chem., Int. Ed., 2003, 42, 4085–4089 CrossRef CAS PubMed
- K. Bhar, S. Das, S. Satapathi, P. Mitra, J. Ribas and B. K. Ghosh, Polyhedron, 2010, 29, 2041–2047 CrossRef CAS PubMed
- M. Albrecht and R. Fröhlich, Bull. Chem. Soc. Jpn., 2007, 80, 797–808 CrossRef CAS
- J. Krzystek, J. Telser, M. J. Knapp, D. N. Hendrickson, G. Aromí, G. Christou, A. Angerhofer and L. C. Brunel, Appl. Magn. Reson., 2001, 21, 571–585 CrossRef CAS
- B. Bocquet, G. Bernardinelli, N. Ouali, S. Floquet, F. Renaud, G. Hopfgartner and C. Piguet, Chem. Commun., 2002, 930–931, 10.1039/B201859D
- Y. Zhang, Q. Wang, Y.-J. Xiao, J. Han and X.-L. Zhao, Polyhedron, 2012, 33, 127–136 CrossRef PubMed
- Y.-L. Miao, J.-L. Liu, J.-Y. Li, J.-D. Leng, Y.-C. Ou and M.-L. Tong, Dalton Trans., 2011, 40, 10229–10236 RSC
- S. Sakaue, A. Fuyuhiro, T. Fukuda and N. Ishikawa, Chem. Commun., 2012, 48, 5337–5339 RSC
- X.-L. Mei, R.-N. Liu, C. Wang, P.-P. Yang, L.-C. Li and D.-Z. Liao, Dalton Trans., 2012, 41, 2904–2909 RSC
- M. Du, C.-P. Li, C.-S. Liu and S.-M. Fang, Coord. Chem. Rev., 2013, 257, 1282–1305 CrossRef CAS PubMed
- B. Joarder, S. Mukherjee, S. Xue, J. Tang and S. K. Ghosh, Inorg. Chem., 2014, 53, 7554–7560 CrossRef CAS PubMed
- M. Du, X.-J. Jiang and X.-J. Zhao, Inorg. Chem., 2006, 45, 3998–4006 CrossRef CAS PubMed
- Y.-W. Li, D.-C. Li, J. Xu, H.-G. Hao, S.-N. Wang, J.-M. Dou, T.-L. Hu and X.-H. Bu, Dalton Trans., 2014, 43, 15708–15712 RSC
- D.-M. Chen, N. Xu, X.-H. Qiu and P. Cheng, Cryst. Growth Des., 2015, 15, 961–965 CAS
- A. A. Schilt, J. Am. Chem. Soc., 1960, 82, 3000–3005 CrossRef CAS
- C. Palomo, M. Oiarbide and J. M. García, Chem. – Eur. J., 2002, 8, 36–44 CrossRef CAS
- I. J. Hewitt, J. Tang, N. T. Madhu, C. E. Anson, Y. Lan, J. Luzon, M. Etienne, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2010, 122, 6496–6500 CrossRef PubMed
- S. K. Langley, B. Moubaraki and K. S. Murray, Inorg. Chem., 2012, 51, 3947–3949 CrossRef CAS PubMed
- A. Caneschi, D. Gatteschi, R. Sessoli, A. L. Barra, L. C. Brunel and M. Guillot, J. Am. Chem. Soc., 1991, 113, 5873–5874 CrossRef CAS
- R. Sessoli, H. L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804–1816 CrossRef CAS
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef CAS PubMed
- D. Gatteschi, A. Caneschi, L. Pardi and R. Sessoli, Science, 1994, 265, 1054–1058 CAS
- P. Zhang, L. Zhang and J. Tang, Dalton Trans., 2015, 44, 3923–3929 RSC
- L. Ungur, S.-Y. Lin, J. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef CAS PubMed
- E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419–5448 CrossRef CAS PubMed
- A. Ardavan, O. Rival, J. J. L. Morton, S. J. Blundell, A. M. Tyryshkin, G. A. Timco and R. E. P. Winpenny, Phys. Rev. Lett., 2007, 98, 057201 CrossRef
- N. Roch, S. Florens, V. Bouchiat, W. Wernsdorfer and F. Balestro, Nature, 2008, 453, 633–637 CrossRef CAS PubMed
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed
- M. Mannini, F. Pineider, C. Danieli, F. Totti, L. Sorace, P. Sainctavit, M. A. Arrio, E. Otero, L. Joly, J. C. Cezar, A. Cornia and R. Sessoli, Nature, 2010, 468, 417–421 CrossRef CAS PubMed
- M. Urdampilleta, N.-V. Nguyen, J.-P. Cleuziou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Int. J. Mol. Sci., 2011, 12, 6656–6667 CrossRef CAS PubMed
- J. D. Rinehart, M. Fang, W. J. Evans and J. R. Long, Nat. Chem., 2011, 3, 538–542 CrossRef CAS PubMed
- K. Katoh, H. Isshiki, T. Komeda and M. Yamashita, Chem. – Asian J., 2012, 7, 1154–1169 CrossRef CAS PubMed
- I. Imaz, M. Rubio-Martinez, J. An, I. Sole-Font, N. L. Rosi and D. Maspoch, Chem. Commun., 2011, 47, 7287–7302 RSC
- J. An, S. J. Geib and N. L. Rosi, J. Am. Chem. Soc., 2009, 131, 8376–8377 CrossRef CAS PubMed
- R. Schibli, R. La Bella, R. Alberto, E. Garcia-Garayoa, K. Ortner, U. Abram and P. A. Schubiger, Bioconjugate Chem., 2000, 11, 345–351 CrossRef CAS PubMed
- F. Pu, X. Liu, B. Xu, J. Ren and X. Qu, Chem. – Eur. J., 2012, 18, 4322–4328 CrossRef CAS PubMed
- N. Metzler-Nolte, Angew. Chem., Int. Ed., 2001, 40, 1040–1043 CrossRef CAS
- Y. Wang, C. Zhang, H. Li, G. Zhu, S.-S. Bao, S. Wei, L.-M. Zheng, M. Ren and Z. Xu, J. Mater. Chem. B, 2015, 3, 296–305 RSC
- R. Tashiro and H. Sugiyama, J. Am. Chem. Soc., 2005, 127, 2094–2097 CrossRef CAS PubMed
- A. S. Stephen and J. L. Donald, Smart Mater. Struct., 2011, 20, 094018 CrossRef
-
P. P. Freitas and H. A. Ferreira, in Handbook of Magnetism and Advanced Magnetic Materials, John Wiley & Sons, Ltd, 2007, DOI:10.1002/9780470022184.hmm428
- X. Liu, L. Li, J. Sun, Y. Yan, X. Shu, B. Liu, W. Sha, H. Feng, S. Sun and J. Zhu, Inorg. Chem., 2012, 51, 188–192 CrossRef CAS PubMed
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC
- K. Tanaka and K. Fukase, Org. Biomol. Chem., 2008, 6, 815–828 CAS
- R. Noguchi, A. Hara, A. Sugie and K. Nomiya, Inorg. Chem. Commun., 2006, 9, 355–359 CrossRef CAS PubMed
- B. Joarder, A. K. Chaudhari, S. S. Nagarkar, B. Manna and S. K. Ghosh, Chem. – Eur. J., 2013, 19, 11178–11183 CrossRef CAS PubMed
- R. Vaidhyanathan, C. A. Bridges, D. Bradshaw and M. J. Rosseinsky, Cryst. Growth Des., 2010, 10, 4348–4356 CAS
- P. Espeau, P. Negrier, H. Allouchi and R. Ceolin, Cryst. Growth Des., 2011, 11, 3418–3423 CAS
- R. Noguchi, A. Sugie, Y. Okamoto, A. Hara and K. Nomiya, Bull. Chem. Soc. Jpn., 2005, 78, 1953–1962 CrossRef CAS
-
G. Abraham and D. Podell, in The Biological Effects of Glutamic Acid and Its Derivatives, ed. V. A. Najjar, Springer, Netherlands, 1981, ch. 11, vol. 1, pp. 181–190 Search PubMed
- D. Chelius, K. Jing, A. Lueras, D. S. Rehder, T. M. Dillon, A. Vizel, R. S. Rajan, T. Li, M. J. Treuheit and P. V. Bondarenko, Anal. Chem., 2006, 78, 2370–2376 CrossRef CAS PubMed
- D. B. Liss, M. S. Paden, E. S. Schwarz and M. E. Mullins, Clin. Toxicol., 2013, 51, 817–827 CrossRef CAS PubMed
- T. Goetze, C. Gansau, N. Buske, M. Roeder, P. Görnert and M. Bahr, J. Magn. Magn. Mater., 2002, 252, 399–402 CrossRef CAS
- M. R. Loebinger, P. G. Kyrtatos, M. Turmaine, A. N. Price, Q. Pankhurst, M. F. Lythgoe and S. M. Janes, Cancer Res., 2009, 69, 8862–8867 CrossRef CAS PubMed
- G. Novitchi, W. Wernsdorfer, L. F. Chibotaru, J.-P. Costes, C. E. Anson and A. K. Powell, Angew. Chem., Int. Ed., 2009, 48, 1614–1619 CrossRef CAS PubMed
- A. K. Chaudhari, B. Joarder, E. Rivière, G. Rogez and S. K. Ghosh, Inorg. Chem., 2012, 51, 9159–9161 CrossRef CAS PubMed
- K. Liu, W. Shi and P. Cheng, Coord. Chem. Rev., 2015, 289–290, 74–122 CrossRef CAS PubMed
- X.-H. Miao, S.-D. Han, S.-J. Liu and X.-H. Bu, Chin. Chem. Lett., 2014, 25, 829–834 CrossRef CAS PubMed
- Y.-Z. Ma, L.-M. Zhang, G. Peng, C.-J. Zhao, R.-T. Dong, C.-F. Yang and H. Deng, CrystEngComm, 2014, 16, 667–683 RSC
- J. Rinck, G. Novitchi, W. Van den Heuvel, L. Ungur, Y. Lan, W. Wernsdorfer, C. E. Anson, L. F. Chibotaru and A. K. Powell, Angew. Chem., Int. Ed., 2010, 49, 7583–7587 CrossRef CAS PubMed
- O. Iasco, G. Novitchi, E. Jeanneau, W. Wernsdorfer and D. Luneau, Inorg. Chem., 2011, 50, 7373–7375 CrossRef CAS PubMed
-
SAINT Plus, (Version 7.03), Bruker AXS Inc., Madison, WI, 2004 Search PubMed
-
G. M. Sheldrick, SHELXTL, Reference manual: version 5.1, Bruker AXS, Madison, WI, 1997 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
-
L. Farrugia, WINGX version 1.80.05, University of Glasgow, 2009 Search PubMed
-
A. L. Spek, PLATON, A multipurpose crystallographic tool, Utrecht University, Utrecht, The Netherlands, 2005 Search PubMed
- O. Kahn , molecular magnetism.
- N. Domingo, P. Gerbier, J. Gómez, D. Ruiz-Molina, D. B. Amabilino, J. Tejada and J. Veciana, Polyhedron, 2003, 22, 2355–2358 CrossRef CAS
- M.-L. Sun, J. Zhang, Q.-P. Lin, P.-X. Yin and Y.-G. Yao, Inorg. Chem., 2010, 49, 9257–9264 CrossRef CAS PubMed
- R. Inglis, F. White, S. Piligkos, W. Wernsdorfer, E. K. Brechin and G. S. Papaefstathiou, Chem. Commun., 2011, 47, 3090–3092 RSC
- G. Novitchi, G. Pilet, L. Ungur, V. V. Moshchalkov, W. Wernsdorfer, L. F. Chibotaru, D. Luneau and A. K. Powell, Chem. Sci., 2012, 3, 1169–1176 RSC
- X.-L. Li, C.-L. Chen, Y.-L. Gao, C.-M. Liu, X.-L. Feng, Y.-H. Gui and S.-M. Fang, Chem. – Eur. J., 2012, 18, 14632–14637 CrossRef CAS PubMed
- G. Novitchi, G. Pilet and D. Luneau, C. R. Chim., 2012, 15, 937–942 CrossRef CAS PubMed
- K. Wang, S. Zeng, H. Wang, J. Dou and J. Jiang, Inorg. Chem. Front., 2014, 1, 167–171 RSC
- N. F. Chilton, R. P. Anderson, L. D. Turner, A. Soncini and K. S. Murray, J. Comput. Chem., 2013, 34, 1164–1175 CrossRef CAS PubMed
- M. L. Kahn, J.-P. Sutter, S. Golhen, P. Guionneau, L. Ouahab, O. Kahn and D. Chasseau, J. Am. Chem. Soc., 2000, 122, 3413–3421 CrossRef CAS
- M. L. Kahn, R. Ballou, P. Porcher, O. Kahn and J.-P. Sutter, Chem. – Eur. J., 2002, 8, 525–531 CrossRef CAS
- J. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Ligand structures for L and D enantiomers, IR and EDX data (for metal analyses) for all three reported compounds, along with crystal data and structure refinement for compound 1L. CCDC 1403754 for 1L. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5qi00090d |
| This journal is © the Partner Organisations 2015 |