
Hydrogen-bond disruption in molecularly engineered Janus evaporators for enhanced solar desalination†
Jie
Zhu‡
 a,
Dong
Wu‡
a,
Xiayun
Huang
*a,
Daoyong
Chen
ab and
Zhihong
Nie
*a
a,
Dong
Wu‡
a,
Xiayun
Huang
*a,
Daoyong
Chen
ab and
Zhihong
Nie
*a
aThe State Key Laboratory of Molecular Engineering of Polymers and Department of Macromolecular Science, Fudan University, Shanghai, 200438, People's Republic of China. E-mail: huangxiayun@fudan.edu.cn; znie@fudan.edu.cn
bThe Key Laboratory of Functional Molecular Solids, Ministry of Education, and Department of Materials Chemistry, School of Chemistry and Materials Science, Anhui Normal University, Wuhu, Anhui 214002, People's Republic of China
First published on 12th February 2025
Abstract
Hydrogels have emerged as effective evaporator platforms, significantly enhancing evaporation rates by disrupting water's hydrogen bond network. Here, we present an advanced strategy to improve hydrogel evaporation performance by tailoring alkyl hydrophobic groups within biparental polyelectrolyte-shell micelles grafted onto the polyvinyl alcohol (PVA) hydrogel surface. Poly(2-(diethylamino)ethyl methacrylate) (PDEAEMA) quaternized with methyl iodide (MeI) or ethyl iodide (EtI) formed the biparental polyelectrolyte shell, while poly(benzyl methacrylate) (PBzMA) constituted the micelle core, creating BE-MeI and BE-EtI micelles, respectively. The molecularly engineered BE-MeI micelles exhibited an optimized configuration of quaternary amines linked to hydrophobic groups, achieving a synergistic balance between water attraction via electrostatic interactions and water repulsion via steric hindrance. This configuration effectively disrupted the water's hydrogen bond network, lowering the water evaporation enthalpy to 1434 J g−1. The BE-MeI micelle-grafted PVA hydrogel achieved a record-breaking evaporation rate of 4.1 kg m−2 h−1 under 1 sun irradiation, surpassing prior benchmarks, including our previously reported poly(4-vinyl pyridine) quaternized by a MeI system. Additionally, the grafted micelle layer exhibited a salt rejection ratio of 99.62%, ensuring excellent desalination performance. The biparental polyelectrolyte-shell micelle grafting strategy is broadly applicable across diverse hydrogel systems, representing a significant advancement in solar-driven desalination technology.
1. Introduction
Solar-driven interfacial evaporation technology for desalination provides a sustainable approach for producing fresh water from seawater.1–7 Recent achievements have focused on developing novel materials and structures to enhance the performance of interfacial evaporators by efficiently harnessing solar energy while minimizing heat loss.8–12 Among these, hydrogels, three-dimensional polymer networks, have gained attention as promising evaporator platforms due to their high hydrophilicity, rapid water transport capabilities, tunable structures and properties across multiple scales (from the molecular to the macroscopic level), and their ability to accelerate water evaporation through interactions with water molecules.13–16 For instance, the water evaporation rate using a polyvinyl alcohol (PVA) hydrogel evaporator reaches 2.3 kg m−2 h−1 under 1 sun (1 kW m−2 solar irradiation),17 compared to merely 0.3 kg m−2 h−1 of bulk water.18 This significant enhancement in water evaporation is attributed to the hydrogel's hydrophilic chains, which disrupt the hydrogen bonding network of water, lower the water evaporation enthalpy, and thereby improve water evaporation efficiency.19–22Our recent research has revealed that biparental groups on the polyelectrolyte, specifically quaternized poly(4-vinyl pyridine) with methyl iodide (P4VP-MeI), significantly disrupted hydrogen bonds in water molecules, lowering the energy required for evaporation and raising the evaporation rate.23,24 Technically, we developed an electric-field-driven polyelectrolyte grafting strategy to evenly and stably graft a monolayer of polyelectrolyte-shell micelles onto the hydrogel, allowing their responsive conformational changes under an electric field to entangle with the hydrogel network. This strategy establishes a Janus evaporator with a high-density polyelectrolyte layer on the evaporation surface, effectively lowering the chemical potential and ensuring sufficient water supply. Consequently, a record-breaking seawater evaporation rate was achieved in the non-photothermal evaporator. Specifically, the polyelectrolyte-shell micelle-grafted PVA hydrogel reached an evaporation rate of 3.7 kg m−2 h−1 under 1 sun, surpassing the highest previously reported non-photothermal evaporation rate by 1.5 times.25–28 Additionally, the concentrated polyelectrolyte salt layer acted as an effective barrier to salt ions, achieving an excellent salt rejection ratio. This strategy is universally applicable to various hydrogel systems, including non-photothermal and photothermal hydrogels, demonstrating its broad potential for enhancing solar water evaporation performance.
In this work, we extended the biparental polyelectrolyte system by covalently linking larger alkyl hydrophobic groups to quaternary amines within the polyelectrolyte-shell micelles. We investigated how the size and configuration of these hydrophobic groups disrupt the surrounding water's hydrogen bond network and their influence on solar-driven water evaporation. Specifically, the micelles (BE-MeI or BE-EtI) consist of poly(benzyl methacrylate) (PBzMA) as the core and quaternized poly(2-(diethylamino)ethyl methacrylate) (PDEAEMA) with MeI (or EtI) as a biparental polyelectrolyte shell. Using a previously developed electric-field-driven polyelectrolyte grafting strategy, BE-MeI (or BE-EtI) micelles were evenly and controllably grafted onto PVA hydrogels.23,24 In the BE-MeI micelles, the quaternary amine attracts water molecules via electrostatic interaction, while the large hydrophobic group—composed of two ethyl groups and one methyl group—repels water through hydrophobic steric repulsion. This synergistic effect allows for strong interactions with water, effectively disrupting the surrounding water's hydrogen bonding network. However, enlarging the hydrophobic group size to three ethyl groups (in the BE-MeI system) did not further enhance the water evaporation rate. The substantial steric hindrance increases the separation distance between the polyelectrolyte and water, thereby weakening their interaction. Moreover, a higher shell grafting density introduces more interactive biparental side groups, increasing the number of interaction sites with water. Consequently, the BE-MeI grafted hydrogel, prepared using micelles with a high shell grafting density, demonstrates a lower water evaporation enthalpy of 1434 J g−1 and a record-breaking evaporation rate of 4.1 kg m−2 h−1, compared to the BE-EtI grafted hydrogel (3.7 kg m−2 h−1) with a similar micelle surface coverage and shell grafting density and our biparental P4VP-MeI system previously reported (3.7 kg m−2 h−1).23,24
2. Materials and methods
2.1. Materials
4-Cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPADB, 98%), 2-(dimethylamino)ethyl methacrylate (DEAEMA, 99%), benzyl methacrylate (BzMA, 98%), 2,2-azobis(2-methylpropionitrile) (AIBN, 98%), s-trioxane (99%), polyvinyl alcohol (PVA, Mn = 88![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, alcoholysis degree = 99.8 mol%), methyl iodide (MeI, 98%), ethyl iodide (EtI, 98%), and simulated seawater were purchased from Adamas. The monomers (DEAEMA and BzMA) were purified by passing them through a basic alumina column to remove the inhibitor. The initiator (AIBN) was recrystallized from methanol. Other reagents were used as received.
000 g mol−1, alcoholysis degree = 99.8 mol%), methyl iodide (MeI, 98%), ethyl iodide (EtI, 98%), and simulated seawater were purchased from Adamas. The monomers (DEAEMA and BzMA) were purified by passing them through a basic alumina column to remove the inhibitor. The initiator (AIBN) was recrystallized from methanol. Other reagents were used as received.
2.2. Synthesis of PBzMA-b-PDEAEMA
The diblock copolymer PBzMA-b-PDEAEMA (denoted as BE) was synthesized via reversible addition–fragmentation chain transfer (RAFT) polymerization. DEAEMA (11.7 g, 63 mmol), CPADB (43 mg, 0.15 mmol), AIBN (2.5 mg, 0.015 mmol), s-trioxane (0.285 g, 3.16 mmol), and 1,4-dioxane (38 mL) were sequentially added to a 100 mL flask. After degassing under argon flow, the solution was then heated at 70 °C for 24 h to achieve 38% conversion (confirmed by 1H NMR), yielding the macroinitiator PDEAEMA-CTA (Mn = 29.9 kg mol−1, PDI = 1.07) after precipitation in hexane 3 times. Next, PDEAEMA-CTA (1.5 mg, 0.05 mmol), BzMA (4.4 g, 25 mmol), AIBN (1.64 mg, 0.01 mmol), and s-trioxane (0.11 g, 1.2 mmol) were dissolved in tetrahydrofuran (THF, 13.5 mL) and degassed under an argon flow. The solution was then carried out at 70 °C for 24 h to obtain the PDEAEMA-b-PBzMA-CTA diblock copolymer (Mn = 81.2 kg mol−1 and PDI = 1.18) with 58% conversion. To ensure polymer stability during storage, the CTA of PDEAEMA-b-PBzMA-CTA was removed by heating the polymer solution in DMF with excess AIBN (nAIBN:nPBzMA-b-PDEAEMA-CTA = 50:1) in an 80 °C oil bath for 4 h. After precipitation in hexane 3 times, the white powder of BE with a cleaved chalcogen chain end was obtained, as confirmed by UV-Vis spectroscopy. The synthetic route, 1H NMR, UV-Vis, and GPC results are shown in Fig. S1 (ESI†).
2.3. Self-assembly and quaternization of BE micelles
BE micelles were prepared using a solvent/non-solvent method. First, 6 mg of BE powder was dissolved in 6 mL of chloroform (CHCl3) to form a BE solution. A certain amount of methanol (MeOH) was then added dropwise to the BE solution at a rate of 3 mL h−1. After dialysis against MeOH 3 times, BE micelles in MeOH were obtained. Subsequently, 200 μL of quaternization reagent (MeI or EtI) was added to the BE micelle suspension in MeOH, followed by heating at 40 °C for 48 h. After dialyzing against water 3 times, BE-MeI or BE-EtI micelles in water were obtained.2.4. Grafting micelles to PVA hydrogels
The electric-field assisted micelle surface modification of the PVA hydrogel was performed based on our previously reported technique.23,24 The PVA hydrogel (18 wt%) was first prepared by a solvent displacement method. The hydrogel was then placed between the anode and cathode chambers of an H-shaped cell. A 0.1 mg mL−1 micelle suspension in water was added to the anode chamber, while a 0.001 M KNO3 solution was added to the cathode chamber. A direct current electric field of 10 V cm−1 was applied for varying durations (30, 60, and 90 minutes) using a Keithley 2400 source meter. After the electric field was removed, the micelle-grafted hydrogel was thoroughly rinsed with water.2.5. Instrumentation
Transmission electron microscopy (TEM) was performed with a Hitachi HT7800 in high-contrast mode. Scanning electron microscopy (SEM) was carried out on a Zeiss Ultra55. Proton nuclear magnetic resonance (1H NMR) spectra were recorded on a 600 MHz Bruker AVANCE NEO. Gel permeation chromatography (GPC) measurements in DMF were conducted with an Agilent 1260 system. X-ray photoelectron spectroscopy (XPS) analysis was performed using a Thermo Scientific K-Alpha spectrometer equipped with a monochromatic Al Kα X-ray source. Dynamic/static light scattering (DLS/SLS) measurements were taken using an ALV/CGS-3 laser light scattering spectrometer paired with an ALV/LSE-5004 multi-τ digital time correlator. Solar evaporation rates were measured under ambient conditions (25 °C, 45% humidity) using a custom-built solar evaporation setup consisting of a Newport Oriel Sol3A solar simulator, a Hikvision K20 thermal infrared meter, and a Mettler Toledo AL204 electronic balance. Differential scanning calorimetry (DSC) was performed using a TA Instruments DSC 250. Inductively coupled plasma mass spectrometry (ICP-MS) measurements were conducted with an Agilent 7500ce.3. Results and discussion
3.1. Preparation of BE-MeI micelles with different shell grafting densities
The diblock copolymer PBzMA-b-PDEAEMA (BE) was synthesized following the procedures illustrated in Fig. S1 (ESI†). First, the PDEAEMA160-CTA macroinitiator with a polydispersity index (PDI) of 1.07 was synthesized by RAFT polymerization. The PDEAEMA160-CTA was then chain-extended with BzMA to produce PDEAEMA160-b-PBzMA291-CTA (PDI = 1.18, Mn,TH = 81.2 kg mol−1, Mn,GPC = 76.3 kg mol−1). To assure the polymer's stability during storage, the CTA of PDEAEMA160-b-PBzMA291-CTA was removed by a radical reaction with AIBN as the initiating species, as evidenced by the disappearance of the 306 nm in the UV-Vis spectra (Fig. S1, ESI†).29,30 Afterwards, BE micelles were prepared using a solvent/non-solvent method (Fig. 1a). BE was first molecularly dissolved in the common solvent chloroform (CHCl3). Micelle formation was subsequently introduced by gradually adding methanol (MeOH) to the BE solution. When the MeOH/CHCl3 volume ratio reached 3:1, the micelles were rapidly quenched by adding micelle suspension to excess MeOH. In the micelles, the aggregated PBzMA block chains form the core, while the PDEAEMA block chains form the shell. The shell-forming PDEAEMA block chains were then fully quaternized with methyl iodide (MeI), confirmed by the completely nitrogen peak shift from 398.5 eV (attributed to the nitrogen in the tertiary amine) to 401.5 eV (attributed to the nitrogen in the quaternary amine) (Fig. 1b).31,32 After dialysis against water, highly charged polycationic BE-MeI (denoted as Q1) micelles with a zeta potential of 51 mV were produced (Fig. S2, ESI†). The quaternization of PDEAEMA introduced substantial electrostatic repulsion between charged Q1 micelles, preventing the micelle aggregation during drying in the TEM sample preparation (Fig. 1c). Moreover, both BE and Q1 micelles had a spherical shape and consistent core size of 63 nm (Fig. 1c and Fig. S3, ESI†); the micelles’ shell is of a low contrast and thus invisible in the TEM images. This suggests that quaternization of the shell-forming PDEAEMA block chains does not alter the core size and aggregation number of the micelles. Dynamic and static light scattering measurements show that Q1 micelles have an average hydrodynamic diameter (〈Dh〉) of 125 nm (PDI = 1.13) in water (Fig. 1d) and an average aggregation number (Nagg, number of polymer chains in the micelles derived from the Zimm plot) of 688 (Fig. 1e). Therefore, the inner shell grafting density, representing the density of shell-forming block chains on the micelle's core, was calculated to be 0.055 nm−2, while the outer shell grafting density, representing the density of shell-forming block chains on the micelle's periphery, was calculated to be 0.014 nm−2 (Fig. 1f).
 | ||
| Fig. 1 (a) Schematic illustration of the preparation of PBzMA-b-PDEAEMA (BE) and BE-MeI micelles and the Tyndall effect of BE-MeI micelle suspension in water. (b) N 1s XPS spectra of BE and BE-MeI micelles. (c) TEM images of BE and BE-MeI micelles. (d) DLS traces of BE-MeI micelles with different shell grafting densities (Qn, n = 1–4) in water. (e) Zimm plots of Q1 micelles in water. (f) Inner and outer shell grafting density of Qn (n = 1–4) micelles. | ||
Since MeOH continuously drives PBzMA aggregation during micelle formation, both the Nagg and shell grafting density of micelles increased by gradual addition of MeOH.33,34 As the MeOH/CHCl3 volume ratio increased from 0.5:1 to 1:1, 2:1 and 3:1 (denoted as Q4, Q3, Q2, and Q1, respectively), the core of the micelles increased from 48 nm (Q4) to 58 nm (Q3), 60 nm (Q2), and 63 nm (Q1) (Fig. S4, ESI†). Further increasing the MeOH/CHCl3 volume ratio from 3:1 to 4:1 does not increase micelle core size (Fig. S5, ESI†), indicating that the PBzMA core begins to freeze, which prevents additional polymer chains from entering the micelles. Accordingly, Nagg of micelles increase from 281 (Q4) to 434 (Q3), 521 (Q2), and 688 (Q1) (Fig. 1e and Fig. S6, ESI†). DLS curves showed that 〈Dh〉 of micelles increased from 95 nm (PDI = 1.11, Q4) to 99 nm (PDI = 1.14, Q3), 119 nm (PDI = 1.16, Q2), and 125 nm (PDI = 1.19, Q1) (Fig. 1d). Therefore, the inner shell grafting density increased from 0.039 nm−2 (Q4), 0.041 nm−2 (Q3), 0.046 nm−2 (Q2), and 0.055 nm−2 (Q1), while the outer shell grafting density increased from 0.010 nm−2 (Q4), to 0.011 nm−2 (Q3), 0.012 nm−2 (Q2), and 0.014 nm−2 (Q1), respectively (Fig. 1f).
3.2. Grafting micelles to the hydrogel surface via an electric field
We start with the Q1 micelle with the highest inner and outer shell grafting density (Fig. 1f). Driven by an electric field, the highly charged polycationic Q1 micelles were firmly grafted onto the surface of PVA hydrogel via extensive shell-forming chain entanglement.23,24 Due to the strong electrostatic repulsion between Q1 micelles, a distinct distributed monolayer of micelles stayed on the PVA hydrogel, as observed by the cryo-microtome TEM image (Fig. 2a). By extending the electric field grafting time from 30 min to 90 min, the micelle surface coverage of the resultant micelle-grafted hydrogel (named Q1-X, where X represents for the electric-field grafting time in minutes) continuously increased (Fig. 2b). The average nearest-micelle distance,35,36 the minimum distance between adjacent dispersed micelles, was 124 nm (PDI = 1.42) for Q1-30, 92 nm (PDI = 1.35) for Q1-60, and 68 nm (PDI = 1.28) for Q1-90 (Fig. 2c). As a result, the micelle surface coverage, calculated by dividing the projected micelle area by the hydrogel's surface area, increased from 22.7% (Q1-30), to 51.8% (Q1-60), and 92.9% (Q1-90) (Fig. 2d).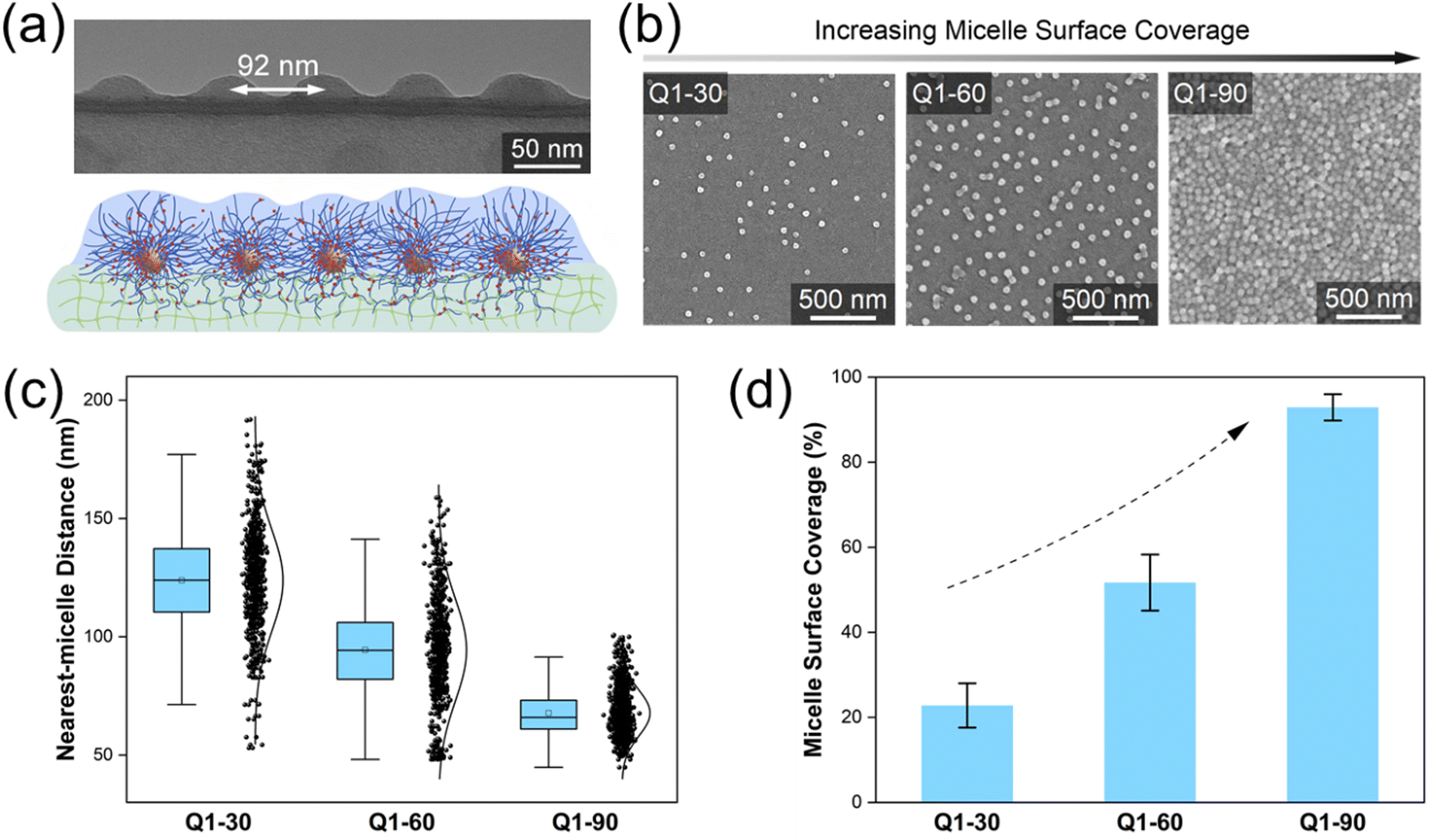 | ||
| Fig. 2 (a) Cross-sectional cryo-microtome TEM image of Q1-60 and the corresponding schematic illustration of a monolayer of micelles on a hydrogel. (b) SEM images of Q1 micelles on a PVA hydrogel surface. The micelle surface coverage increased by extending the electric field grafting time from 30 min to 90 min. (c) Nearest-micelle distance, and (d) micelle surface coverage of Q1-30, Q1-60 and Q1-90. | ||
3.3. Solar water evaporation under 1 sun
To evaluate the solar water evaporation performance, both the PVA hydrogel and micelle-grafted hydrogels floating on water were subjected to solar radiation under 1 sun using the setup shown in Fig. 3a. As water evaporated from the evaporator's surface, the mass change over time for all evaporators exhibited a linear relationship at a rate of 3.1 kg m−2 h−1 for Q1-30, 4.1 kg m−2 h−1 for Q1-60, and 2.4 kg m−2 h−1 for Q1-90, which is significantly higher than those of pure water (0.33 kg m−2 h−1) and the PVA hydrogel (1.5 kg m−2 h−1) (Fig. 3b). Q1-60 with a micelle surface coverage of ∼50% showed the lowest equilibrium temperature (33.5 °C) at the hydrogel surface and highest water evaporation rate during solar water evaporation (Fig. 3b and c). The lowest equilibrium temperature was attributed to the rapid evaporation of surface water molecules to effectively dissipate localized heat. Accordingly, Q1-60 demonstrated the highest evaporation efficiency of 79%, as estimated by conventional dark room experiments (Fig. 3d and Note S1, Fig. S7, ESI†). As the micelle surface coverage increased, the surface area expanded significantly by 42% for Q1-30, 125% for Q1-60, and 208% for Q1-90 compared to the PVA hydrogel surface (Fig. 3d). This increase provides a larger surface area beneficial for solar water evaporation. However, despite Q1-90 exhibiting the highest surface area, its evaporation rate and efficiency were lower than those of Q1-60. This reduction is attributed to the high surface coverage of PBzMA core's hydrophobic regions (92% for Q1-90), which restricted adequate water supply to the surface. | ||
| Fig. 3 (a) Schematic illustration of the solar water evaporation system. (b) The mass change of water over time. (c) The equilibrium temperature at the hydrogel surface and bulk water. Insert images are the IR images of the equilibrium temperature of Q1-60. (d) The surface area enhancement and evaporation efficiency of Q1-30, Q1-60 and Q1-90. (e) Differential scanning calorimetry (DSC) curves showing the fitting peaks representing IW and FW in Q1-30, Q1-60 and Q1-90. (f) The water evaporation enthalpy of water in Q1-30, Q1-60 and Q1-90. | ||
According to the literature,13 intermediate water (IW), produced by disrupting a certain number of free water (FW)'s hydrogen bond networks, evaporates more efficiently, and a higher IW/FW ratio is always associated with a faster evaporation rate. In the polyelectrolyte, quaternary amine attracts water molecules through electrostatic interaction, whereas the large hydrophobic group—comprising two ethyl groups and one methyl group—repels water via hydrophobic steric repulsion. The high density polyelectrolyte, radially distributed on the shell, adopted a stretched conformation via electrostatic repulsion between the polyelectrolyte chains. This structures ensures adequate exposure of biparental side groups to water, enhancing interaction and water adsorption efficiency.23,24 As the micelle surface coverage increased, the IW/FW ratio increased from 1.6 (Q1-30, ∼20%) to 3.7 (Q1-60, ∼50%) (Fig. 2d and 3e). Further increasing the micelle surface coverage to 90% results in the formation of the overlapping polyelectrolyte chains between adjacent micelles. Although the concentrated polyelectrolyte layer rapidly absorbs water from the hydrogel to the surface, the overlapping polyelectrolyte chains between adjacent micelles hinder their extensive interaction with water. This reduces the IW/FW ratio and water evaporation enthalpy, ultimately limiting the evaporation rate (Fig. 3e). Consequently, Q1-60 exhibited the lowest water evaporation enthalpy of 1434 J g−1, significantly lower than that of PVA (2200 J g−1) (Fig. 3f). Therefore, an optimized micelle surface coverage of ∼50% maximizes the solar water evaporation rate and efficiency by effectively disrupting hydrogen bonds in the water network.
To investigate the effect of micelles’ polyelectrolyte shell grafting density on solar water evaporation, BE-MeI micelles with varying shell grafting densities (Q1, Q2, Q3, and Q4) were grafted onto the hydrogels at a fixed grafting time of 60 minutes to achieve a consistent micelle surface coverage of ∼50% (Fig. 4a and b). Accordingly, the surface area enhancement for Q1-60 to Q4-60 is 125%, 118%, 112%, and 110%, which are close to the averaged value of 116% (Fig. 4b). Since the polyelectrolyte shell-forming chains extend radially outward from the core, the inner shell grafting density represents the highest local polyelectrolyte salt density, which is crucial for facilitating efficient water supply. In the BE-MeI micelles, Q1 exhibits the highest inner shell grafting density of 0.055 nm−2. As mentioned earlier, the biparental polyelectrolytes in the micelles’ shell adopted a stretched conformation, ensuring adequate exposure of biparental side groups to water. A higher shell grafting density introduces more interactive biparental side groups, increasing the number of interaction sites with water. This enhanced number of interactions effectively disrupts hydrogen bonds in the water network. Consequently, Q1, with the highest number of interactive sites, enabled Q1-60 to achieve the highest evaporation rate (4.1 kg m−2 h−1), the highest IW/FW ratio (3.7) and lowest water evaporation enthalpy (1434 J g−1) (Fig. 4c–e).
 | ||
| Fig. 4 (a) SEM images of Qn (n = 1–4) micelles on the PVA hydrogel surface. The electric field grafting time was fixed at 60 min. (b) The micelle surface coverage and chain grafting density of Qn-60 (n = 1–4). (c) The evaporation rate of micelle-grafted hydrogel evaporators by varying shell grafting density of micelles. (d) DSC curves showing the fitting peaks representing IW and FW in Qn-60 (n = 1–4). (e) The water evaporation enthalpy of water in Q1-60, Q2-60, Q3-60 and Q4-60. | ||
To understand the superior efficiency of BE-MeI in disrupting the hydrogen bonds in the water network, we synthesized three types of micelles: BE (with a pKa of 7.0–7.5,37 partially quaternized in water), BE-MeI (fully quaternized with MeI), and BE-EtI (fully quaternized with EtI). These micelles all have the same grafting density as Q1, which is equivalent to BE-MeI in this context. The molecular states of water, BE, BE-MeI, and BE-EtI in pure water systems, were analysed using molecular dynamics (MD) simulations (Fig. 5a and Note S2, ESI†). For simplicity, the simulation focused solely on the interaction between the polyelectrolyte chain and water. Compared to pure water, the average number of hydrogen bonds (H-bond) decreased significantly by the introduction of polymer chains. Quaternizing the tertiary amine groups in PDMAEMA with MeI (BE-MeI) or EtI (BE-EtI) further enhanced hydrogen bond disruption through a synergistic effect: the quaternary amine attracts water molecules electrostatically, while the hydrophobic groups that covalently linked to the amine cause steric hindrance, repelling water molecules. This combined disruption reduced the average number of H-bonds to 0.93 per water molecule (Fig. 5b). Consequently, the kinetic energy of water molecules, reflected by the diffusion coefficient, increased. The diffusion coefficient of water molecules increased from 2.6 × 10−9 m2 s−1 in pure water to 3.1 × 10−9 m2 s−1 in the BE system, and further to ∼8.4 × 10−9 m2 s−1 in the BE-MeI and BE-EtI systems (Fig. 5c and Fig. S8, ESI†).
 | ||
| Fig. 5 (a) A simulation snapshot of BE-MeI in water. (b) Average number of hydrogen bonds and (c) diffusion coefficient of water molecules in four systems: water to water/BE/BE-MeI/BE-EtI. (d) Optimized molecular structures showing interactions between water and water/BE/BE-MeI/BE-EtI, along with the corresponding interaction energies. (e) Raman spectra of water, BE-MeI micelles, and BE-EtI micelles in water at a concentration of 0.1 mg mL−1. (f) IW/FW ratio, (g) water evaporation enthalpy of water shown in DSC curves, and (h) evaporation rate of BE, BE-MeI, and BE-EtI micelles grafted PVA hydrogel (electric field grafting time is fixed at 60 min). | ||
Furthermore, the interaction energy between the polyelectrolyte and water was calculated (with more negative values indicating stronger interactions) to explore the influence of the size and configuration of the alkyl groups on the quaternized PDEAEMA. The interaction energies between water–water and BE–water were −6.26 kcal mol−1 and −5.87 kcal mol−1, respectively. Upon quaternization, the interaction between the polymer and water was significantly enhanced due to the electrostatic interaction between the N+ in the quaternary amine and water molecules (Fig. 5d). In the BE-MeI system, the N+ in the quaternary amine is linked to the hydrophobic group consisting of two ethyl groups and one methyl group, exhibiting the strongest interaction with water, with an interaction energy of −14.99 kcal mol−1. When the hydrophobic group consists of three ethyl groups (as in the BE-EtI system), the interaction between the polyelectrolyte and water (−12.28 kcal mol−1) is weakened. This is because the configuration of three ethyl groups hinders the N+–water interaction, as evidenced by the longer interaction distance of 5.98 Å in the BE-EtI system, compared to 4.19 Å in the BE-MeI system. Stronger interactions between the polyelectrolyte and water (BE-MeI system) suggest a greater capacity for disrupting hydrogen bonds in water. The Raman peaks at 3233 cm−1 and 3401 cm−1 correspond to strongly hydrogen-bonded water molecules, while those at 3514 cm−1 and 3630 cm−1 are associated with weakly hydrogen-bonded water molecules.13 As shown in Fig. 5e, the content of weakly hydrogen-bonded water molecules increased by ∼95% when BE-MeI micelles were introduced to water, indicating that the hydrogen bonds in water were significantly disrupted. When the BE-MeI micelles were replaced with BE-EtI micelles, the content of weakly hydrogen-bonded water molecules decreased by ∼40%, suggesting a reduction in hydrogen-bond disruption. Accordingly, the BE-MeI system exhibited a higher IW/FW ratio and a lower water evaporation enthalpy, when the micelles were grafted onto the hydrogel (Fig. 5f and g). As a result, the BE-MeI system (Q1-60) has a higher water evaporation rate of 4.1 kg m−2 h−1 (Fig. 5h).
3.4. Durability of the evaporator
We further evaluated the durability of the Q1-60 evaporator in simulated seawater. When Q1-60 was exposed to the simulated seawater for continuous 24-hour evaporation under 1 sun, all the mass change over time curves demonstrated a good linearity; the 1st and 24th hour mass change over time curves are shown in Fig. 6a (inert image). The evaporation rate consistently maintained at a high value with slight fluctuation over time—from 4.0 kg m−2 h−1 in the 1st hour to 3.9 kg m−2 h−1 by the 24th hour—indicating potential for long-term desalination (Fig. 6a). Consistent with previous studies, Q1-60 displayed excellent salt rejection capabilities via a concentrated quaternized PDEAEMA salt barrier, as evidenced by a two-order-of-magnitude decrease in the concentrations of four primary ions in simulated seawater (Na+, Mg2+, K+, and Ca2+) (Fig. 6b). The ion rejection ratio, calculated as the total ion concentration in the desalinated water divided by that in the simulated seawater, showed only a slight decrease from 99.62% in the 1st hour to 99.50% by the 24th hour, both of which meet international drinking standards.38 Additionally, a 5-day-night cycle evaporation test in simulated seawater was conducted to confirm the evaporator's stability under simulated day-night cycles under 1 sun for 9 hours each day. As shown in Fig. 6c and d, the evaporation rate of Q1-60 remained above 3.8 kg m−2 h−1, with a total daily evaporation rate approaching 35 kg m−2 day−1, while the ion rejection ratio remained above 99.49%, demonstrating excellent cycling evaporation performance. | ||
| Fig. 6 (a) The evaporation rate of Q1-60 during 24 h evaporation test in simulated seawater under 1 sun and (b) the salinity of desalination after 1 h and 24 h evaporation test. (c) Mass change curves, evaporation rate and (d) ion rejection ratio of Q1-60 in simulated seawater during the 5-day-night cycling experiment, with 9 hours of exposure to 1 sun every day. | ||
4. Conclusions
We have prepared two biparental polyelectrolyte-shell micelle-grafted hydrogel systems by covalently linking larger alkyl hydrophobic groups of varying sizes and configurations to quaternary amines within the polyelectrolyte shell: (1) quaternary amine linked to two ethyl groups and one methyl group (BE-MeI system), and (2) quaternary amine linked to three ethyl groups (BE-EtI system). Among these, the BE-MeI micelles exhibited an optimized size and configuration, achieving a synergistic balance between water attraction via electrostatic interactions and water repulsion through steric hindrance. This synergistic effect allows for strong interactions with water, effectively disrupting the surrounding water's hydrogen bonding network, lowering the water evaporation enthalpy to 1434 J g−1. Conversely, enlarging the hydrophobic group to three ethyl groups introduced significant steric hindrance, increasing the separation distance between the polyelectrolyte and water, weakening their interaction and raising the water evaporation enthalpy. As a result, the BE-MeI micelle-grafted hydrogel system demonstrated superior performance, achieving an exceptional evaporation rate of 4.1 kg m−2 h−1 under 1 sun irradiation with a 99.62% salt rejection ratio. This biparental polyelectrolyte-shell micelle grafting strategy is broadly applicable across diverse hydrogel systems, representing a significant advancement in solar-driven desalination technology.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors are grateful for the financial support from the National Natural Science Foundation of China (52125308, 52293473, 22071032, and 51721002) and the MOST (2022YFA1203001). The authors also thank Prof. Huisheng Peng's group for their support with the solar simulator and Ms Yi Feng for his help in microtome sample preparation.References
- J. Lord, A. Thomas, N. Treat, M. Forkin, R. Bain, P. Dulac, C. H. Behroozi, T. Mamutov, J. Fongheiser, N. Kobilansky, S. Washburn, C. Truesdell, C. Lee and P. H. Schmaelzle, Nature, 2021, 598, 611–617 CrossRef PubMed.
- Y. Song, S. Fang, N. Xu, M. Wang, S. Chen, J. Chen, B. Mi and J. Zhu, Science, 2024, 385, 1444–1449 CrossRef CAS PubMed.
- H. Li, W. Zhang, X. Liao and L. Xu, Sci. Adv., 2024, 10, eado1019 CrossRef.
- Q. Xia, Y. Pan, B. Liu, X. Zhang, E. Li, T. Shen, S. Li, N. Xu, J. Ding, C. Wang, C. D. Vecitis and G. Gao, Sci. Adv., 2024, 10, eadj3760 CrossRef CAS PubMed.
- S. Li, J. Liu, C. Li, X. Fang, X. Wang, L. Tian, Z.-Z. Yu and X. Li, ACS Appl. Mater. Interfaces, 2024, 16, 66560–66570 CrossRef CAS.
- M.-Y. Yu, J. Wu, G. Yin, F.-Z. Jiao, Z.-Z. Yu and J. Qu, Nano-Micro Lett., 2024, 17, 48 CrossRef.
- T. Zhang, J. Qu, J. Wu, F.-Z. Jiao, C. Li, F.-L. Gao, J. Liu, Z.-Z. Yu and X. Li, Adv. Funct. Mater., 2024, 34, 2403505 CrossRef CAS.
- P. Tao, G. Ni, C. Song, W. Shang, J. Wu, J. Zhu, G. Chen and T. Deng, Nat. Energy, 2018, 3, 1031–1041 CrossRef.
- B. Yang, Z. Zhang, P. Liu, X. Fu, J. Wang, Y. Cao, R. Tang, X. Du, W. Chen, S. Li, H. Yan, Z. Li, X. Zhao, G. Qin, X.-Q. Chen and L. Zuo, Nature, 2023, 622, 499–506 CrossRef CAS PubMed.
- C. Dang, Y. Cao, H. Nie, W. Lang, J. Zhang, G. Xu and M. Zhu, Nat. Water, 2024, 2, 115–126 CrossRef CAS.
- Y. Xu, J. Xu, J. Zhang, X. Li, B. Fu, C. Song, W. Shang, P. Tao and T. Deng, Nano Energy, 2022, 93, 106882 CrossRef CAS.
- M. Jiang, Q. Shen, J. Zhang, S. An, S. Ma, P. Tao, C. Song, B. Fu, J. Wang, T. Deng and W. Shang, Adv. Funct. Mater., 2020, 30, 1910481 CrossRef CAS.
- X. Zhou, F. Zhao, Y. Guo, B. Rosenberger and G. Yu, Sci. Adv., 2019, 5, eaaw5484 CrossRef CAS.
- C. Li, S. Cao, J. Lutzki, J. Yang, T. Konegger, F. Kleitz and A. Thomas, J. Am. Chem. Soc., 2022, 144, 3083–3090 CrossRef CAS.
- F. Zhao, Y. Guo, X. Zhou, W. Shi and G. Yu, Nat. Rev. Mater., 2020, 5, 388–401 CrossRef.
- N. Xu, J. Li, C. Finnerty, Y. Song, L. Zhou, B. Zhu, P. Wang, B. Mi and J. Zhu, Nat. Water, 2023, 1, 494–501 CrossRef.
- H. Li, W. Zhang, J. Liu, M. Sun, L. Wang and L. Xu, Adv. Funct. Mater., 2023, 33, 2308492 CrossRef CAS.
- Z. Sun, C. Han, S. Gao, Z. Li, M. Jing, H. Yu and Z. Wang, Nat. Commun., 2022, 13, 5077 CrossRef CAS PubMed.
- X. Zhou, Y. Guo, F. Zhao and G. Yu, Acc. Chem. Res., 2019, 52, 3244–3253 CrossRef CAS PubMed.
- Y. Tu, J. Zhou, S. Lin, M. Alshrah, X. Zhao and G. Chen, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2312751120 CrossRef CAS PubMed.
- X. Zhou, F. Zhao, Y. Guo, Y. Zhang and G. Yu, Energy Environ. Sci., 2018, 11, 1985–1992 RSC.
- A. R. Pati, Y.-S. Ko, C. Bae, I. Choi, Y. J. Heo and C. Lee, Soft Matter, 2024, 20, 4988–4997 RSC.
- J. Zhu, X. Zhao, N. Wu, D. Wu, X. Huang, Z. Nie and D. Chen, Macromolecules, 2024, 57, 9811–9822 CrossRef CAS.
- J. Zhu, Z. Xiao, F. Song, X. Huang, D. Chen and Z. Nie, J. Mater. Chem. A, 2024, 12, 17142–17150 RSC.
- X. Lin, P. Wang, R. Hong, X. Zhu, Y. Liu, X. Pan, X. Qiu and Y. Qin, Adv. Funct. Mater., 2022, 32, 2209262 CrossRef CAS.
- P. Liu, Y.-b Hu, X.-Y. Li, L. Xu, C. Chen, B. Yuan and M.-L. Fu, Angew. Chem., Int. Ed., 2022, 61, e202208587 CrossRef CAS PubMed.
- X. Hao, H. Yao, P. Zhang, Q. Liao, K. Zhu, J. Chang, H. Cheng, J. Yuan and L. Qu, Nat. Water, 2023, 1, 982–991 CrossRef CAS.
- H. Liang, Y. Mu, M. Yin, P.-P. He and W. Guo, Sci. Adv., 2023, 9, eadj1677 CrossRef CAS.
- S. Perrier, P. Takolpuckdee and C. A. Mars, Macromolecules, 2005, 38, 2033–2036 CrossRef CAS.
- Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2007, 40, 4446–4455 CrossRef CAS.
- J. S. Stevens, S. J. Byard, C. C. Seaton, G. Sadiq, R. J. Davey and S. L. M. Schroeder, Phys. Chem. Chem. Phys., 2014, 16, 1150–1160 RSC.
- X. Du, S.-Y. Wang, M. Wei, J.-R. Zhang, G. Ge and W. Hua, Phys. Chem. Chem. Phys., 2022, 24, 8196–8207 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS.
- H. Shen and A. Eisenberg, J. Phys. Chem. B, 1999, 103, 9473–9487 CrossRef CAS.
- X. Zhao, D. Li, J. Zhu, Y. Fan, J. Xu, X. Huang, Z. Nie and D. Chen, ACS Macro Lett., 2024, 13, 882–888 CrossRef CAS PubMed.
- J. Zhu, N. Wu, X. Zhao, Y. Fan, X. Huang and D. Chen, Acta Polym. Sin., 2024, 55, 900 CAS.
- C. Yang, J. Xiao, W. Xiao, W. Lin, J. Chen, Q. Chen, L. Zhang, C. Zhang and J. Guo, RSC Adv., 2017, 7, 27564–27573 RSC.
- I. Martinez-Morata, B. C. Bostick, O. Conroy-Ben, D. T. Duncan, M. R. Jones, M. Spaur, K. P. Patterson, S. J. Prins, A. Navas-Acien and A. E. Nigra, Nat. Commun., 2022, 13, 7461 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sm01484g |
| ‡ J. Z. and D. W. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |