DOI:
10.1039/D4SM01325E
(Paper)
Soft Matter, 2025,
21, 2124-2132
Stereoisomerism-dependent gelation and crystal structures of glycosylated N-methylbromomaleimide-based supramolecular hydrogels†
Received
9th November 2024
, Accepted 8th February 2025
First published on 12th February 2025
Abstract
In this study, we developed glycosylated N-methylbromomaleimide-based supramolecular hydrogels exhibiting colour change along with gel–sol transition and found that the stereoisomerism of the saccharide residue affects their gelation ability. Single-crystal X-ray diffraction analysis revealed the molecular packing and hydrogen-bonding networks contributed by the saccharide residues. Interestingly, it was found that water molecules were incorporated into the hydrogen-bonding network in the crystals of the compounds that showed gelation ability.
Introduction
Low molecular weight (LMW) supramolecular hydrogels1 formed by self-assembly of LMW amphiphiles (gelators) have attracted increased interest due to their high responsiveness to external stimuli, such as pH,2 chemical/biological additives3 and temperature.4 The high stimulus responsiveness due to the weak non-covalent interactions (e.g. hydrogen-bonding, π–π stacking and hydrophobic interactions) enables the self-assembly of LMW gelators and, therefore, fine-tuning of the self-assembled structures and non-covalent interactions of LMW gelators is of primary importance. As user-friendly sensing materials that do not require special equipment, LMW hydrogels that exhibit colour change (i.e. chromism) in response to specific external stimuli are useful. However, the number of reports on chromic supramolecular hydrogels is still limited, except for the appearance of a visible absorption band upon complexation with transition metal cations.5,6 Our group focuses on supramolecular hydrogel systems based on N-alkyl-2-anilino-3-halogenomaleimide-type chromophores, whose colour changes depending on the molecular assembly state.6 For example, glycolipid-type gelators such as glycosylated N-alkyl-2-anilino-3-chloromaleimide (MS-AAC-C11-COOH (MS (monosaccharide) = α/β-D-glucose (Glc), α/β-D-galactose (Gal) or α-D-mannose (Man)); Fig. 1A) exhibited chromism from yellow (hydrogel state) to orange (solution state) upon gel–sol transition.6a The hypsochromic shift of the absorption in the hydrogel state compared with the relevant solution (sol) state indicates that the MS-AAC moieties in the gelators were stacked in an H-type arrangement.7 In our chromic hydrogel system, the efficient induction of the gelator stacking and self-assembly is an important process. The main factor determining the self-assembly ability of glycolipid-type gelators is the stereoisomerism (epimeric and anomeric isomers) of the saccharide moiety.8 The multiple hydroxy groups in the saccharide moiety act as hydrogen-bonding sites, forming a unique hydrogen-bonding network that significantly affects gelation ability and gel stability. Therefore, the elucidation of the hydrogen-bonding network pattern contributed by saccharide residues is expected to provide a useful guide for the molecular design of gelators.
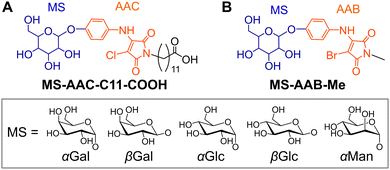 |
| | Fig. 1 Chemical structures of glycolipid-type amphiphiles. (A) MS-AAC-C11-COOH (previous work)6a and (B) MS-AAB-Me (this work). | |
In this study, novel glycolipid-type amphiphiles MS-AAB-Me with a glycosylated N-alkyl-2-anilino-3-bromomaleimide (AAB)-based chromophore (Fig. 1B) were designed and synthesised, and the effects of the stereoisomerism of the saccharide moiety on the gelation ability with colour change behaviour were investigated. Furthermore, single crystals of several glycolipids, αGal-AAB-Me, αGlc-AAB-Me and βGlc-AAB-Me, were obtained from aqueous media, and their crystal structures were successfully elucidated by single-crystal X-ray diffraction (XRD) analysis. The molecular packing with hydrogen-bonding networks of a LMW gelator is quite important for the design of LMW gelators.9 There is, however, a trade-off relationship between gelation and crystallisation abilities. Therefore, the single-crystal XRD analysis of the packing mode and hydrogen-bonding networks of LMW gelators is generally difficult, and only one single-crystal XRD analysis of a glycolipid-type gelator has been reported by Hamachi and co-workers.9a The obtained crystal structures of the LMW gelators, in which the saccharide moiety contributes to the hydrogen-bonding networks, are informative for the design of the glycolipid-type LMW gelators.
Results and discussion
Molecular design and synthesis of gelators
MS-AAB-Me amphiphiles (Fig. 1B) possessing a hydrophobic core consisting of an AAB-based chromophore and a hydrophilic monosaccharide (MS) moiety were designed. In our previous study, all the MS-AAC-C11-COOH amphiphiles (Fig. 1A) formed supramolecular hydrogels, and the stereoisomerism of the saccharide moiety had little effect on the gelation ability.6a This phenomenon was thought to be due to the participation of both the carboxy (COOH) group at the end of the flexible C11 chain and the saccharide moiety in the hydrogen-bonding network. In this study, we therefore replaced the COOH-terminated C11 chain with a short methyl (Me) group to reduce the flexibility and hydrophilicity of the N-alkyl moiety. In addition, we expected that the introduction of the Me group would increase the fraction of the saccharide moiety in the molecule, thereby enhancing the effect of the stereoisomerism of the saccharide moiety on the gelation ability. We also changed the halogen species of the chromophore from chlorine to bromine to promote stronger halogen-bonding interactions between the bromomaleimide moieties.10 The molecular library was obtained from 2,3-dibromo-N-methylmaleimide (1) and the corresponding 4-aminophenyl pyranoside in good yields, as shown in Scheme 1.
 |
| | Scheme 1 Synthesis of MS-AAB-Me. | |
Gelation ability and gel stability
The gelation ability, critical gelation concentrations (CGCs) and gel-to-sol phase transition temperature (Tgel) of MS-AAB-Me in 200 mM HEPES–KOH buffer (pH 6.7) were investigated using the tube inversion method11 (Table 1, Fig. 2 and Fig. S1, ESI†). The gelation ability can be influenced by the pH and salt strength. Here, HEPES–KOH buffer which is one of Good's buffers with a neutral pH was used. αGal-AAB-Me, βGal-AAB-Me and βGlc-AAB-Me formed stable hydrogels above the CGC. In particular, βGlc-AAB-Me showed a high gelation ability (CGC = 0.32 wt%). The transparency and colour of the obtained gels were slightly different from each other (Fig. 2 and Fig. S2, ESI†). The αGal-AAB-Me and βGlc-AAB-Me hydrogels were turbid and bright yellow (λmax = 409 nm and 405 nm, respectively), whereas the βGal-AAB-Me hydrogel was transparent and orange (λmax = 411 nm with a shoulder at 460 nm, Fig. S2B, ESI†). The colour difference indicates a difference in molecular packing. In addition, the βGlc-AAB-Me hydrogel also had higher thermal stability (Tgel = 81 °C) than αGal-AAB-Me and βGal-AAB-Me hydrogels (Tgel = 76 and 54 °C, respectively) at the same concentration (2.2 wt%). The gelation and chromism of the hydrogels were thermally reversible (Fig. 3, typically for βGlc-AAB-Me). Conversely, αGlc-AAB-Me and αMan-AAB-Me underwent precipitation. These results suggest that the gelation ability is related to the epimeric and anomeric isomerism of the molecules, induced by a difference in the orientation of the hydroxy groups. In addition to hydrogen-bonding, hydrophobic interactions contribute to the self-assembly ability of the molecule. The hydrophobicity of unprotected monosaccharides, based on their solubility in water decreases in the order of Gal > Glc > Man.12 In practice, αMan-AAB-Me, which has the most hydrophilic saccharide residue, exhibited high solubility and hardly precipitated even at high concentration (<0.80 wt%) compared to its Gal or Glc counterparts (0.25, 0.49, 0.26 and 0.20 wt% for αGal-AAB-Me, βGal-AAB-Me, αGlc-AAB-Me and βGlc-AAB-Me, respectively). These results suggest that the hydrogen-bonding networks and the hydrophobic–hydrophilic balance of the saccharide moiety contribute to the molecular aggregation ability. Furthermore, the compounds bearing β-anomers exhibited higher gelation ability than the corresponding α-anomers with the same epimeric saccharide (Gal and Glc). The theory that the hydrogen-bonding mode of the β-configuration is preferred during the molecular stacking process supports these results.8,13
Table 1 Gelation ability (G: gel, P: precipitation), critical gelation concentrations (CGCs), absorption maxima (λmax) at room temperature (around 23 °C) and gel-to-sol phase transition temperature (Tgel) of MS-AAB-Me. Conditions: 200 mM HEPES–KOH buffer (pH 6.7)
| Entry |
Compound |
|
CGC [wt%] |
T
gel
[°C] |
λ
max
,
[nm] |
|
[MS-AAB-Me] = 2.2 wt%.
Fig. S2, ESI.
Not measurable due to sample inhomogeneity.
|
| 1 |
αGal-AAB-Me
|
G |
1.7 |
76 |
409 |
| 2 |
βGal-AAB-Me
|
G |
1.4 |
54 |
411 |
| 3 |
αGlc-AAB-Me
|
P |
— |
— |
—c |
| 4 |
βGlc-AAB-Me
|
G |
0.32 |
81 |
405 |
| 5 |
αMan-AAB-Me
|
P |
— |
— |
—c |
 |
| | Fig. 2 Photographs of MS-AAB-Me. Condition: [αGal-AAB-Me] = 1.9 wt%, [βGal-AAB-Me] = 1.9 wt%, [αGlc-AAB-Me] = 2.0 wt%, [βGlc-AAB-Me] = 1.9 wt%, [αMan-AAB-Me] = 2.1 wt% in 200 mM HEPES–KOH buffer (pH 6.7). | |
 |
| | Fig. 3 Photographs and schematics of βGlc-AAB-Me hydrogel exhibiting reversible thermal gel–sol transition and thermochromism. Condition: [βGlc-AAB-Me] = 1.7 wt% in 200 mM HEPES–KOH buffer (pH 6.7). | |
Morphology of the hydrogels
Insight into the morphology of the self-assembled structures was obtained by transmission electron microscopy (TEM) on the αGal-AAB-Me, βGal-AAB-Me and βGlc-AAB-Me hydrogels (Fig. 4 and Fig. S3, ESI† (other views of Fig. 4)). TEM images showed that αGal-AAB-Me (Fig. 4A and Fig. S3A, ESI†) and βGal-AAB-Me (Fig. 4B and Fig. S3B, ESI†) self-assembled into thin one-dimensional (1D) nanofibers or two-dimensional (2D) nanosheets. On the other hand, βGlc-AAB-Me self-assembled into longer and thick 1D nanofibers, thereby facilitating the formation of a highly entangled three-dimensional (3D) network (Fig. 4C, D and Fig. S3C, ESI†). These conventional TEM images are not cryo-TEM and thus there is the potential influence of the sample preparation process (especially during the drying process) on the observed morphologies. Nevertheless, we speculate that the highly entangled 3D network formed by the longer and thicker nanofibers of βGlc-AAB-Me may be one of the reasons for the stable and mechanically tough hydrogel formation (vide infra).
 |
| | Fig. 4 Representative TEM images of αGal-AAB-Me (A), βGal-AAB-Me (B) and βGlc-AAB-Me (C) and (D) hydrogels transferred on an elastic carbon-coated grid. The scale bar represents 500 nm. Conditions: [αGal-AAB-Me] = [βGal-AAB-Me] = [βGlc-AAB-Me] = 2.2 wt% in 200 mM HEPES–KOH buffer (pH 6.7). | |
Rheological measurements of hydrogels
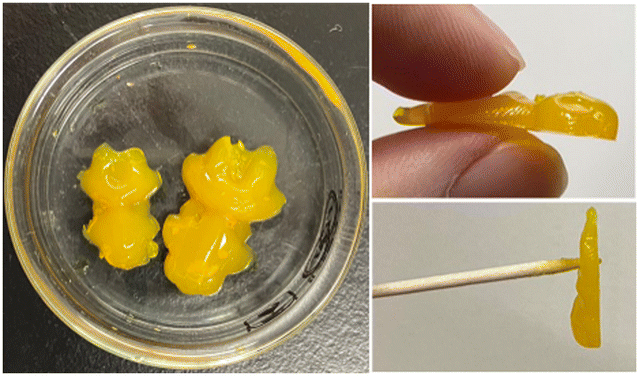
Interestingly, the βGlc-AAB-Me hydrogel prepared at a high concentration (1.4 wt%) showed an agar-like texture. The obtained hydrogel was able to maintain a complex shape, could be held by hand or a toothpick and showed unusually high strength for a supramolecular hydrogel (Fig. 5). Most LMW supramolecular hydrogels do not have mechanical toughness comparable to polymer-based hydrogels, and the lack of mechanical toughness unfortunately limits their usefulness as functional materials. The development of mechanically tough LMW supramolecular hydrogels has therefore become a research objective.14 Given the above research background, the high strength properties of the βGlc-AAB-Me hydrogel is intriguing and are expected to be useful as functional materials.
 |
| | Fig. 5 Photographs of βGlc-AAB-Me hydrogel prepared at high concentration. Condition: [βGlc-AAB-Me] = 1.4 wt% in 200 mM HEPES–KOH buffer (pH 6.7). | |
The mechanical strength of the αGal-AAB-Me, βGal-AAB-Me and βGlc-AAB-Me hydrogels (1.9 wt%) was evaluated by rheological measurements (Fig. 6). The frequency sweep measurements revealed that the storage modulus (G′) was greater than the loss modulus (G′′) throughout the measurement range, demonstrating typical gel properties. The maximum values of G′ for αGal-AAB-Me, βGal-AAB-Me and βGlc-AAB-Me hydrogels were about 13, 4 and 83 kPa, respectively. The G′ value of βGlc-AAB-Me (83 kPa) is remarkably high for LMW supramolecular hydrogels reported so far.14 One of the factors that may have contributed to the extremely high strength of βGlc-AAB-Me is the highly entangled 3D network shown in the TEM observation described above (Fig. 4C, D and Fig. S3C, ESI†). In addition, the stability of the fibre structure may also have contributed. The mechanism behind the high strength of the βGlc-AAB-Me hydrogel requires further investigation.
 |
| | Fig. 6 Frequency sweep (G′ (closed symbols); G′′ (open symbols)) of αGal-AAB-Me (green data), βGal-AAB-Me (blue data) and βGlc-AAB-Me (red data) hydrogels. Conditions: [αGal-AAB-Me] = [βGal-AAB-Me] = [βGlc-AAB-Me] = 1.9 wt% in 200 mM HEPES–KOH buffer (pH 6.7). The measurements were carried out at a strain of 0.20%, 25 °C. | |
Hydrogen-bonding networks revealed by single-crystal XRD analyses
Diluted solutions of MS-AAB-Me in 200 mM HEPES–KOH buffer (pH 6.7) at concentrations below CGC gave precipitates. Polarized optical microscopic images revealed that all the precipitates except for that of αMan-AAB-Me were micrometre-scale crystals, with αGlc-AAB-Me exhibiting plate-like crystals and the others crystallising in the form of needle-like crystals (Fig. S4, ESI†). In addition, single crystals suitable for X-ray structural analysis were successfully obtained for αGal-AAB-Me, αGlc-AAB-Me and βGlc-AAB-Me and the results are shown in Fig. 7 and Fig. S5, ESI† (other views of Fig. 7), and the corresponding data are summarised in Table 2. It should be noted that the structures of αGlc-AAB-Me and βGlc-AAB-Me were determined with large errors due to poor crystal quality. Moreover, the structural analysis of the βGal-AAB-Me crystals could not be performed due to a significant loss of crystallinity after collection from the solution. The crystal structure of αGal-AAB-Me is shown in Fig. 7A and Fig. S5A, ESI.† Each unit cell contains two crystallographically equivalent αGal-AAB-Me molecules and six water molecules. The water molecules are connected via hydrogen bonds and the individual water molecules are surrounded by the NH and carbonyl groups in the AAB backbone and the hydroxy groups at the 2-, 4- and 6-positions in the αGal residue. This kind of hydrogen-bonding network, which encapsulates water, is thought to be important for the formation of stable hydrogels. It is also worth noting that the two C–Br⋯Br angles between two AAB moieties in a unit cell are 177.2(2)° and 95.3(2)°, suggesting the presence of type-II halogen bonds according to our molecular design. In addition, the AAB moiety is stacked with the AAB moiety of a molecule in the neighbouring unit cell, supporting the hypothesis that the colour change mechanism of this chromic supramolecular hydrogel is due to the formation of H-aggregates.7 A similar hydrogen-bonding network with a halogen-bonded and π-stacked AAB system was obtained for the crystal of βGlc-AAB-Me (Fig. 7C and Fig. S5C, ESI†). The crystal structure contains 2.5 water molecules per one βGlc-AAB-Me molecule and the individual water molecules are surrounded by the NH and carbonyl groups in the AAB backbone and the hydroxy groups at the 2-, 4- and 6-positions in the βGlc residue. Furthermore, the hydroxy groups at the 3- and 6-positions also form hydrogen bonds between neighbouring βGlc-AAB-Me molecules. In contrast with the crystal structures of gel-forming αGal-AAB-Me and βGlc-AAB-Me, which contain more than two water molecules, the crystals of αGlc-AAB-Me, which showed no gelation ability, contain no water molecule (Fig. 7B and Fig. S5B, ESI†). Although the results represent the mode of interaction in the crystalline state and it is questionable whether they fully reflect those in the gel state, we have succeeded in elucidating the hydrogen-bonding network pattern in this molecular system. These results suggest that a unique hydrogen-bonding network is formed depending on the stereoisomerism of the saccharide residues, and that this network may determine the properties of the nanostructure that is the component of the hydrogels.10a
 |
| | Fig. 7 Single-crystal X-ray structures of αGal-AAB-Me (A), αGlc-AAB-Me (B) and βGlc-AAB-Me (C) (left: overview with 50% ellipsoids, right: schematic illustration). Colour code: grey, carbon; red, oxygen; blue, nitrogen; orange, bromine; white, hydrogen; green, water. Red dots represent hydrogen bonds. The crystallographic data are summarised in Table 2. | |
Table 2 Crystallographic data for αGal-AAB-Me, αGlc-AAB-Me and βGlc-AAB-Me
|
R
1 = R = ∑‖Fo| − |Fc‖/∑|Fo|.
wR2 = [(∑w(|Fo|2 − |Fc|2)2)/∑w(Fo2)2]1/2.
|
| Compound |
αGal-AAB-Me
|
αGlc-AAB-Me
|
βGlc-AAB-Me
|
| Formula |
C17H19BrN2O8·3H2O |
C17H19BrN2O8 |
C17H19BrN2O8·2.5H2O |
| Formula weight |
513.30 |
459.25 |
503.79 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21 |
P21 |
P1 |
|
a/Å |
12.8648(7) |
6.0927(6) |
9.4507(9) |
|
b/Å |
4.7385(2) |
7.2170(6) |
12.7474(14) |
|
c/Å |
18.1370(10) |
20.9754(18) |
17.6136(11) |
|
a/deg. |
90 |
90 |
76.759(7) |
|
β/deg. |
104.231(5) |
95.482(9) |
89.921(6) |
|
c/deg. |
90 |
90 |
88.668(8) |
|
V/Å3 |
1071.70(10) |
918.09(14) |
2065.0(3) |
|
Z
|
2 |
2 |
4 |
| Crystal size/mm3 |
0.639 × 0.028 × 0.021 |
0.17 × 0.053 × 0.019 |
0.688 × 0.032 × 0.019 |
|
T/K |
103.15 |
103.15 |
103.15 |
|
D
c/g cm−3 |
1.591 |
1.661 |
1.620 |
|
F
000
|
528.0 |
468.0 |
1034 |
|
λ/Å |
0.71073 |
0.71073 |
0.71073 |
|
μ (Mo Kα)/mm−1 |
1.980 |
2.289 |
2.051 |
|
R
1 [I > 2.00σ(I)]a |
0.0353 |
0.1026 |
0.1029 |
|
R (all reflections)a |
0.0481 |
0.1299 |
0.1784 |
| wR2 (all reflections)b |
0.0752 |
0.2580 |
0.3162 |
| GOF |
1.028 |
1.075 |
0.945 |
| Number of observations |
7763 |
5291 |
15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 537 537 |
| Number of variables |
3816 |
3231 |
10448 |
| CCDC number |
2370385
|
— |
— |
To investigate the impact of the hydrogen-bonding networks triggered by the hydroxy groups of the saccharide residues on the formation of the hydrogels, we performed Fourier-transform infrared (FT-IR) measurements for the solvent-retained αGal-AAB-Me, βGal-AAB-Me and βGlc-AAB-Me hydrogels (Fig. S6, ESI†). We prepared the hydrogel samples (1.9 wt%) using distilled water as the solvent instead of the HEPES–KOH buffer to avoid complicatedness of analysis due to the presence of buffer-derived signals. In the 2700–3700 cm−1 region, the bending vibrations of the O–H moiety in the saccharide residues and water molecules could be observed. In particular, the βGlc-AAB-Me hydrogel, which showed high gelation ability and crystallised with water molecules, exhibited a strong and narrow band at around 3290 cm−1. Furthermore, the band at ∼1640 cm−1 was also narrow compared with that of H2O. These spectroscopic characteristics strongly suggest the existence of the hydrogen-bonding network in the βGlc-AAB-Me hydrogel.15 Similar behaviours were also observed for the αGal-AAB-Me hydrogel, of which the crystal structure was successfully analysed. Thus, the hydrogen-bonding networks between the hydroxy groups of the saccharide residues and water molecules in the hydrogel state were experimentally detected.
Experimental
Generals
Chemical reagents were purchased from Tokyo Chemical Industry Co., Ltd, FUJIFILM Wako Pure Chemical Co., or Watanabe Chemical Industries, Ltd, and used without further purification. Thin layer chromatography (TLC) was performed on TLC silica gel 60F254 (Merck). Column chromatography was performed on silica gel 60 N (Kanto Chemical Co., Inc., spherical neutral, 63–210 μm). 1H and 13C NMR spectra in CDCl3, CD3OD, or DMSO-d6 were recorded on a JEOL ECA500 spectrometer, and chemical shifts were determined by tetramethylsilane (TMS) or residual non-deuterated solvents as an internal reference. Multiplicities are abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, dd = double doublet, and br = broad. ESI-MS analyses were carried out using a Bruker amaZon SL mass spectrometer. FT-IR spectra (for chemical structure identification of compounds) were recorded on a JEOL FT/IR-4100 spectrometer using KBr pellets in the range of 4000 to 500 cm−1, with a resolution of 4 cm−1.
Preparation of hydrogels
Powders of compounds (typically, 2 mg) were suspended in 200 mM HEPES–KOH buffer (pH 6.7, 20–500 μL) in a glass screw neck vial, sealed with a polypropylene cap and Teflon packing (Mighty vial, Maruemu, Japan). The suspensions were heated until homogeneous solutions were obtained. The solutions were solidified into hydrogels after incubating for several minutes at room temperature.
Determination of Tgel of hydrogels
T
gel was determined by a tube inversion method (Fig. S1, ESI†).11 The vial containing the hydrogel (2.2 wt% in 200 mM HEPES–KOH buffer (pH 6.7)) was hung upside down in an oil bath, and the temperature of the oil bath was gradually increased (1 °C intervals, from 25 °C to 81 °C). The temperature at which the gel collapsed was defined as the Tgel.
Measurements of the UV-visible absorption spectra of hydrogels
Hydrogel samples (2.2 wt% in 200 mM HEPES–KOH buffer (pH 6.7)) were heated to form a homogeneous solution. The hot solution (100 μL) was transferred into an assembled quartz cell (path length: 0.1 mm (GL Sciences Inc., cat. no. AB10-UV-0.1 with cell adaptor, GL Sciences Inc., cat. no. CAS-10-1)) and stored at room temperature for 10 min. The absorption spectra at room temperature were recorded using a Shimadzu UV-1280 spectrophotometer.
Rheological measurement of hydrogels
Dynamic frequency sweep experiments were carried out on a TA instruments AR-G2 rheometer using a 20-mm stainless steel parallel plate (the temperature of the plate was controlled at 25 °C by Peltier system) at the gap of 2000 μm. Hydrogel samples (1.9 wt% in 200 mM HEPES–KOH buffer (pH 6.7)) were placed on the plate. Typically, a freshly prepared solution (800 μL) of MS-AAB-Me, as described above in a glass vial, was immediately transferred into a mold, which was prepared by cutting the top off a 20 mL (2 cm diameter) plastic syringe and setting it up vertically with its plunger. The top of the syringe mold was covered with parafilm and allowed to stand for 24 h at room temperature. The hydrogel was ejected and transferred onto the bottom plate of the rheometer by carefully pushing the plunger. All gels exhibited an almost linear viscoelastic regime up to 1.0% strain (frequency: 1.0 rad s−1). Thus, a frequency sweep (0.1–100 rad s−1) was performed under 0.2% strain. The sampling and measurements were according to previously reported methods.9e,16
TEM analysis of hydrogels
Hydrogel samples (2.2 wt% in 200 mM HEPES–KOH buffer (pH 6.7), 5 μL) were dropped on a copper TEM grid covered by an elastic carbon-support film (20–25 nm) with filter paper underneath and the excess solution was blotted with the filter paper immediately. The TEM grids were washed with H2O (5 μL) three times and dried under a reduced pressure for 6 h prior to TEM observation. The TEM images were acquired using a Hitachi H-7100 (accelerating voltage: 100 kV) equipped with a CCD camera and AMTV600 software.
Single-crystal X-ray diffraction analyses
Powders of compounds (typically, 2 mg) were suspended in 200 mM HEPES–KOH buffer (pH 6.7) in a mighty vial (1.6 wt%, 2.8 wt% and 0.20 wt% for αGal-AAB-Me, αGlc-AAB-Me and βGlc-AAB-Me, respectively). The suspensions were heated until homogeneous solutions were obtained. The solutions were incubated at room temperature for several days to form crystals. One crystal was picked up under an optical microscope, and then, single-crystal XRD measurements were performed using a Rigaku XtaLAB Synergy-S/Mo system (Mo Kα; λ = 0.71073 Å). X-Ray diffraction data were collected at 103.15 K by a HyPix-600HE photon counting detector and analysed using an Olex2 crystallographic software package.17 The structure was solved by direct methods (ShelXT18) and refined through full-matrix least-squares techniques on F2 using ShelXL.19 All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were placed at calculated positions and refined “riding” on their corresponding carbon, nitrogen or oxygen atom. The crystal data are summarized in Table 2. Final cif files for all the crystals are listed in the ESI.† Deposition number 2370385 for αGal-AAB-Me, contains the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service https://www.ccdc.cam.ac.uk/structures.
FT-IR measurement of hydrogels
FT-IR spectra of hydrogels were recorded on a Jasco FT/IR-4600 spectrometer with an attenuated total reflectance (ATR) attachment (ATR-PRO-ONE) in the range of 4000 to 500 cm−1, with a resolution of 4 cm−1. Hydrogel samples (1.9 wt% in distilled water) were prepared for the measurement. Spectra of the corresponding powdery samples which were obtained by drying the methanolic solutions and H2O were also measured as references.
Synthesis
Aniline derivatives with a saccharide residue (MS-Ph-NH2) were synthesized according to previously reported methods.8b1H NMR spectra of the compounds are shown in Fig. S7–S11, ESI.†
Synthesis of αGal-AAB-Me.
To a solution of 1 (108 mg, 0.4 mmol) in dry N,N-dimethylformamide (DMF, 1 mL) was added αGal-Ph-NH2 (120 mg, 0.44 mmol, 1.1 equiv.) and N,N-diisopropylethylamine (DIEA, 77 μL, 0.44 mmol, 1.1 equiv.), and the mixture was stirred at room temperature overnight under ambient atmosphere. The solvent was then evaporated, and residue was washed with diethyl ether and H2O. The resulting product was dried under vacuum to give αGal-AAB-Me (150 mg, 82%) as a yellow powder. 1H NMR (500 MHz, CD3OD): δ = 3.01 (s, 3H), 3.68–3.70 (m, 2H), 3.92–3.97 (m, 4H), 5.49 (d, J = 1.8 Hz, 1H), 7.17 ppm (dd, J1 = 8.9 Hz, J2 = 18 Hz, 4H). 13C NMR (125 MHz, DMSO-d6): δ = 24.27, 53.66, 60.32, 68.03, 68.57, 69.45, 72.33, 77.65, 98.54, 116.60, 126.19, 130.37, 141.93, 155.16, 166.07, 167.89 ppm. LRMS (ESI, positive mode): Calcd. for [M(C17H19BrN2O8) + Na]+: m/z = 481.0; found: 481.0. FT-IR (KBr pellet): v = 3355.5, 3031.6, 2958.3, 2935.1, 1762.6, 1712.5, 1658.5, 1508.1, 1446.4, 1388.5, 1311.4, 1226.5, 1149.4, 1091.5, 1037.52, 983.5, 960.4, 860.1, 833.1, 771.4, 740.5, 705.8, 667.3, 582.4, 536.1 cm−1.
Synthesis of βGal-AAB-Me.
To a solution of 1 (108 mg, 0.4 mmol) in DMF (1 mL) was added βGal-Ph-NH2 (120 mg, 0.44 mmol, 1.1 equiv.) and DIEA (77 μL, 0.44 mmol, 1.1 equiv.), and the mixture was stirred at room temperature overnight under ambient atmosphere. The solvent was then evaporated, and the residue was washed with diethyl ether and H2O. The resulting product was dried under vacuum to give βGal-AAB-Me (138 mg, 75%) as a yellow powder. 1H NMR (500 MHz, CD3OD): δ = 3.01 (s, 3H), 3.58 (dd, J1 = 3.5 Hz, J2 = 9.7 Hz, 1H), 3.68–3.71 (m, 1H), 3.74–3.82 (m, 3H), 3.90 (d, J = 3.5 Hz, 1H), 4.85–4.92 (m, 1H (overlap with water)), 7.14 ppm (dd, J1 = 8.9 Hz, J2 = 18 Hz, 4H). 13C NMR (125 MHz, DMSO-d6): δ = 24.25, 53.64, 60.42, 68.16, 70.30, 73.28, 75.53, 77.59, 101.15, 115.72, 126.17, 130.26, 141.90, 155.36, 166.05, 167.88 ppm. LRMS (ESI, positive mode): Calcd. for [M(C17H19BrN2O8) + Na]+: m/z = 481.0; found: 481.0. FT-IR (KBr pellet): v = 3509.8, 3371.0, 3239.8, 3016.1, 2946.7, 2881.1, 1762.6, 1712.5, 1654.6, 1616.1, 1508.1, 1442.5, 1384.6, 1307.5, 1234.2, 1137.8, 1083.8, 1052.9, 887.1, 829.2, 744.4, 698.1, 640.3, 574.7, 524.5 cm−1.
Synthesis of αGlc-AAB-Me.
To a solution of 1 (108 mg, 0.4 mmol) in DMF (1 mL) was added αGlc-Ph-NH2 (121 mg, 0.44 mmol, 1.1 equiv.) and DIEA (77 μL, 0.44 mmol, 1.1 equiv.), and the mixture was stirred at room temperature overnight under ambient atmosphere. The solvent was then evaporated, and residue was washed with diethyl ether and H2O. The resulting product was dried under vacuum to give αGlc-AAB-Me (160 mg, 87%) as a yellow powder. 1H NMR (500 MHz, CD3OD): δ = 3.01 (s, 3H), 3.42 (t, J = 9.2 Hz, 1H), 3.57 (dd, J1 = 3.5 Hz, J2 = 9.7 Hz, 1H), 3.64–3.76 (m, 3H), 3.85 (t, J = 9.2 Hz, 1H), 5.48 (d, J = 3.5 Hz, 1H), 7.17 ppm (dd, J1 = 9.5 Hz, J2 = 15 Hz, 4H). 13C NMR (125 MHz, DMSO-d6): δ = 24.24, 60.73, 69.95, 71.61, 73.06, 73.80, 77.69, 98.20, 116.57, 126.19, 130.46, 141.90, 154.97, 166.05, 167.86 ppm. LRMS (ESI, positive mode): Calcd. for [M(C17H19BrN2O8) + Na]+: m/z = 481.0; found: 481.0. FT-IR (KBr pellet): v = 3390.2, 3278.4, 2950.6, 2912.0, 1774.2, 1704.8, 1643.1, 1511.9, 1446.4, 1384.6, 1272.8, 1214.9, 1153.2, 1099.2, 1022.1, 929.5, 864.0, 837.0, 528.4 cm−1.
Synthesis of βGlc-AAB-Me.
To a solution of 1 (107 mg, 0.4 mmol) in DMF (1 mL) was added βGlc-Ph-NH2 (120 mg, 0.44 mmol, 1.1 equiv.) and DIEA (77 μL, 0.44 mmol, 1.1 equiv.), and the mixture was stirred at room temperature overnight under ambient atmosphere. The solvent was then evaporated, and residue was washed with diethyl ether and H2O. The resulting product was dried under vacuum to give βGlc-AAB-Me (167 mg, 91%) as a yellow powder. 1H NMR (500 MHz, CD3OD): δ = 3.01 (s, 3H), 3.44–3.49 (m, 4H), 3.70 (dd, J1 = 5.8 Hz, J2 = 12.0 Hz, 1H), 3.90 (dd, J1 = 2.3 Hz, J2 = 15 Hz, 1H), 4.86–4.91 (m, 1H (overlap with water)), 7.13 ppm (dd, J1 = 9.5 Hz, J2 = 15 Hz, 4H). 13C NMR (125 MHz, DMSO-d6): δ = 24.24, 53.63, 60.69, 69.69, 73.25, 76.59, 77.04, 77.54, 100.54, 115.69, 126.14, 130.34, 141.88, 155.24, 166.05, 167.87 ppm. LRMS (ESI, positive mode): Calcd. for [M(C17H19BrN2O8) + Na]+: m/z = 481.0; found: 481.0. FT-IR (KBr pellet): v = 3367.1, 3293.8, 2919.7, 1877.3, 1762.6, 1712.5, 1650.8, 1511.9, 1450.2, 1388.5, 1295.9, 1230.4, 1076.1, 1388.5, 1295.9, 1230.4, 1076.1, 1018.2, 891.0, 829.2, 744.4, 644.1, 582.4, 528.4 cm−1.
Synthesis of αMan-AAB-Me.
To a solution of 1 (107 mg, 0.4 mmol) in DMF (1 mL) was added αMan-Ph-NH2 (119 mg, 0.44 mmol, 1.1 equiv.) and DIEA (77 μL, 0.44 mmol, 1.1 equiv.), and the mixture was stirred at room temperature overnight under ambient atmosphere. The solvent was then evaporated, and the residue was purified by column chromatography (SiO2, CH2Cl2/MeOH = 12/1 to 6/1 to 1/1 (v/v)) and was then evaporated. The residue was washed with diethyl ether. The resulting product was dried under vacuum to give αMan-AAB-Me (95 mg, 52%) as a yellow powder. 1H NMR (500 MHz, CD3OD): δ = 3.01 (s, 3H), 3.58–3.61 (m, 1H), 3.69–3.78 (m, 3H), 3.89 (dd, J1 = 3.5 Hz, J2 = 5.70 Hz, 1H), 3.99–4.01 (m, 1H), 5.46 (d, J = 3.5 Hz, 1H), 7.14 ppm (dd, J1 = 8.7 Hz, J2 = 16 Hz, 4H). 13C NMR (125 MHz, DMSO-d6): δ = 24.24, 61.04, 66.71, 70.09, 70.67, 75.14, 99.19, 117.83, 126.19, 130.54, 141.88, 154.18, 166.04, 167.85 ppm. LRMS (ESI, positive mode): Calcd. for [M(C17H19BrN2O8) + Na]+: m/z = 481.0; found: 481.0. FT-IR (KBr pellet): v = 3394.1, 3293.8, 2939.0, 1712.5, 1643.1, 1508.1, 1446.4, 1384.6, 1303.6, 1226.5, 1122.4, 1060.66, 1022.1, 983.5, 914.1, 887.1, 833.1, 744.4, 675.0, 644.1, 574.7, 505.3 cm−1.
Conclusions
We developed novel glycolipid-type amphiphiles, MS-AAB-Me, and found that the stereoisomerism of the saccharide residue affects their gelation ability. The single-crystal XRD analysis revealed the presence of hydrogen-bonding networks and molecular packing in the crystals of some compounds. Interestingly, water molecules were incorporated into the hydrogen-bonding networks in the crystals of the gel-forming compounds. These results indicate that the stereoisomerism of the saccharide residue has a marked effect on the formation of the hydrogen-bonding networks, as well as on the gelation ability and gel stability and are expected to provide important insights not only for the design of glycolipid-type supramolecular hydrogels but also for their application as functional materials. Interestingly, the βGlc-AAB-Me hydrogel prepared at a high concentration (1.4 wt%) showed an agar-like texture and sufficient mechanical toughness for easy handling. The TEM observation suggests that βGlc-AAB-Me can form stable hydrogels by forming a 3D structure with long and thick 1D nanofibers. We would like to investigate the mechanical properties and mechanism in more detail.
Author contributions
Conceptualisation: R. O. and K. Y.; synthesis and structural analysis: K. Y. and M. I. (M. Ishida); data curation (self-assembly properties): K. Y.; single-crystal XRD analysis: A. I.; rheological measurement: Y. S. and M. I. (M. Ikeda); absorption spectra measurements: K. Y.; polarised light microscopy experiments: K. Y. and Y. S.; TEM analysis: S. H. and K. Y.; FT-IR measurement: R. O. and A. I.; supervision: M. I. (M. Ikeda) and M. I. (M. Izumi); writing – original draft: K. Y. and R. O.; writing – review and editing: R. O., A. I., K. Y., M. I. (M. Ikeda), S. H. and M. I. (M. Izumi).
Data availability
The data supporting this article have been included as part of the ESI† and cif files in the Supplementary files. Additional data are available from the corresponding author, Rika Ochi, upon reasonable request.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was partially supported by the JSPS KAKENHI Grant Number JP20K15416 and JP24K08637 and the Assisted Joint Research Program (Exploration Type and Acceleration Type) of the J-GlycoNet cooperative network, which is accredited by the Minister of Education, Culture, Sports, Science and Technology, MEXT, Japan, as a Joint Usage/Research Center. We would like to thank MARUZEN-YUSHODO Co., Ltd (https://kw.maruzen.co.jp/kousei-honyaku/) for the English language editing.
Notes and references
-
(a) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS PubMed;
(b) M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615 CrossRef CAS;
(c) R. V. Ulijn and A. M. Smith, Chem. Soc. Rev., 2008, 37, 664 RSC;
(d) M. Ikeda, R. Ochi and I. Hamachi, Lab Chip, 2010, 10, 3325 RSC;
(e) A. Dawn, T. Shiraki, S. Haraguchi, S. Tamaru and S. Shinkai, Chem. – Asian J., 2011, 6, 266 CrossRef CAS PubMed;
(f) S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973 CrossRef CAS PubMed;
(g) X. Du, J. Zhou, J. Shi and B. Xu, Chem. Rev., 2015, 115, 13165 CrossRef CAS PubMed;
(h) B. O. Okesola and D. K. Smith, Chem. Soc. Rev., 2016, 45, 4226 RSC;
(i) H. Shigemitsu and I. Hamachi, Acc. Chem. Res., 2017, 50, 740 CrossRef CAS PubMed;
(j) K. Sato, M. P. Hendricks, L. C. Palmer and S. I. Stupp, Chem. Soc. Rev., 2018, 47, 7539 RSC;
(k) N. Mehwish, X. Dou, Y. Zhao and C.-L. Feng, Mater. Horiz., 2019, 6, 14 RSC;
(l) T. Shimizu, W. Ding and N. Kameta, Chem. Rev., 2020, 120, 2347 CrossRef CAS PubMed;
(m) M. Yokoya, S. Kimura and M. Yamanaka, Chem. – Eur. J., 2021, 27, 5601 CrossRef CAS PubMed;
(n) S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165 RSC;
(o) D. K. Smith, Soft Matter, 2024, 20, 10 RSC.
-
(a) S. L. Zhou, S. Matsumoto, H. D. Tian, H. Yamane, A. Ojida, S. Kiyonaka and I. Hamachi, Chem. – Eur. J., 2005, 11, 1130 CrossRef CAS PubMed;
(b) S. Ray, A. K. Das and A. Banerjee, Chem. Mater., 2007, 19, 1633 CrossRef CAS;
(c) A. Shome, S. Debnath and P. K. Das, Langmuir, 2008, 24, 4280 CrossRef CAS PubMed;
(d) J. Nanda, A. Biswas and A. Banerjee, Soft Matter, 2013, 9, 4198 RSC;
(e) T. J. Moyer, J. A. Finbloom, F. Chen, D. J. Toft, V. L. Cryns and S. I. Stupp, J. Am. Chem. Soc., 2014, 136, 14746 CrossRef CAS PubMed;
(f) B. O. Okesola, Y. Wu, B. Derkus, S. Gani, D. Wu, D. Knani, D. K. Smith, D. Adams and A. Mata, Chem. Mater., 2019, 31, 7883 CrossRef CAS PubMed;
(g) G. Zhang, J. Li, W. Cai, Y. Kong and Z. Z. Yin, Microchem. J., 2023, 193, 109160 CrossRef CAS.
-
(a) M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960 CrossRef CAS PubMed;
(b) J. Hu, G. Zhang and S. Liu, Chem. Soc. Rev., 2012, 41, 5933 RSC;
(c) A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540 RSC;
(d) M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086 RSC;
(e) J. Zhou and B. Xu, Bioconjugate Chem., 2015, 26, 987 CrossRef CAS PubMed;
(f) M. Ikeda, Polym. J., 2019, 51, 371 CrossRef CAS;
(g) J. Gao, J. Zhan and Z. Yang, Adv. Mater., 2019, 1805798 Search PubMed;
(h) A. N. Shy, B. J. Kim and B. Xu, Matter, 2019, 1, 1127 CrossRef PubMed;
(i) R. Kuosmanen, K. Rissanen and E. Sievänen, Chem. Soc. Rev., 2020, 49, 1977 RSC;
(j) S. Kimura, M. Yokoya and M. Yamanaka, Chem. Lett., 2021, 50, 459 CrossRef CAS;
(k) S. Panja and D. J. Adams, Chem. – Eur. J., 2021, 27, 8928 CrossRef CAS PubMed;
(l) J. Chen, H. Wang, F. Long, S. Bai and Y. Wang, Chem. Commun., 2023, 59, 14236 RSC;
(m) N. Das and C. Maity, ACS Catal., 2023, 13, 5544 CrossRef CAS;
(n) R. Kubota and I. Hamachi, Adv. Sci., 2024, 11, 2306830 CrossRef CAS PubMed.
-
(a) D. J. Pochan, J. P. Schneider, J. Kretsinger, B. Ozbaas, K. Rajagopal and L. Haines, J. Am. Chem. Soc., 2003, 125, 11802 CrossRef CAS PubMed;
(b) K.-S. Moon, H.-J. Kim, E. Lee and M. Lee, Angew. Chem., Int. Ed., 2007, 46, 6807 CrossRef CAS PubMed;
(c) Z. Chu and Y. Feng, Chem. Commun., 2011, 47, 7191 RSC;
(d) M. Ikeda, R. Ochi, Y. Kurita, D. J. Pochan and I. Hamachi, Chem. – Eur. J., 2012, 18, 13091 CrossRef CAS PubMed;
(e) R. Ochi, T. Nishida, M. Ikeda and I. Hamachi, J. Mater. Chem. B, 2014, 2, 1464 RSC;
(f) S. Xian and M. J. Webber, J. Mater. Chem. B, 2020, 40, 9197 RSC;
(g) J. P. Fan, F. H. Tao, X. H. Zhang, T. T. Yuan, C. F. Xie, H. P. Chen and H. L. Peng, Colloids Surf., A, 2022, 652, 129839 CrossRef CAS.
-
(a) S. Wang, W. Shen, Y. L. Feng and H. Tian, Chem. Commun., 2006, 1497 RSC;
(b) Z. Qiu, H. Yu, J. Li, Y. Wang and Y. Zhang, Chem. Commun., 2009, 3342 RSC;
(c) F. Rodrıguez-Llansola, B. Escude, J. F. Miravet, D. Hermida-Merino, I. W. Hamley, C. J. Cardinb and W. Hayes, Chem. Commun., 2010, 46, 7960 RSC;
(d) P. K. Sukul, P. K. Singh, S. K. Maji and S. Malik, J. Mater. Chem. B, 2013, 1, 153 RSC;
(e) Y. Cai, Y. Shi, H. Wang, J. Wang, D. Ding, L. Wang and Z. Yang, Anal. Chem., 2014, 86, 2193 CrossRef CAS PubMed;
(f) X. Yu, X. Ge, H. Lan, Y. Li, L. Geng, X. Zhen and T. Yi, ACS Appl. Mater. Interfaces, 2015, 7, 24312 CrossRef CAS PubMed;
(g) T. Xu, C. Liang, S. Ji, D. Ding, D. Kong, L. Wang and Z. Yang, Anal. Chem., 2016, 88, 7318 CrossRef CAS PubMed;
(h) T. Yuan, Y. Xu, C. Zhu, Z. Jiang, H.-J. Sue, L. Fang and M. A. Olson, Chem. Mater., 2017, 29, 9937 CrossRef CAS;
(i) S. Dhiman, R. Ghosh and S. J. George, ChemSystemsChem, 2019, 1, e1900042 Search PubMed;
(j) P. Singh, S. Misra, A. Das, S. Roy, P. Datta, G. Bhattacharjee, B. Satpati and J. Nanda, ACS Appl. Bio Mater., 2019, 2, 4881 CrossRef CAS PubMed;
(k) X. Xu, X. Zhou, L. Qu, L. Wang, J. Song, D. Wu, W. Zhou, X. Zhou, H. Xiang, J. Wang and J. Liu, ACS Appl. Bio Mater., 2020, 3, 7236 CrossRef CAS PubMed;
(l) B. C. Roy and T. S. Mahapatra, Soft Matter, 2023, 19, 1854 RSC;
(m) A.-L. Leistner, M. M. Most and Z. L. Pianowski, Chem. – Eur. J., 2023, 29, e202302295 CrossRef CAS PubMed.
-
(a) R. Ochi, K. Kurotani, M. Ikeda, S. Kiyonaka and I. Hamachi, Chem. Commun., 2013, 49, 2115 RSC;
(b) R. Oosumi, M. Ikeda, A. Ito, M. Izumi and R. Ochi, Soft Matter, 2020, 16, 7274 RSC;
(c) N. Tsutsumi, A. Ito, A. Ishigamori, M. Ikeda, M. Izumi and R. Ochi, Int. J. Mol. Sci., 2021, 22, 1860 CrossRef CAS PubMed;
(d) Y. Chabatake, T. Tanigawa, Y. Hirayama, R. Taniguchi, A. Ito, K. Takahashi, S. Noro, T. Akutagawa, T. Nakamura, M. Izumi and R. Ochi, Soft Matter, 2024, 20, 8170 RSC.
-
(a) M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371 CAS;
(b) F. C. Spano, Acc. Chem. Res., 2010, 43, 429 CrossRef CAS PubMed.
-
(a) T. Shimizu and M. Masuda, J. Am. Chem. Soc., 1997, 119, 2812 CrossRef CAS;
(b) J. H. Jung, S. Shinkai and T. Shimizu, Chem. – Eur. J., 2002, 8, 2684 CrossRef CAS PubMed;
(c) S. Kiyonaka, S. Shinkai and I. Hamachi, Chem. – Eur. J., 2003, 9, 976 CrossRef CAS PubMed;
(d) T. Shimizu, M. Masuda and H. Minamikawa, Chem. Rev., 2005, 105, 1401 CrossRef CAS PubMed;
(e) R. Roytman, L. Adler-Abramovich, K. S. A. Kumar, T.-C. Kuan, C.-C. Lin, E. Gazit and A. Brik, Org. Biomol. Chem., 2011, 9, 5755 RSC;
(f) S. Datta and S. Bhattacharya, Chem. Soc. Rev., 2015, 44, 5596 RSC;
(g) M. Delbianco, P. Bharate, S. Varela-Aramburu and P. H. Seeberger, Chem. Rev., 2016, 116, 1693 CrossRef CAS PubMed;
(h) T. Tsuzuki, M. Kabumoto, H. Arakawaa and M. Ikeda, Org. Biomol. Chem., 2017, 15, 4595 RSC;
(i) L. Latxague, A. Gaubert and P. Barthélémy, Molecules, 2018, 23, 89 CrossRef PubMed;
(j) J. Morris, J. Bietsch, K. Bashaw and G. Wang, Gels, 2021, 7, 24 CrossRef CAS PubMed;
(k) A. Brito, S. Kassem, R. L. Reis, R. V. Ulijn, R. A. Pires and I. Pashkuleva, Chem, 2021, 7, 2943 CrossRef CAS;
(l) L. Su, S. I. S. Hendrikse and E. W. Meijer, Curr. Opin. Chem. Biol., 2022, 69, 102171 CrossRef CAS PubMed;
(m) R. Tyagi, K. Singh, N. Srivastava and R. Sagar, Mater. Adv., 2023, 4, 3929 RSC.
-
(a) S. Kiyonaka, K. Sada, I. Yoshimura, S. Shinkai, N. Kato and I. Hamachi, Nat. Mater., 2004, 3, 58 CrossRef CAS PubMed;
(b) Y. Wang, L. Tang and J. Yu, Cryst. Growth Des., 2008, 8, 884 CrossRef CAS;
(c) M. Ikeda, T. Tanida, T. Yoshii and I. Hamachi, Adv. Mater., 2011, 23, 2819 CrossRef CAS PubMed;
(d) R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519 CrossRef CAS PubMed;
(e) Y. Shintani, H. Katagiri and M. Ikeda, Adv. Funct. Mater., 2024, 34, 2312999 CrossRef CAS.
-
(a) P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789 CrossRef CAS PubMed;
(b) P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114 CrossRef CAS PubMed;
(c) F. Meyer and P. Dubois, CrystEngComm, 2013, 15, 3058 RSC;
(d) A. Mukherjee, S. Tothadi and G. R. Desiraju, Acc. Chem. Res., 2014, 47, 2514 CrossRef CAS PubMed;
(e) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118 CrossRef CAS PubMed;
(f) A. M. S. Riel, R. K. Rowe, E. N. Ho, A.-C. C. Carlsson, A. K. Rappé, O. B. Berryman and P. S. Ho, Acc. Chem. Res., 2019, 52, 2870 CrossRef CAS PubMed;
(g) L. Turunen and M. Erdélyi, Chem. Soc. Rev., 2020, 49, 2688 RSC.
- C. K. Han and Y. H. Bae, Polymer, 1998, 39, 2809 CrossRef CAS.
- M. C. Gray, A. O. Converse and C. E. Wyman, Biotechnol. Appl. Biochem., 2003, 105–108, 179 CrossRef CAS PubMed.
- R. Roytman, L. Adler-Abramovich, K. S. A. Kumar, T.-C. Kuan, C.-C. Lin, E. Gazit and A. Brik, Org. Biomol. Chem., 2011, 9, 5755 RSC.
-
(a) Y. Zhang, Z. Yang, F. Yuan, H. Gu, P. Gao and B. Xu, J. Am. Chem. Soc., 2004, 126, 15028 CrossRef CAS PubMed;
(b) S. E. Paramonov, H.-W. Jun and J. D. Hartgerink, J. Am. Chem. Soc., 2006, 128, 7291 CrossRef CAS PubMed;
(c) E. T. Pashuck, H. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041 CrossRef CAS PubMed;
(d) M. Ikeda, R. Ochi, A. Wada and I. Hamachi, Chem. Sci., 2010, 1, 491 RSC;
(e) H. Komatsu, M. Ikeda and I. Hamachi, Chem. Lett., 2011, 40, 198 CrossRef CAS;
(f) H. Komatsu, S. Tsukiji, M. Ikeda and I. Hamachi, Chem. – Asian J., 2011, 6, 2368 CrossRef CAS PubMed;
(g) J. H. Lee, J. Park, J.-W. Park, H.-J. Ahn, J. Jaworski and J. H. Jung, Nat. Commun., 2015, 6, 6650 CrossRef CAS PubMed;
(h) S. V. Jadhav, P. Amabili, H.-G. Stammler and N. Sewald, Chem. – Eur. J., 2017, 23, 10352 CrossRef CAS PubMed;
(i) H. Sawada and M. Yamanaka, Chem. – Asian J., 2018, 13, 929 CrossRef CAS PubMed.
-
(a) Y. Shen and P. Y. Wu, J. Phys. Chem. B, 2003, 107, 4224 CrossRef CAS;
(b) W. Li, B. Sun and P. Wu, Carbohydr. Polym., 2009, 78, 454 CrossRef CAS;
(c) F. Mashingaidze, Y. E. Choonara, P. Kumar, L. C. du Toit, V. Maharaj, E. Buchmann, V. M. K. Ndesendo and V. Pillay, J. Biomed. Mater. Res., Part A, 2013, 101A, 3616 CrossRef CAS PubMed;
(d) Z. Wang, H. Yang and Z. Zhu, Polymer, 2019, 163, 144 CrossRef CAS.
- A. M. Castilla, M. Wallace, L. L. E. Mears, E. R. Draper, J. Doutch, S. Rogers and D. J. Adams, Soft Matter, 2016, 12, 7848 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S11 and cif files. CCDC 2370385. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sm01325e |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bc,
Masashi
Ishida
d,
Yuki
Shintani
e,
Masato
Ikeda
*bc,
Masashi
Ishida
d,
Yuki
Shintani
e,
Masato
Ikeda