
VHL-independent degradation of hepatitis B virus e antigen (HBeAg) by VHL-binding chimeric small molecules†
Liam T.
Hales‡
 a,
Simon J.
Mountford
a,
Mina
Takawy
b,
Danni
Colledge
b,
Belinda
Maher
cd,
Jake
Shortt
cde,
Philip E.
Thompson
*a,
Sam A.
Greenall
*c and
Nadia
Warner
*b
a,
Simon J.
Mountford
a,
Mina
Takawy
b,
Danni
Colledge
b,
Belinda
Maher
cd,
Jake
Shortt
cde,
Philip E.
Thompson
*a,
Sam A.
Greenall
*c and
Nadia
Warner
*b
aMedicinal Chemistry, Monash Institute of Pharmaceutical Sciences, Monash University, Parkville, 3052, Australia. E-mail: philip.thompson@monash.edu
bVictorian Infectious Diseases Reference Laboratory, Royal Melbourne Hospital at The Peter Doherty, Institute for Infection and Immunity, Melbourne, 3000, Australia. E-mail: nadia.warner@vidrl.org.au
cBlood Cancer Therapeutics Laboratory, Department of Medicine, School of Clinical Sciences at Monash Health, Monash University, Clayton, 3168, Australia. E-mail: sameer.greenall@monash.edu
dMonash Haematology, Monash Health, Clayton, 3168, Australia
eSir Peter MacCallum Department of Oncology, The University of Melbourne, Parkville, 3010, Australia
First published on 28th March 2025
Abstract
Hepatitis B virus (HBV) is a leading cause of liver cancer worldwide, with current treatment options unable to provide lasting efficacy against chronic infection. A key viral protein, HBV e antigen (HBeAg), plays an important role in suppressing the cellular and humoral immune response during infection and its loss is a precursor to clearance of chronic HBV infection. Its structural similarity to capsid forming HBV core protein antigen (HBcAg) makes it an intriguing, yet understudied target for pharmaceutical intervention. Recently, targeted protein degradation has been successfully applied against several viral proteins. This work investigates the targeting of HBeAg using heterobifunctional degraders derived from reported HBcAg ligands known to interact with HBeAg. Multiple compounds designed to recruit the VHL E3 ligase were found to be capable of reducing recombinant HBeAg protein levels in a HiBiT reporter assay system. Surprisingly, this decrease was found to be independent of VHL recruitment but driven by structural motifs of the VHL recruiting ligand, VH032. Virological assessment of these compounds against wildtype virus revealed an equipotent capability to reduce secreted HBeAg compared to the parental inhibitor, however increased efficacy was observed against an inhibitor resistant strain. Together, this work provides an initial description of the feasibility of converting HBV capsid-targeting ligands into degraders and provides evidence that such degraders may harbour improved activity against mutated forms of target which are resistant to parental compounds.
Introduction
Hepatitis B virus (HBV) infection is a global health issue. Of particular concern is chronic HBV infection (CHB) due to the severe damage that ongoing infection and inflammation causes to the liver, often leading to liver cirrhosis and hepatocellular carcinoma (HCC). According the WHO's 2017 Global Hepatitis report, 257 million people live with CHB, with the disease especially prevalent in the African and Western Pacific regions.1HBV DNA is transcribed into four mRNAs which code for seven key proteins involved in the HBV infection and replication cycle: three HBV surface antigens (HBsAg), HBV X protein (HBx), viral DNA polymerase (Pol), HBV core protein antigen (HBcAg) and HBV e protein (HBe). While current treatment options targeting these proteins exist, such as the nucleos(t)ide Pol inhibitors, latent reservoirs of HBV covalent closed circle DNA (cccDNA) lead to viral rebound following withdrawal of treatment.2
Drugs that target the closely related HBcAg and HBeAg are of particular interest. Despite the functional differences of these proteins, they are structurally similar. The HBcAg forms the icosahedral nucleocapsid, predominantly via the oligomerisation of 120 sets of HBcAg dimers.3 After packaging the pre-genomic RNA fused to Pol, the mature nucleocapsid is coated with envelope protein, then secreted as a virion, highlighting the essential role HBcAg plays in viral replication. Mature nucleocapsids can also enter the nucleus and refill the pool of cccDNA, implicating HBcAg in maintenance of HBV infection.4 HBeAg differs from HBcAg by a 10 amino acid N-terminal sequence leader sequence. This sequence directs the protein to the Golgi apparatus,5 preparing HBeAg for secretion by inducing an alternate dimerization mode compared to HBcAg, which allows for the secretion of HBeAg.6–8
HBeAg secretion has been shown to disrupt the humoral immune response, including pathogen recognition and cytokine production.9,10 Evidence has also suggested that HBeAg functions as a tolerogen for immune T-cells,11,12 acting as a “decoy” protein to reduce the immune response toward the nucleocapsids, composed of antigenically similar HBcAg.13 This tolerogenic role, along with the aforementioned interactions with various immune signalling pathways, suggest that successful removal of HBeAg from systemic circulation may aid the humoral immune response to infection.
HBeAg and its precursors (p25 and p22) are also partially retained intracellularly.14–16 Here they contribute to dampening of the local hepatocyte immune response and inhibit apoptosis pathways responsible for hepatocyte death during HBV infection.10,17 In this way, HBeAg contributes to the survival of these infected cells, promoting viral replication and chronic infection. HBeAg has also been linked to the pathogenesis of HCC through upregulation of miR-106b, a protein known to accelerate cell-cycle progression and increase cell proliferation in hepatocytes.10,18 HBeAg is produced during chronic HBV infection, however can be lost in later stages of infection. HBeAg-negative individuals are more likely to progress to HBsAg loss and eventual functional cure of HBV infection. Loss of HBeAg could therefore be considered an important precursor for clearance of HBV.19
HBcAg has been targeted using a class of small molecules termed core protein allosteric modulators (CpAMs). The heteroaryldihydropyrimidine (HAP) chemotype has had particular success with multiple examples having entered clinical trials.20 This class of HBV inhibitors work by altering the kinetics of capsid assembly, with the increased rate leading to the formation of aberrant capsids or non-capsid polymers.21 These proteins are then degraded via the macroautophagy-lysosomal pathway.22,23 Interestingly, HAP CpAMs have also been shown to reduce secreted HBeAg in cell and mouse models and cause an accumulation of precore proteins in the nucleus.24
Over the last decade, small molecule degraders of disease-causing proteins have risen to the forefront of drug discovery.25 These molecules have predominantly fallen into the categories of proteolysis targeting chimeras (PROTACs) or molecular glues.26 Both compound classes function by manipulating the ubiquitin proteasome system (UPS), inducing proximity between an E3 ubiquitin ligase (E3L) and a target protein, facilitating the ubiquitination of the protein of interest, and signalling it for degradation via the proteasome.27 Recently, additional classes of small molecule degraders have emerged, including hydrophobic tagged (HyT) degraders, which initiate the protein quality control response (PQR) within the cell.28
While oncological protein targets have dominated the literature to-date, the same advantages in efficacy and specificity associated with targeted protein degradation (TPD) have also encouraged application of this approach in the pursuit of antivirals. Examples include degraders against SARS-CoV-2, hepatitis C virus (HCV), human immunodeficiency virus (HIV) and dengue virus.29–34 Several of these studies have also suggested that TPD can generate additional efficacy against drug resistant strains of virus.30–32
The progression of HBV infection is reliant upon HBeAg due to a combination of its intracellular function and the humoral effects of the secreted portion. Abrogation of both sets of effect is highly desirable, making it an attractive target for degradation via the TPD approach. Successful degradation of HBeAg and reduction of its immunosuppressive effects, described above, may restore the ability of the infected cell, tissue, or body to combat viral infection. Additionally, due to the structural similarity of HBeAg and HBcAg, HAP-based degraders could engage both HBcAg and HBeAg,24 potentially enabling successful small molecule induced degradation of both proteins. This may result in an enhanced antiviral effect with potential to overcome issues with drug resistance exhibited by the antiviral drugs from which these degraders are derived.29
Here, we have described the elaboration of HAP HBV inhibitor NVR-010-001-E2,35 into bifunctional compounds containing E3L-recruiting moieties. Biochemical testing of these compounds in a HiBiT tagged HBeAg cell model revealed that several VHL-recruiting compounds caused the anticipated reductions in secreted HBeAg. Notably, we observed that these compounds sharply decreased intracellular HBeAg levels, in contrast to treatment with the parental inhibitor. We determined that degradation was largely VHL-independent using pharmacological and genetic controls. Next, we prepared analogues bearing hydrophobic tags and truncated VHL-recruiting scaffolds, with our results suggesting that a hydrophobic tag mechanism may be responsible for the observed degradation. Finally, using a transfected model of viral infection, we demonstrated that compound LH-3 and its cis epimer were active in reducing HBeAg secretion from wild-type HBV to a comparable level to the parental inhibitor, and exhibited superior activity in preventing HBeAg secretion from a HAP-resistant strain of HBV.
Results
Design and synthesis of degrader candidates
We began by designing a screening set of PROTAC-like compounds linked to the HAP scaffold after inspection of the co-crystal structure of HBcAg and inhibitor NVR-010-001-E2 (Fig. 1A).35Analysis of this structure revealed a solvent exposed morpholine group (Fig. 1B), which had been adapted to piperazine for the inclusion of fluorophores (Fig. 1C),36 or substituted in second and third generation HBV inhibitors without interrupting essential binding interactions.37,38 A variety of alkyl and PEG linkers with high flexibility and diverse length were selected to aid in determining the ideal distance between the two recruiting elements (Fig. 2A). | ||
| Fig. 1 A. Crystal structure of NVR-010-001-E2 bound to HBcAg N-domain (Y132A mutant, PDB: 5E0I35), B. Structure of second-generation HAP, NVR-010-001-E2, C. Structure of fluorophore-linked HAP, HAP-ALEX.36 | ||
 | ||
| Fig. 2 A. Chemical structure of degrader candidates, composed of a heteroaryldihydropyridine (HAP) HBeAg recruiting domain, varied linkers, and one of three E3L recruiters. B. HEK293THiBiT-HBeAg-NS cells were treated with candidates at 10 μM for 24 h, then luminescence detected and normalised to the DMSO control. Compounds with a statistically significant increase in potency compared to parental compound 2 are marked, where **** represents P < 0.0001, *** P ≤ 0.001, as determined by one-way ANOVA. C. A matched ViaLight™ assay was performed to determine the effect of treatment on cell viability. | ||
The compounds were prepared by adaptation of literature procedures from the commercially available starting material (Scheme 1). Bromination yielded key intermediate, 1,38 followed by the installation of various heterocycles via nucleophilic substitution to generate parental inhibitor and reference compound, 2 along with key intermediates. Full details are provided in the ESI.†
 | ||
| Scheme 1 Synthesis of key HAP intermediates and initial series of HBeAg degrader candidates. a) N-Bromosuccinimide, DCM, r.t, 1 h, 64%, b) morpholine, MeOH, 3 h, r.t., 40%. | ||
E3 ligase recruiter (E3R)-linker conjugates were prepared with carboxylate, piperazinyl, azido or amino terminal functionality (ESI,† Schemes S2–S8). Three E3R ligands were selected for incorporation into the initial series of degrader candidates. Canonical VHL-recruiter, VH032, was coupled with linkers via the terminal amine of the tert-leucine residue. To recruit CRBN we utilised 4-hydroxythalidomide, pomalidomide (Pom) or DGY- a ligand which has been previously used to develop Hepatitis C virus targeting PROTACs.31 Various strategies were employed to prepare the final degrader candidates including amide or urea coupling, Cu(I)-catalysed azide–alkyne cycloaddition (CuAAC) or nucleophilic substitution. Collectively, 13 candidate compounds were prepared, spanning three E3Rs and including linkers ranging from 2 to 14 heavy atoms (Fig. 2A).
Biochemical assessment of HBeAg levels using a HiBiT assay system
To assay HBeAg levels following treatment with degrader candidates, a HEK293T cell line with doxycycline (DOX) inducible HiBiT-tagged HBeAg expression was established. This assay format has been widely used in degrader development to assess protein levels in both cell lysate and live cells.39–42 Two cell lines were developed to enable the assessment of the compounds in two ways. Both contained the appropriate C-terminal deletion to match the sequence of endogenously secreted HBeAg, however the first non-secretory cell line, HEK293THiBiT-HBeAg-NS, lacked the N-terminal leader sequence required for secretion of HBeAg.14 In doing so, compounds were assessed for their holistic effect on HBeAg levels, rather than differentiating between secreted and intracellular levels of HBeAg. A second secretory construct, HEK293THiBiT-HBeAg-S, contained the N-terminal leader sequence required for secretion, enabling differentiation between intracellular and secreted HBeAg levels.Compounds were first assessed using the HiBiT assay. The non-secretory HEK293THiBiT-HBeAg-NS cell line was treated with each test compound at 10 μM for 24 h and then subjected to a Nano-Glo HiBiT lytic detection assay with no washout of the media, providing a measure of total luminescence (Fig. 2B). A no DOX control group was included to ensure luminescence was a result of DOX induced HiBiT-HBeAg expression.
Compared to the parental inhibitor 2, several candidates demonstrated a significant (P ≤ 0.001) reduction in luminescence. Corresponding to a reduction in total HiBiT-HBeAg-NS protein. VH032-linked compounds LH-1, LH-2, LH-3 and LH-6 appeared to be the most active, reducing HBeAg levels to less than 30% of the DMSO control. As expected, the parental inhibitor 2 caused an overall reduction of HBeAg consistent with literature reports (59% of control levels).24 CRBN recruiting compounds were notably less active, indicating a preference for the VHL-recruiting scaffold. Interestingly, SM-3, and more markedly SM-4, caused an accumulation of total HBeAg, potentially indicating stabilisation of the protein. Note that a matched ViaLight™ assay showed no evidence of compound cytotoxicity (Fig. 2C).
Of the most active molecules, there was no apparent pattern in terms of linker composition. LH-2 and LH-3 represent members of a sub-series defined by amide coupling between an alkyl linker and the piperazine of the HAP component and differ by two linker carbons. Compounds LH-1 and LH-6 were prepared by amine alkylation resulting in a tertiary amine linkage motif, however LH-1 contains a minimal methylene linker, while LH-6 contains an additional triazole ring and a hexyl linker.
We next used the secretory HEK293THiBiT-HBeAg-S cell line to assess secreted and intracellular HBeAg using LH-1 and LH-3. The two protein populations were determined by extracting the cell media present during drug treatment from the cells after 24 h and reading the luminescence signal for secreted HBeAg. The cells were then washed with fresh media (to remove trace amounts of secreted HBeAg) and assayed separately using a lytic HiBiT assay for determination of intracellular HBeAg.
These assays revealed a markedly different phenotypic response to LH-3 compared to parental HAP compound 2. While both treatments decreased levels of secreted HBeAg in a concentration dependent manner (<7% of DMSO at 20 μM), the effect of the two compounds on intracellular HBeAg levels was markedly different (Fig. 3A). While treatment with 2 caused an accumulation of intracellular HBeAg, LH-3 caused a significant reduction in intracellular HBeAg at concentrations above 1 μM, with luminescence reduced to 46% of the untreated control at 20 μM (Fig. 3A). Both treatments decreased levels of secreted HBeAg in a concentration dependent manner (<7% of DMSO at 20 μM) The same trend was observed for related compound LH-1 (Fig. S1†).
 | ||
| Fig. 3 A. HEK293THiBiT-HBeAg-S cells were titrated with compounds 2 and LH-3 for a 24 h treatment period. B. The HEK293THiBiT-HBeAg-S cell line was treated with compounds 2, LH-1, cisLH-1, LH-3 and cisLH-3 at 10 μM for 24 h. C. Regular or VHL KO HEK293THiBiT-HBeAg-S cell lines were treated with compounds 2, LH-1 or LH-3 at 10 μM for 24 h. Secreted HBeAg levels were reduced in all treatment groups, with minimal difference observed between cell lines. VHL KO was also unable to rescue intracellular HBeAg levels. **** represents P < 0.0001, *** P ≤ 0.001, ** P ≤ 0.01, * P ≤ 0.05, ns = P > 0.05 as determined by one-way ANOVA. | ||
Given that these compounds were all designed to recruit VHL we aimed to assess if the reduction in HBeAg levels caused by LH-1 and LH-3 was due to VHL-dependent proteasomal degradation. We therefore conducted parallel experiments using negative control compounds cisLH-1 and cisLH-3 alongside LH-3 and LH-1 (Fig. 3B). If a VHL-dependent PROTAC-like mechanism was responsible for degradation of HBeAg, treatment with cis compounds should cause a substantial reduction in activity compared to the trans counterparts. At 10 μM, the negative control compounds both showed a slightly reduced ability to reduce secreted HBeAg compared to their trans counterparts (LH-1vs.cisLH-1, 7% vs. 31% and LH-3vs.cisLH-3, 7% vs. 20%, P < 0.0001), indicating some contribution from VHL recruitment.
Intracellular HBeAg levels were only modestly affected with the cis compounds. The LH-1/cisLH-1 pair exhibited a small decrease in activity (48% vs. 59%, P = 0.006), while the difference between the LH-/cisLH-3 pair (44% vs. 38%, P = 0.135) was not significant. We confirmed abrogation of VHL binding by immunoblotting where LH-1 and LH-3 exhibited stabilisation of VHL while the cis epimers did not (Fig. S2†). These results suggested that while a PROTAC-like mechanism may have a minor contribution to the reduction of intracellular HBeAg, the primary mechanism of degradation is independent of VHL recruitment.
For further confirmation, a VHL knockout (KO) cell line (HEK293TVHL KO HiBiT-HBeAg-S) was generated for use in the HiBiT assay. Treating these two cell lines with compounds 2, LH-1 and LH-3 revealed a similar trend to the investigation of the cis/trans pairs. We compared the VHL-expressing cell line to VHL KO cells by HiBiT assay (Fig. 3C) which were further confirmed by western blot (Fig. S3B and C†). There was no significant difference in secreted HBeAg levels between treatment groups following incubation with 10 μM of 2 (7% vs. 10%). LH-1 (20% vs. 32%, P < 0.0001) and LH-3 (7% vs. 12% P = 0.0177) showed statistically significant, albeit minor, rescue of secreted HBeAg levels. We were again interested in intracellular HBeAg levels, which increased in all treatment groups when moving from the parental HEK293THiBiT-HBeAg-S to the VHL KO cell line (2: 116% vs. 144% P < 0.0001, LH-1: 38% vs. 50% P = 0.0074, LH-3: 44% vs. 56% P = 0.0126). This increase in luminescence for all drug treatment groups (including parental compound 2) potentially indicates a general VHL-dependent depletion of HBeAg, rather than a true dependence on VHL recruitment for the mechanism of LH-1/LH-3 induced HBeAg degradation.
Together, these data obtained through chemical and biological interrogation indicate that despite being designed to recruit VHL, the observed degradation of HBeAg induced by LH-1 and LH-3 is both largely independent of VHL, yet distinct from the activity of the HAP parental inhibitor, 2. We attempted to probe the role of the proteasome and Cullin-RING E3 ligase activation on the observed degradation of HBeAg via co-administration of LH-3 with proteasome inhibitor bortezomib and neddylation inhibitor MLN4924. Unfortunately, these treatments at typically used concentrations (bortezomib: 10 nM, MLN4924: 500 nM) led to reduced cell viability over the 24 h incubation, which confounded attempts to measure reversal of proteasome dependent degradation.
Preparation and assessment of truncated LH-3 derivatives
Having established that LH-1 and LH-3 function via a largely VHL-independent mechanism, we next investigated the molecular features responsible for degradation to aid in determining mechanism. Structurally, parental inhibitor 2 and LH-1/LH-3 differ by the addition of a linker and the VH032 motif yet have markedly different effects on intracellular HBeAg levels. Therefore, to determine the important moieties for this effect, we prepared a series of compounds in which the VHL-recruiting scaffold of LH-3 is truncated at five locations or replaced with an alternative E3 ligase recruiter (Fig. 4A). This included the consecutive removal of the 4-methylthiazole, benzylamine, trans-hydroxyproline and tert-leucine motifs to give LH-3-T1, LH-3-T2, LH-3-T3, LH-3-T4 respectively. Additionally, a des-hydroxy variant, LH-3-desHyd, was prepared where proline replaced trans-hydroxyproline, and LH-3-Pom, which replaced the VHL-recruiter with cereblon-recruiter, pomalidomide. These compounds were prepared in analogous fashion to LH-3, but also included solid-phase synthesis routes in some examples (see ESI† Fig. S5).43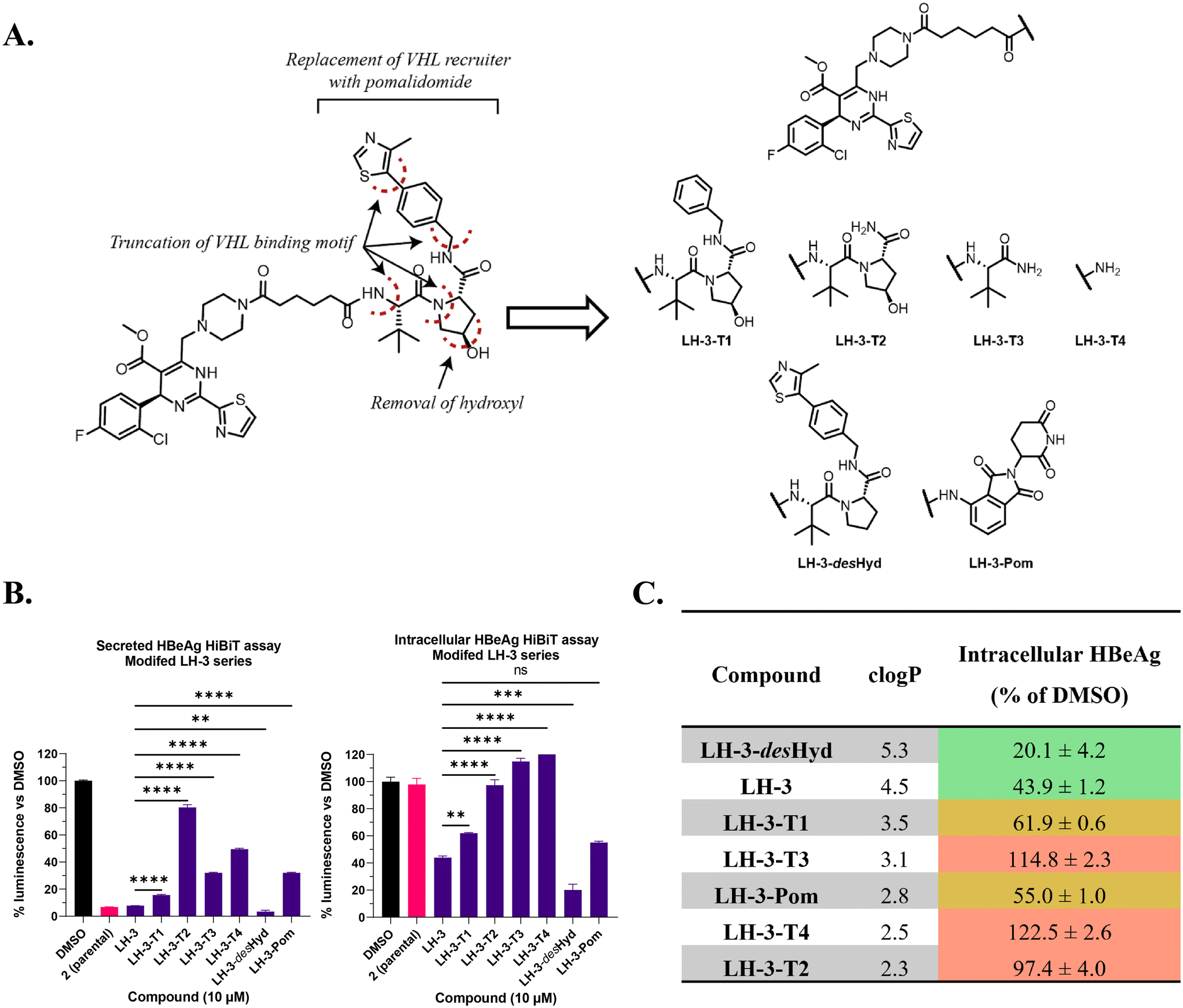 | ||
Fig. 4
A. A series of LH-3 analogues were prepared bearing truncated or altered E3L recruiting domains. B and C. HEK293THiBiT-HBeAg-S cells were treated with LH-3 derivatives at 10 μM for 24 h then levels of secreted (B) and intracellular (C) HBeAg levels were assessed via HiBiT assay. Varying levels of activity against secreted HBeAg were observed, while only LH-3-T1, LH-3-desHyd and LH-3-Pom retained activity against intracellular HBeAg. **** represents P < 0.0001, *** P ≤ 0.001, ** P ≤ 0.01, * P ≤ 0.05, ns = P > 0.05 as determined by one-way ANOVA. D. Consensus log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P of LH-3 and derivatives were calculated using Swiss ADME,44 and compared to activity against intracellular HBeAg. Relative activity is colour coded as high (green), moderate (yellow), or low (red). P of LH-3 and derivatives were calculated using Swiss ADME,44 and compared to activity against intracellular HBeAg. Relative activity is colour coded as high (green), moderate (yellow), or low (red). | ||
The truncated derivatives of LH-3 were assessed in the HiBiT assay at 10 μM (Fig. 4B). LH-3-T1 (16%) and LH-3-desHyd (3%) reduced secreted HBeAg to similar levels as LH-3 (8%). Other derivatives caused a more modest reduction in HBeAg, with LH-3-T2 (80%) exhibiting the most dramatic reduction in potency compared to LH-3.
Monitoring intracellular levels of HBeAg following compounds treatment revealed key motifs within the VH032 scaffold (Fig. 4C). LH-3-desHyd (20%) exhibited an apparent improvement in activity compared to LH-3 (44%) despite it lacking the hydroxyl group essential for VHL binding. However, this compound also exhibited some cytotoxicity at 10 μM in the parallel ViaLight® assay (Fig. S4†). Further analysis illustrated that extensive truncations caused the compounds to shift from reducing intracellular HBeAg levels, to causing an accumulation, closer to the effect of a HAP inhibitor such as 2. This is particularly evident when LH-3-T1 (active, 62% of DMSO levels) and LH-3-T2 (inactive, 97% of DMSO levels) are compared.
These data indicated that the full VH032 moiety is not entirely essential for activity, but drastic alteration hinders the ability of these compounds to act as HBeAg degraders. LH-3-T1 retained activity in both assays, revealing that the 4-methylthiazole is not required for the novel intracellular degradation mechanism or for inhibition of HBeAg secretion. In contrast, removal of the benzylamine group and subsequent truncations reduced the potency of the compounds as inhibitors of HBeAg secretion and caused the compounds to exhibit limited effect on intracellular HBeAg. Treatment with LH-3-T3 (115%) and LH-3-T4 (122%) led to an accumulation of intracellular HBeAg compared to control level, similar to the phenotype observed for cells treated with the parental compound in previous assays (see Fig. 3). In this way, truncates LH-3-T2, LH-3-T3 and LH-3-T4, which failed to reduce intracellular HBeAg levels, can be considered HAP compounds with reduced potency, rather than HBeAg degraders. Therefore, it is the properties of the 4-methylthiazole and benzyl group which seem to be important for this mechanistic switch. Interestingly, LH-3-Pom (55%) was also able to reduce intracellular HBeAg levels and retain the distinct degradation effect in contrast to 2.
To determine if any link between activity and lipophilicity was present, the logP of LH-3 and its derivatives was calculated using the SIB Swiss ADME web tool (Fig. 4D).44 In considering these data, a trend emerges whereby LH-3 and LH-3-desHyd, which exhibit the highest activity, also have the highest clogP values of 4.5 and 5.3, respectively. As clogP decreases towards 2, activity is heavily reduced. LH-3-Pom is a notable exception to this trend, with comparable activity to LH-3 despite a clogP of 2.8. This suggests that other properties, such as aromaticity, may also be important.
Preparation and assessment of hydrophobic tagged LH-3 derivatives
Having considered these results and the apparent importance of lipophilicity, we hypothesised that degradation may be occurring via a hydrophobic-tagged mechanism where the VH032 moiety is acting as HyT and triggering the cellular PQR.27 To investigate this hypothesis, three HyT-HAP analogues of LH-3 were prepared which replaced the VHL recruiting motif with known HyT groups (Fig. 5A) and utilised a similar synthetic strategy as for the preparation of LH-3 (see ESI† for full details).28 | ||
| Fig. 5 A. Compounds were prepared based on LH-3 where the VH032 motif was replaced with reported hydrophobic tags. B and C. HEK293THiBiT-HBeAg-S cells were titrated with HyT deriviatives for 24 h and assessed for secreted (B) and intracellular HBeAg levels (C). | ||
We assessed the activity of LH-3-adam and LH-3-menth and LH-3-fluor alongside LH-3 using the established HiBiT assay (Fig. 5B). All HyT compounds exhibited a similar trend against secreted levels of HBeAg when compared to LH-3. At 10 μM LH-3 reduced HBeAg to 9% of DMSO levels, while LH-3-adam1 (27%) most potently reduced HBeAg levels amongst the HyT compounds.
Assessment of intracellular HBeAg levels revealed that the HyT analogues were less potent than LH-3 (Fig. 5C). Consistent with previous assays, at 10 μM LH-3 reduced intracellular levels to 45% of control. In comparison, the activity of LH-3-adam (93%) and LH-3-menth (77%) was significantly reduced, while LH-3-fluor (110%) caused an accumulation of HBeAg. These results suggest that certain properties of the VH032 scaffold are more suited inducing HBeAg degradation than any of the selected HyTs. However, the reduction of intracellular HBeAg by LH-3-menth demonstrates that it is possible to replicate the LH-3 intracellular HBeAg depletion phenotype by replacing the VH032 moiety with a HyT. This provides preliminary evidence for HyT-induced degradation, however further work is required to confirm the mechanism responsible.
Virological assessment of LH-3 related compounds
Having identified several compounds of interest, including compound LH-3, we sought to assess its activity in a viral infection model. We were particularly interested to determine if the mechanistic difference observed for LH-3 compared to the parental inhibitor in HEK293T cells would be recapitulated in an infection model.As LH-3 incorporates a ligand capable of binding both HBeAg and HBcAg, we assessed the effect on both protein populations following compound treatment. HBV expressing AD38 cells were treated with test compounds for 6 days. Cell lysates were probed using an anti-HBV core protein antibody (Fig. 6C). LH-3 and cisLH-3 both caused a strong reduction in HBcAg (p21) relative to DMSO, although less intensely than observed for the parental compound, 2. Low assay sensitivity prevented the detection of HBeAg.
 | ||
| Fig. 6 Virological assessment of degrader candidates. A. HepAD38 cells treated with compounds at 10 μM for 6 days before removal of media and western blot analysis of cell lysate using anti-core protein antibody, B. Antiviral activity of LH-3/cisLH-3 was assessed in Huh7 cells transfected with either WT or T109I mutant strain of HBV. The cells were treated with compound at 10 μM for 24 h and media levels of HBeAg quantified. * P ≤ 0.05, ns = P > 0.05 as determined by two-way ANOVA. | ||
A more sensitive clinical assay was utilised to determine if LH-3 and cisLH-3 were able to reduce secreted HBeAg in both WT and HAP-resistant strains of HBV. For this study, Huh7 cells were transfected with the entire genome (1.3mer) of either wild-type (WT) or a mutant strain of HBV encoding the T109I mutation in the core protein. The T109I HBV strain is known to confer resistance to HAP HBV inhibitors, reducing their binding affinity and causing a 10 to 20-fold reduction in EC50 as measured by inhibition of HBV replication.35,45
Following incubation with parental HAP inhibitor, 2, lead compound LH-3, or control compounds cisLH-3 at 10 μM for 24 h, secreted HBeAg levels were assessed using the Diasorin Liaison® system. The collected data is summarised in Fig. 6B and highlights the following key findings. Treatment of the WT expressing cells with both 2 and LH-3 decreased secreted HBeAg levels to ∼8% of the untreated levels, indicating that LH-3 is effective at curtailing virally produced WT HBeAg, supporting our findings from the transfected HiBiT screening cell line.
When considering the T109I mutant HBV LH-3 demonstrated increased efficacy compared to the parental inhibitor. As anticipated, given the resistance mutation, the efficacy of 2 was reduced by 2 to 3-fold when moving from WT to resistant strain (8% vs. 23.5% ± 5.5) which was significantly inferior to its activity against WT strain (P = 0.0438). In comparison, heterobifunctional compound LH-3 reduced HBeAg secretion to a similar extent to the WT transfection model (11.5% ± 1.5). These data suggest that the conversion of 2 into a bifunctional molecule imparts increased efficacy against a HAP resistant strain of HBV.
Treatment of WT HBV expressing cells with cisLH-3 reduced HBeAg secretion to 17.5% ± 4.5 as compared to LH-3 (8.5% ± 1.5). This is consistent with the trend observed during HiBiT assay assessment where activity against secreted HBeAg is reduced, but not lost, upon inverting the stereochemistry of the hydroxyproline residue (see Fig. 3C). This indicates that the antiviral activity of LH-3 may be partially reliant on VHL binding. In the resistant T109I expressing cells cisLH-3 reduced HBeAg secretion to ∼22%, which was not significantly different to the activity against the WT group, providing evidence that the VHL-independent degrader activity was also crucial to maintaining activity against the mutated T109I target.
Discussion
The work presented here describes the identification of PROTAC-like molecules capable of degrading HBeAg, an important HBV protein. These compounds were derived from a HAP scaffold closely related to preclinical inhibitor NVR-010-001-E2 and display an interesting and diverse set of phenotypes across both transfected and viral infection models.Biochemical testing using a transfected HiBiT reporter system enabled rapid assessment of both secreted and intracellular HBeAg levels following drug treatment, which facilitated the identification of multiple candidates, including LH-1 and LH-3, capable of reducing HBeAg levels in a distinct manner from the parental inhibitor, presumably via the degradation of HBeAg. Following the application of negative control compounds and a VHL KO cell line, we were particularly surprised to observe that HBeAg degradation was largely VHL-independent, despite designing the compounds to recruit VHL.
These findings serve to highlight the importance of a thorough investigation of the mechanism unpinning observed degradation. This has become an increasingly important discussion in the PROTAC literature due to a notable report of an unexpected E3L being responsible for degradation,46 and the appearance of false positives due to secondary cellular effects or assay artefacts.47
While an unexpected mechanism of degradation has been reported for a PROTAC recruiting the aryl hydrocarbon receptor (AhR),48 to our knowledge, our findings represent the first report of a VHL-independent degrader designed to recruit VHL. Given the wide range of proteins targeted using this approach, this finding is of note and raises the question of how many other reported degraders are acting in unexpected ways. Our investigation into the role of the structural motifs present within the VHL recruiting domain demonstrated the importance of the hydrophobic para-benzylamine for the observed degradation. Given the abundance of this functionality in VHL-recruiting PROTACs, it is possible that it may be a contributing factor to other successful degraders. The installation of known hydrophobic tags generated compounds LH-3-menth and LH-3-adam capable of recapitulating the effects of LH-3, albeit less potently. Thus, if LH-3 is facilitating degradation via a HyT mechanism, it appears that compared to the HyTs utilised, the structure of the VHL recruiting moiety is more appropriately configured to initiate misfolding and induce the PQR mechanism responsible for HyT-mediated degradation. It is also important to acknowledge that the data presented here does not consider the effect of the modifications on factors such as cell permeability or target occupancy. Consequently, it is difficult to entirely resolve the intricacies of the structure–activity relationship of the VH032 or HyT modifications.
Alongside understanding the important structural motifs, it is crucial to more extensively investigate the cellular pathways involved in degradation, particularly those involving the proteasome. Attempts to probe this pathway using standard methods such as administration of test compounds with proteasome inhibitor bortezomib and neddylation inhibitor MLN4924 were inconclusive due to interference with the transfection system as compared to reported mammalian constructs.49,50 We had similar experience with HiBiT-Mpro constructs.42 Other strategies using an alternate experimental set up may be necessary to study these processes. One previously successful approach involved a CRISPR-Cas9 screen of gene dependency during the identification of DCAF16 as the E3L responsible for the activity of a bivalent molecular glue.46 A similar strategy could be utilised here to specifically probe the role of Cullin-RING E3L in the observed degradation. Any advances made towards identifying key proteins will serve to improve understanding of this system, as well as additional forms of degradation, HyT-driven or otherwise.
Assessment of these compounds in cellular HBV infection models confirmed activity of the heterobifunctional compounds against virally produced protein. Application of viral infection models is crucial and differs from the HiBiT reporter assay for several reasons. We firstly note that HBeAg was the primary target in this investigation and the sole overexpressed viral protein in the transfected cells. In the viral infection cellular context, the ligand would be expected to bind both HBeAg and HBcAg and exert its antiviral effect through a binary mechanism including the disruption of capsid formation. As a result, there is an expected background level of antiviral activity which contributes to a reduction in overall protein levels, including HBeAg. Second, it would be expected that the different compound structures with modified linkers are likely to have different affinities for the various protein forms, hence exhibit varied levels of background antiviral activity. Third, the assays are conducted over 6 days, allowing for a more pronounced effect of protein turnover (synthesis, secretion and degradation) than in the 24 h treatment of the transfection model.
Through immunoblotting we were able to assess HBcAg levels and confirmed activity of LH-3 and cisLH-3 against core protein albeit less effectively than the parental compound. Unfortunately, the assay format failed to detect HBeAg levels due to a lack of sensitivity. However, upon moving to a more sensitive assay format, we demonstrated that LH-3 shows a unique antiviral phenotype against a HAP resistant strain of HBV. The consistent efficacy of LH-3 across the WT and HAP-resistant HBV strains suggests that the binding affinity of the HAP component of the molecule may be less crucial to its efficacy than for the parental HAP compound. This feature could be attributed to the TPD mechanism where PROTACs and other heterobifunctional degraders are less reliant on tight binding to their target, due to their catalytic mechanism of degradation.51 Harnessing these benefits by further optimising anti-HBV degraders is therefore an attractive option in efforts to overcome drug resistant strains of the virus.
A TPD approach to the treatment of HBV infection is an exciting, yet underexplored field of research, with this study serving as the first report of small-molecule degrader of HBeAg, albeit one which functions via an ambiguous mechanism. When considering this unexpected mechanism, this work also highlights the complexity of TPD, particularly in the field of antiviral drug discovery. Additionally, the data presented here contributes evidence to the growing body of work that indicates targeted degraders of viral proteins have additional antiviral benefits over the parental compound. While significant optimisation and mechanistic investigation is required to develop a potent degrader of HBeAg, we are encouraged to continue investigating protein degraders of HBeAg along with other viral protein targets.
Methods
Synthetic chemistry
Full details can be found in the ESI.†Computational determination of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P
P
The SMILES strings of the compounds of interested were input into the Swiss ADME Web tool (https://www.swissadme.ch/) and results exported.44 The consensus logP values were collated and compared.
Cell line generation
:1, Fugene:DNA) and grown for 72 h before flow cytometry sorting of the eGFP expressing cell population. After cell recovery and expansion, cells were frozen for archive and passaged for HBeAg-HiBiT reporter cell line generation. Loss of VHL target was verified by Western blotting vs. HEK293T parental cells using an anti-VHL rabbit polyclonal antibody (Cell Signaling).
HiBiT assay
For DOX induction of target and monitoring degrader activity against secreted HBeAg target by HiBiT assay, 96 well white wall plates were seeded with 20000 cells per well in triplicate overnight. The next day, various concentrations of DOX were added and cells incubated for 24 hours at 37 °C (for degrader treatments – degraders were added at the same time as DOX induction and then incubated for another 24 h to degrade target). The next day, 50 μL of media per well was removed and transferred to a fresh white walled 96-well plate and were subjected to a Nano-Glo® HiBiT Extracellular Detection System assay as per manufacturer's instructions. In the source plate, the remaining media was removed and wells carefully washed once in DMEM + 10% FBS before a fresh 50 μL of media was then added back in – the level of intracellular HBeAg was then measured using the Nano-Glo® HiBiT Lytic Detection System assay as per manufacturer's instructions.
Western blot detection of HBeAg-HiBiT protein
For verification of target identity and degrader activity on target by Western blot, cells were plated at 500000 per well in a 6-well plate overnight. The next day, 1 μg mL−1 final concentration of DOX was added and cells were incubated for 24 h. Degraders were added at the same time as DOX and incubated for another 24 h. After incubation, for HBeAg, supernatant samples were taken before cells were harvested, washed once in ice cold PBS and then lysed in RIPA buffer supplemented with protease/phosphatase inhibitor cocktail (Pierce – HALT cocktail). Lysates were clarified by centrifugation at 21000 × g for 20 min at 4 °C and then subjected to a BCA assay to determine protein concentration. 50 μg of each lysate or 20 μL of undilute supernatant mixed with 4 × LDS reducing sample buffer were then run in MES buffer on 4–12% Bis-Tris SDS-PAGE gels at 150 V for 1 h and transferred to PVDF membranes using an iBlot apparatus at P3 for 7 min. After 1 h of blocking the membrane in Intercept blocking buffer (Licor), membranes were probed with anti-HiBiT mouse monoclonal antibody at 1 μg mL−1 (Promega) and with anti-HiBiT tag mouse monoclonal antibody at 1:2000 (Promega), anti-HBcAg mouse monoclonal antibody at 1:500 (Santa Cruz Biotechnology) or anti-GAPDH rabbit monoclonal antibody at 1:10000 (Cell Signaling) overnight at 4 °C, all diluted in Intercept buffer + 0.1% Tween-20. After three washes in TBS + 0.1% Tween-20, membranes were probed for 1 h at room temperature with anti-mouse AF680 and anti-rabbit AF800 secondary antibodies (Thermo Fisher) diluted 1:10000 in Intercept buffer + 0.1% Tween-20 followed by three final washes. Membranes were then scanned on the Odyssey XF infrared scanning system (Licor) for image acquisition.
HepAD38 culture and western blot
HepAD38 cells52 (kindly provided by Gilead Sciences USA) were cultured in DMEM/F-12 (ThermoFisher) supplemented with 10% FCS, penicillin/streptomycin (ThermoFisher) and 500 μg mL−1 geneticin (ThermoFisher) at 37 °C, 5% CO2.To examine the effect of compound treatment on intracellular core protein levels, HepAD38 cells were seeded into 12-well plates and cultured in the presence of each compound at 10 μM with media replenished at day 3. After 6 days of culture, media was aspirated, cells washed with PBS, and cells lysed in NP40 lysis buffer (1% NP-40, 150 mM NaCl, 50 mM Tris, pH 7.5 with protease inhibitor (Pierce)) for 20 min at 4 °C with gentle rocking. Lysates were then clarified at 5000 g for 5 min at 4 °C.
For Western blot analysis, clarified lysates were diluted in Laemmli buffer (Biorad) and boiled for 5 min prior to loading on Mini-PROTEAN TGX Precast Protein Gels (Any kD, Biorad) alongside MagicMark XP Western Protein Standard (ThermoFisher) and electrophoresed using Biorad MINI-Protean electrophoresis apparatus in 1× Tris/Glycine/SDS buffer (Biorad). After electrophoresis, samples were transferred onto nitrocellulose membranes using semi-dry transfer apparatus (Biorad Trans Blot Turbo system). Membranes were then blocked overnight in 3% skim milk powder in PBS-Tween (0.1%) at 4 °C. After washing with PBS-Tween (0.1%) at room temp, membranes were probed with primary mouse anti-hepatitis B virus core antigen antibody [C1] (Abcam) at 1:1000 for 3 h, and secondary rabbit anti-mouse-HRP (Sigma) for 1 h, and detection of bands using Western Lightning Plus chemiluminescent substrate (Perkin Elmer) and Biorad ChemiDoc and Image lab software.
Virological HBeAg secretion assay
Huh7 cells (maintained on DMEM + 10% FCS + penicillin/streptomycin) were transfected with 1.3mer hepatitis B virus infectious clones encoding either wildtype HBV (1.3_HBVgenD-wt) or HBV encoding the core_T109I mutation (1.3_HBVgenD_c_T109I), using Fugene 6 Transfection reagent (Promega) according to manufacturer's instructions, and incubated at 37 °C, 5% CO2.At 24 hours post-transfection, cell culture media was removed and replaced with fresh media containing each compound at a final concentration of 10 μM, or vehicle control (DMSO). After a further 24 hours incubation, cell culture supernatant was collected, and HBeAg quantified using the Diasorin Liaison® diagnostic platform.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There is no conflict of interest to declare.Acknowledgements
L. T. H. was the recipient of a Research Training Program scholarship from the Australian Government.References
- WHO, Global Hepatitis Report 2017, Geneva, 2017 Search PubMed.
- S. K. Fung and A. S. F. Lok, Nat. Clin. Pract. Gastroenterol. Hepatol., 2004, 1, 90–97 CrossRef CAS PubMed.
- U. Viswanathan, N. Mani, Z. Hu, H. Ban, Y. Du, J. Hu, J. Chang and J. T. Guo, Antiviral Res., 2020, 182, 104917 CrossRef CAS PubMed.
- A. Zlotnick, B. Venkatakrishnan, Z. Tan, E. Lewellyn, W. Turner and S. Francis, Antiviral Res., 2015, 121, 82–93 CrossRef CAS PubMed.
- F. Messageot, S. Salhi, P. Eon and J.-M. Rossignol, J. Biol. Chem., 2003, 278, 891–895 CrossRef CAS PubMed.
- X. Hong, L. Luckenbaugh, M. Mendenhall, R. Walsh, L. Cabuang, S. Soppe, P. A. Revill, D. Burdette, B. Feierbach, W. Delaney and J. Hu, J. Virol., 2021, 95 DOI:10.1128/jvi.01695-01620.
- D. Milich and T. J. Liang, Hepatology, 2003, 38, 1075–1086 CrossRef CAS PubMed.
- M. A. DiMattia, N. R. Watts, S. J. Stahl, J. M. Grimes, A. C. Steven, D. I. Stuart and P. T. Wingfield, Structure, 2013, 21, 133–142 CrossRef CAS PubMed.
- T. Lang, C. Lo, N. Skinner, S. Locarnini, K. Visvanathan and A. Mansell, J. Hepatol., 2011, 55, 762–769 CrossRef CAS PubMed.
- K. Padarath, A. Deroubaix and A. Kramvis, Viruses, 2023, 15, 857 CrossRef CAS PubMed.
- M. T. Chen, J.-N. Billaud, M. Sällberg, L. G. Guidotti, F. V. Chisari, J. Jones, J. Hughes and D. R. Milich, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14913–14918 CrossRef CAS PubMed.
- M. Chen, M. Sällberg, J. Hughes, J. Jones, G. Guidotti Luca, V. Chisari Francis, J.-N. Billaud and R. Milich David, J. Virol., 2005, 79, 3016–3027 CrossRef CAS PubMed.
- I. Lazarevic, A. Banko, D. Miljanovic and M. Cupic, Viruses, 2019, 11, 778 CrossRef CAS PubMed.
- P. D. Garcia, J. H. Ou, W. J. Rutter and P. Walter, J. Cell Biol., 1988, 106, 1093–1104 CrossRef CAS PubMed.
- X. Hong, N. Bianchini Emily, C.-Y. Wang Joseph and J. Hu, mBio, 2023, 14, e03501–e03522 CrossRef PubMed.
- D. R. Milich, Hum. Vaccines Immunother., 2019, 15, 2187–2191 CrossRef PubMed.
- J. Wu, Z. Meng, M. Jiang, R. Pei, M. Trippler, R. Broering, A. Bucchi, J. P. Sowa, U. Dittmer, D. Yang, M. Roggendorf, G. Gerken, M. Lu and J. F. Schlaak, Hepatology, 2009, 49, 1132–1140 CrossRef CAS PubMed.
- J. Samal, M. Kandpal and P. Vivekanandan, Sci. Rep., 2017, 7, 14371 CrossRef PubMed.
- A. M. Mohareb, A. F. Liu, A. Y. Kim, P. A. Coffie, M. G. Kouamé, K. A. Freedberg, A. Boyd and E. P. Hyle, J. Infect. Dis., 2022, 226, 1761–1770 CrossRef CAS PubMed.
- P. K. Parikh, N. H. Parikh, B. Mahalakshmi, K. M. Ranch, S. H. S. Boddu, B. R. Jayachandra and A. K. Tiwari, Arab. J. Chem., 2023, 16, 105013 CrossRef.
- C. R. Bourne, M. G. Finn and A. Zlotnick, J. Virol., 2006, 80, 11055–11061 CrossRef CAS PubMed.
- C. Bourne, S. Lee, B. Venkataiah, A. Lee, B. Korba, M. G. Finn and A. Zlotnick, J. Virol., 2008, 82, 10262–10270 CrossRef CAS PubMed.
- J. Lin, L. Yin, X.-Z. Xu, H.-C. Sun, Z.-H. Huang, X.-Y. Ni, Y. Chen and X. Lin, PLoS Pathog., 2022, 18, e1010204 CrossRef CAS PubMed.
- Z. Yan, D. Wu, H. Hu, J. Zeng, X. Yu, Z. Xu, Z. Zhou, X. Zhou, G. Yang, J. A. T. Young and L. Gao, Hepatology, 2019, 70, 11–24 CrossRef CAS PubMed.
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed.
- G. Dong, Y. Ding, S. He and C. Sheng, J. Med. Chem., 2021, 64, 10606–10620 CrossRef CAS PubMed.
- J. M. Tsai, R. P. Nowak, B. L. Ebert and E. S. Fischer, Nat. Rev. Mol. Cell Biol., 2024, 25, 740–757 CrossRef CAS PubMed.
- S. Xie, J. Zhu, J. Li, F. Zhan, H. Yao, J. Xu and S. Xu, J. Med. Chem., 2023, 66, 10917–10933 CrossRef CAS PubMed.
- A. Chakravarty and P. L. Yang, Antiviral Res., 2023, 210, 105480 CrossRef CAS PubMed.
- Y. R. Alugubelli, J. Xiao, K. Khatua, S. Kumar, L. Sun, Y. Ma, X. R. Ma, V. R. Vulupala, S. Atla, L. R. Blankenship, D. Coleman, X. Xie, B. W. Neuman, W. R. Liu and S. Xu, J. Med. Chem., 2024, 67, 6495–6507 CrossRef CAS PubMed.
- M. de Wispelaere, G. Du, K. A. Donovan, T. Zhang, N. A. Eleuteri, J. C. Yuan, J. Kalabathula, R. P. Nowak, E. S. Fischer, N. S. Gray and P. L. Yang, Nat. Commun., 2019, 10, 3468 CrossRef PubMed.
- Z. Xu, X. Liu, X. Ma, W. Zou, Q. Chen, F. Chen, X. Deng, J. Liang, C. Dong, K. Lan, S. Wu and H.-B. Zhou, Cell Insight, 2022, 1, 100030 CrossRef PubMed.
- S. Cheng, Y. Feng, W. Li, T. Liu, X. Lv, X. Tong, G. Xi, X. Ye and X. Li, Eur. J. Med. Chem., 2024, 275, 116629 CrossRef CAS PubMed.
- Z. Li, H.-Y. Liu, Z. He, A. Chakravarty, R. P. Golden, Z. Jiang, I. You, H. Yue, K. A. Donovan, G. Du, J. Che, J. Tse, I. Che, W. Lu, E. S. Fischer, T. Zhang, N. S. Gray and P. L. Yang, bioRxiv, 2024, preprint, DOI:10.1101/2024.06.01.596987.
- K. Klumpp, A. M. Lam, C. Lukacs, R. Vogel, S. Ren, C. Espiritu, R. Baydo, K. Atkins, J. Abendroth, G. Liao, A. Efimov, G. Hartman and O. A. Flores, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 15196 CrossRef CAS PubMed.
- S. Nair, L. Li, S. Francis, W. W. Turner, M. VanNieuwenhze and A. Zlotnick, J. Am. Chem. Soc., 2018, 140, 15261–15269 CrossRef CAS PubMed.
- W. Zhang, L. Guo, H. Liu, G. Wu, H. Shi, M. Zhou, Z. Zhang, B. Kou, T. Hu, Z. Zhou, Z. Xu, X. Zhou, Y. Zhou, X. Tian, G. Yang, J. A. T. Young, H. Qiu, G. Ottaviani, J. Xie, A. V. Mayweg, H. C. Shen and W. Zhu, J. Med. Chem., 2023, 66, 4253–4270 CrossRef CAS PubMed.
- Z. Qiu, X. Lin, W. Zhang, M. Zhou, L. Guo, B. Kocer, G. Wu, Z. Zhang, H. Liu, H. Shi, B. Kou, T. Hu, Y. Hu, M. Huang, S. F. Yan, Z. Xu, Z. Zhou, N. Qin, Y. F. Wang, S. Ren, H. Qiu, Y. Zhang, Y. Zhang, X. Wu, K. Sun, S. Zhong, J. Xie, G. Ottaviani, Y. Zhou, L. Zhu, X. Tian, L. Shi, F. Shen, Y. Mao, X. Zhou, L. Gao, J. A. T. Young, J. Z. Wu, G. Yang, A. V. Mayweg, H. C. Shen, G. Tang and W. Zhu, J. Med. Chem., 2017, 60, 3352–3371 CrossRef CAS PubMed.
- M. K. Schwinn, T. Machleidt, K. Zimmerman, C. T. Eggers, A. S. Dixon, R. Hurst, M. P. Hall, L. P. Encell, B. F. Binkowski and K. V. Wood, ACS Chem. Biol., 2018, 13, 467–474 CrossRef CAS PubMed.
- K. M. Riching, S. Mahan, C. R. Corona, M. McDougall, J. D. Vasta, M. B. Robers, M. Urh and D. L. Daniels, ACS Chem. Biol., 2018, 13, 2758–2770 CrossRef CAS PubMed.
- K. M. Riching, S. D. Mahan, M. Urh and D. L. Daniels, High-Throughput Cellular Profiling of Targeted Protein Degradation Compounds Using HiBiT CRISPR Cell Lines, 1940-087X, 2020.
- B. Pan, S. Mountford, M. Kiso, D. Anderson, G. Papadakis, K. Jarman, D. Tilmais, B. Maher, T. Tran, J. Shortt, S. Yamayoshi, Y. Kawaoka, P. Thompson, S. Greenall and N. Warner, Research Square, 2024 DOI:10.21203/rs.3.rs-3932156/v1.
- L. T. Hales and P. E. Thompson, Chem. – Eur. J., 2023, 29, e202301975 CrossRef CAS PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- A. D. Huber, D. L. Pineda, D. Liu, K. N. Boschert, A. T. Gres, J. J. Wolf, E. M. Coonrod, J. Tang, T. G. Laughlin, Q. Yang, M. N. Puray-Chavez, J. Ji, K. Singh, K. A. Kirby, Z. Wang and S. G. Sarafianos, ACS Infect. Dis., 2019, 5, 750–758 CrossRef CAS PubMed.
- O. Hsia, M. Hinterndorfer, A. D. Cowan, K. Iso, T. Ishida, R. Sundaramoorthy, M. A. Nakasone, H. Imrichova, C. Schätz, A. Rukavina, K. Husnjak, M. Wegner, A. Correa-Sáez, C. Craigon, R. Casement, C. Maniaci, A. Testa, M. Kaulich, I. Dikic, G. E. Winter and A. Ciulli, Nature, 2024, 627, 204–211 CrossRef CAS PubMed.
- V. Vetma, L. C. Perez, J. Eliaš, A. Stingu, A. Kombara, T. Gmaschitz, N. Braun, T. Ciftci, G. Dahmann, E. Diers, T. Gerstberger, P. Greb, G. Kidd, C. Kofink, I. Puoti, V. Spiteri, N. Trainor, H. Weinstabl, Y. Westermaier, C. Whitworth, A. Ciulli, W. Farnaby, K. McAulay, A. B. Frost, N. Chessum and M. Koegl, ACS Chem. Biol., 2024, 19, 1484–1494 CrossRef CAS PubMed.
- T. Shoda, N. Ohoka, G. Tsuji, T. Fujisato, H. Inoue, Y. Demizu, M. Naito and M. Kurihara, Pharmaceuticals, 2020, 13, 34 CrossRef CAS PubMed.
- X. Qu, H. Liu, X. Song, N. Sun, H. Zhong, X. Qiu, X. Yang and B. Jiang, Eur. J. Med. Chem., 2021, 218, 113328 CrossRef CAS PubMed.
- Y.-Q. Li, W. G. Lannigan, S. Davoodi, F. Daryaee, A. Corrionero, P. Alfonso, J. A. Rodriguez-Santamaria, N. Wang, J. D. Haley and P. J. Tonge, J. Med. Chem., 2023, 66, 7454–7474 CrossRef CAS PubMed.
- D. P. Bondeson, A. Mares, I. E. D. Smith, E. Ko, S. Campos, A. H. Miah, K. E. Mulholland, N. Routly, D. L. Buckley, J. L. Gustafson, N. Zinn, P. Grandi, S. Shimamura, G. Bergamini, M. Faelth-Savitski, M. Bantscheff, C. Cox, D. A. Gordon, R. R. Willard, J. J. Flanagan, L. N. Casillas, B. J. Votta, W. den Besten, K. Famm, L. Kruidenier, P. S. Carter, J. D. Harling, I. Churcher and C. M. Crews, Nat. Chem. Biol., 2015, 11, 611–617 CrossRef CAS PubMed.
- S. K. Ladner, M. J. Otto, C. S. Barker, K. Zaifert, G. H. Wang, J. T. Guo, C. Seeger and R. W. King, Antimicrob. Agents Chemother., 1997, 41, 1715–1720 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5md00118h |
| ‡ Current address: MRC Protein Phosphorylation and Ubiquitylation Unit, Sir James Black Centre, School of Life Sciences, University of Dundee, Dundee, DD1 5EH, UK. |
| This journal is © The Royal Society of Chemistry 2025 |