
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of spiroimidazopyridineoxindole, spiropyridopyrimidineoxindole and spiropyridodiazepineoxindole derivatives based on heterocyclic ketene aminals via a four-component reaction†
Fatemeh Rahimi,
Mohammad Bayat * and
Hajar Hosseini
* and
Hajar Hosseini
Department of Chemistry, Faculty of Science, Imam Khomeini International University, Qazvin, Iran. E-mail: bayat_mo@yahoo.com; m.bayat@sci.ikiu.ac.ir; Tel: +98 28 33780040
First published on 24th May 2019
Abstract
Here, we have described the synthesis of novel spiropyridineoxindole derivatives containing a pyridone ring via a four-component reaction between various diamines, 1,1-bis(methylthio)-2-nitroethylene, isatin derivatives and Meldrum's acid in the presence of p-toluenesulfonic acid. This protocol has some advantages such as the availability of starting materials, good yields, facile separation of products, the use of ethanol as an environmentally benign solvent and easy formation of three new bonds in one operation.
Introduction
The indole moiety is the most well-known heterocycle and a common and important feature of a variety of natural products and medicinal agents.1 Furthermore, it has been reported that the addition of the indole 3-carbon atom in the formation of spiroindoline derivatives highly enhances biological activity.2Spirooxindoles are a key structural element in a wide range of natural products with biological activities.3 They have attracted significant attention due to their useful pharmacological properties and biological activities including anti-microbial,4 anti-tumor,5 anti-tubercular,6 anti-inflammatory,7 anti-HIV,8 anti-fungal,9 the action as inhibitors of the human NK-1 receptor,10 anti-cancer,11 antibiotic,12 and anti-malarial.13
Spirocyclic oxindoles containing a six-membered moiety, especially a six-membered piperidine structure at the C-3 position, have a wide spectrum of biological activities; examples include the non-peptidyl growth hormone secretagogue MK-0677 (ref. 14) and potent non-peptide MDM2 inhibitors,15 which may have utility as anticancer agents (Fig. 1).
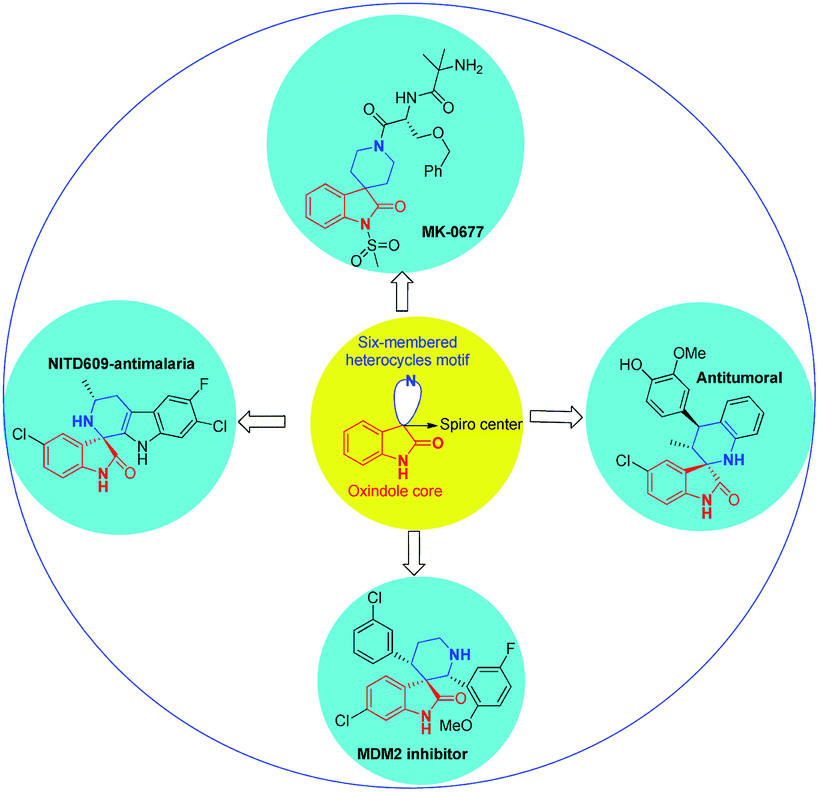 | ||
| Fig. 1 Bioactive and medicinally important compounds containing a spiropyridineoxindole skeleton. | ||
During the last years, there has been considerable interest in the synthesis of spirooxindoles fused in the 3-position from three-membered to seven-membered spiro-rings16 and other fused heterocyclic compounds, which have multi-ring structures similar to spirooxindole dihydropyridines and spirooxindolepyrans.17
In general, the synthesis of spirooxindole frameworks containing six-membered nitrogen rings has more limitations than that of five-membered heterocyclic moieties. In the last decade, synthetic methods for the generation of spiropyridineoxindoles via multicomponent reactions have been abundantly developed.18 Previous approaches for producing these structures relied on utilizing isatin, various C–H acids and a wide variety of different enamines as starting materials. Among these strategies, similar cases in which ketene aminals have been used as enamines are reported here (Fig. 2).
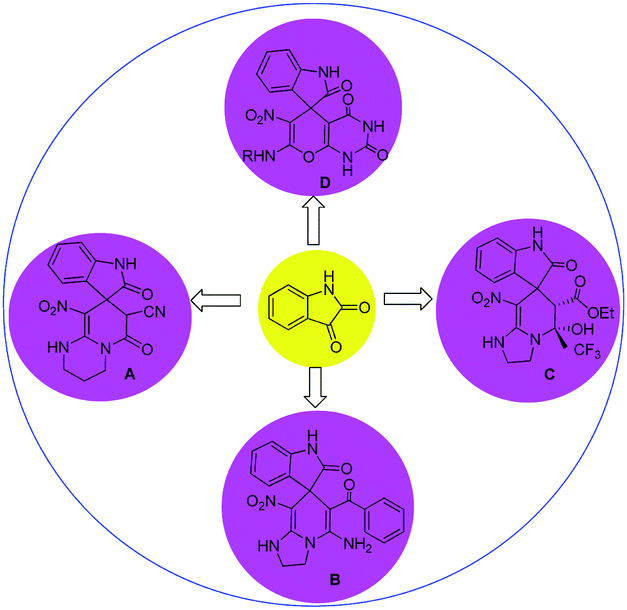 | ||
| Fig. 2 Summary of previous works of spiropyridineoxindole synthesis. | ||
In 2015, a green approach to the synthesis of the spirooxoindole derivative A was described in water in the presence of a catalytic amount of NaCl by using ethylcyanoacetate and heterocyclic ketene aminals.19 In 2014, another reaction was reported using benzoylacetonitrile and 2-(nitromethylene)imidazoline in ethanol and trimethylamine, which led to the corresponding product B.20
In 2013, a method was developed using a mixture of ethyl trifluoroacetate, HKA and different isatins in the presence of piperidine in ethanol to give product C.21 In 2019, the four-component reaction of various amines and nitroketene dithioacetal with isatin and barbituric acid derivatives in water afforded spirooxindole D.22
As a part of our program on the study of developing new multi-component reactions for the synthesis of heterocyclic compounds, we report the efficient synthesis of novel spiroimidazopyridineoxindole, spiropyridopyrimidineoxindole and spiropyridodiazepineoxindole structures via a one-pot, four-component reaction of nitro ketene aminals derived from the addition of various 1,n-diamines to 1,1-bis(methylthio)-2-nitroethylene, isatin and its derivatives and Meldrum's acid in the presence of p-TSA. To the best of our knowledge, these structures have not been synthesized so far and there are no reports on the preparation of spirooxindoles from Meldrum's acid.
Results and discussion
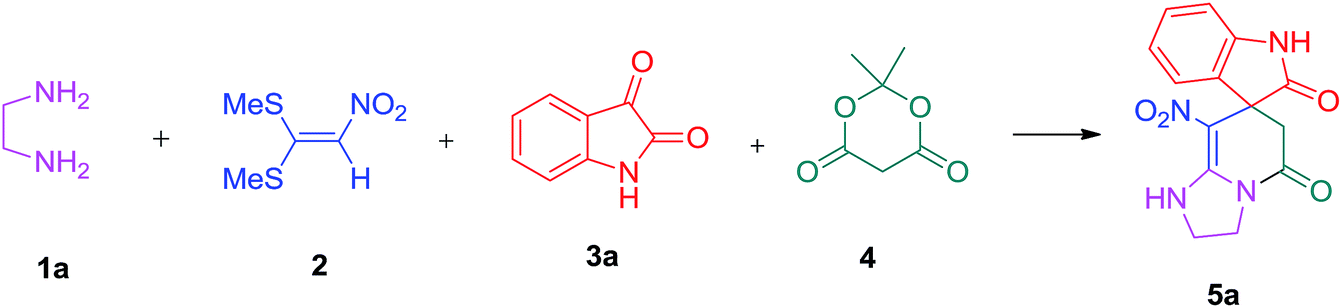
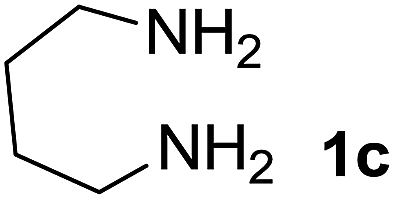
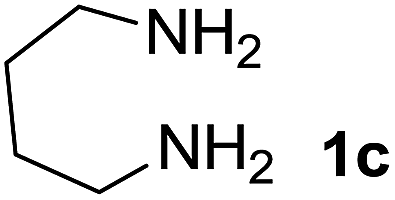
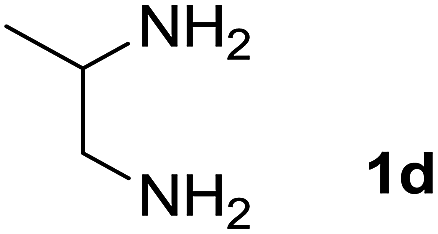
We prepared the spiropyridineoxindole derivative 5 via one-pot four-component condensation of diamine 1, 1,1-bis(methylthio)-2-nitroethylene 2, isatin 3 and Meldrum's acid 4 in the presence of p-TSA catalyst in ethanol under reflux conditions (Scheme 1). The reaction was completed after 0.15–6 h to afford the corresponding heterocyclic systems 5a–j in moderate to good yields (69–87%).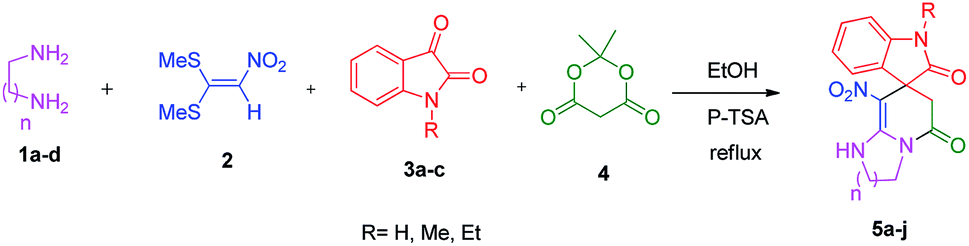 | ||
| Scheme 1 Synthetic scheme for the generation of products 5a–j. | ||
For our initial investigation, the reaction of diamine 1a (1 mmol), dithioacetal 2 (1 mmol), isatin 3a (1 mmol) and Meldrum's acid 4 (20% mmol) was considered as the model reaction. The effects of various catalysts, solvents and temperatures were monitored (Table 1). These results indicated that the best reaction conditions for the synthesis of spirooxindole 5a were obtained in ethanol as the solvent and by using p-TSA as the catalyst at reflux conditions. This procedure provided the highest yield of 87% and the shortest reaction time (Table 1, entry 7).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst (mol%) | Time (h) | Temp. (°C) | Yieldb (%) |
| a The reaction was performed using 1a, 2, 3a, 4 (1 mmol), catalyst (0.2 mmol), and solvent (10 mL).b Isolated yield based on 5a. | |||||
| 1 | EtOH | NEt3 | 1 | 80 | 75 |
| 2 | EtOH | Piperidine | 24 | 80 | None |
| 3 | EtOH | — | 5 | r.t | 60 |
| 4 | H2O/EtOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) :1, v/v) |
— | 6 | 80 | 65 |
| 5 | H2O/EtOH (3:1, v/v) |
— | 7 | 80 | 40 |
| 6 | EtOH | p-TSA | 24 | r.t | Trace |
| 7 | EtOH | p-TSA | 0.15 | 80 | 87 |
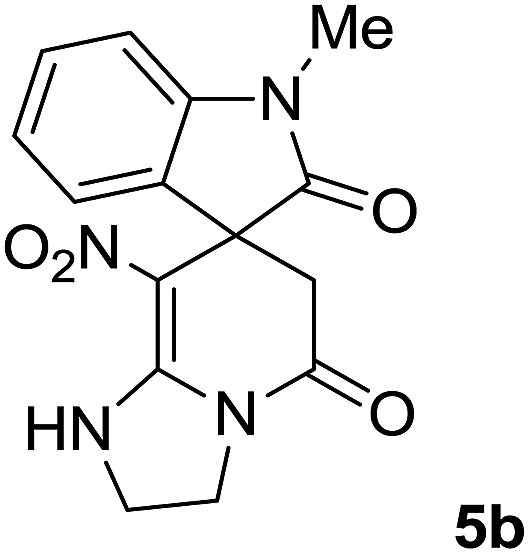
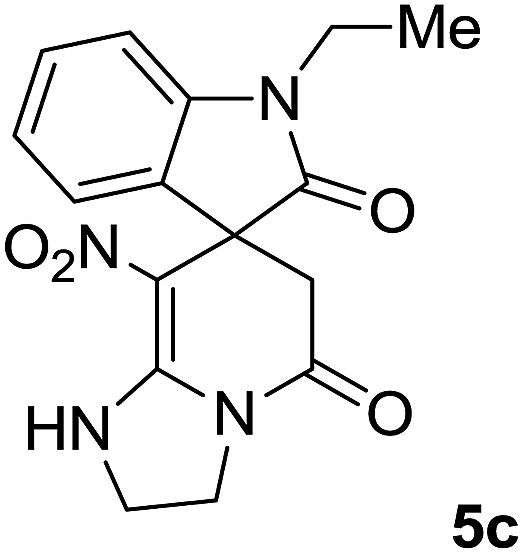
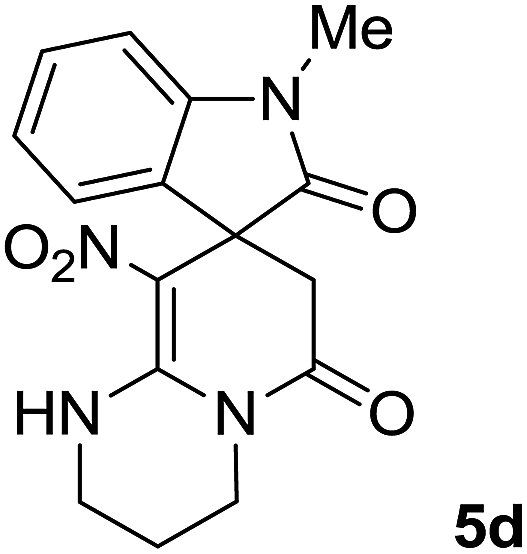
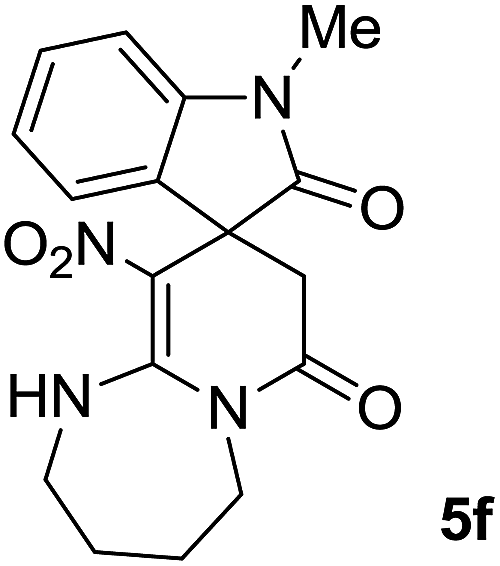
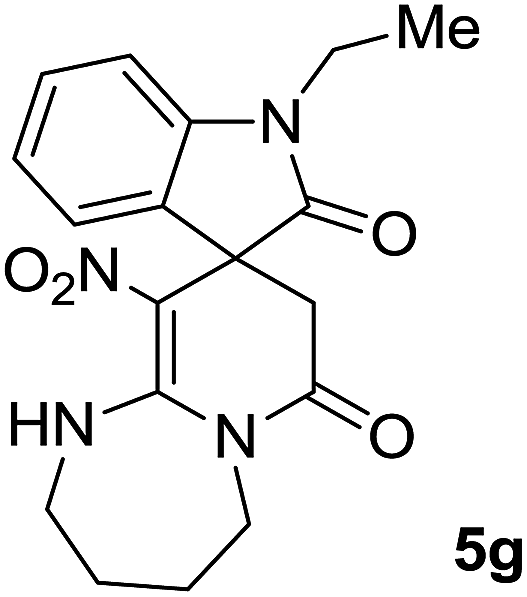
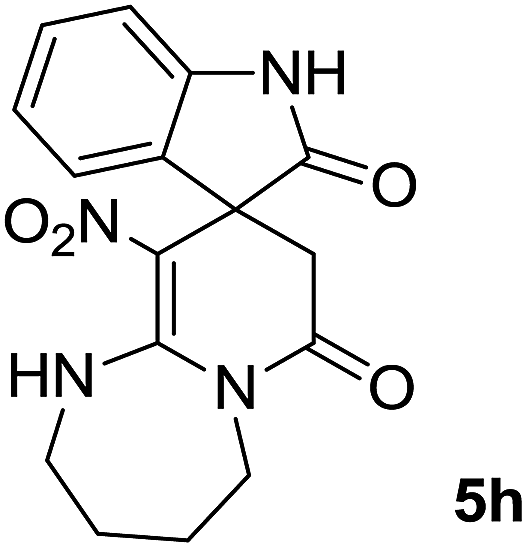
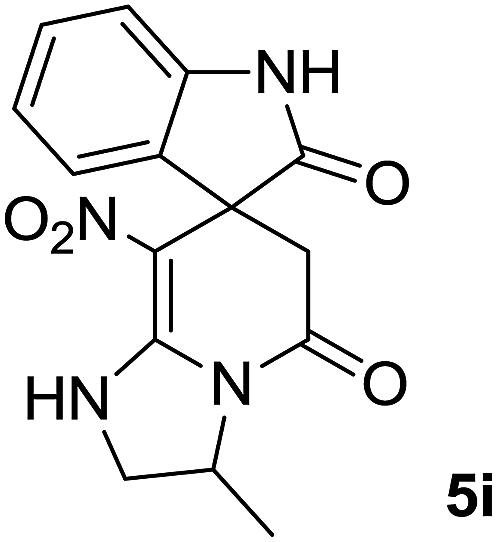
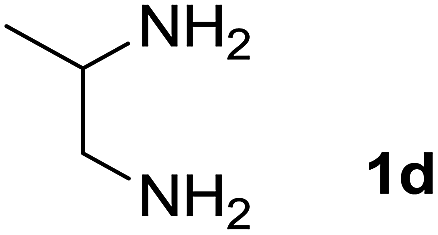
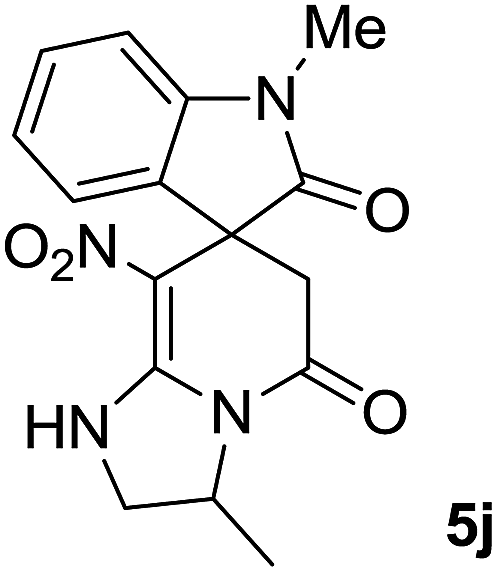
We explored the scope of this reaction by varying the structures of diamine 1a–d and isatin 3a–c components. The reaction proceeded to afford a series of spiropyridineoxindole derivatives 5a–j in 66–87% yields. The results are summarized in Table 2. This reaction was performed with other derivatives of isatin (N-benzyl and N-butyl isatin) under the same conditions but did not result in the product. Additionally, the reaction of N-ethyl isatin with 1,2-diaminopropane and 1,3-diaminopropane did not produce a product. Also, when 1,2-diaminocyclohexane was used for the synthesis of ketene aminal, no reaction occurred. We also tried using 1,2-diaminophenyl and aromatic amines in the reaction conditions. All the reactions were very slow and did not result in the desired products. The structures of compounds 5a–j were deduced from their mass, IR, 1H NMR, and 13C NMR spectroscopic data (ESI†).
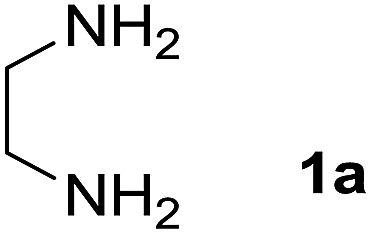
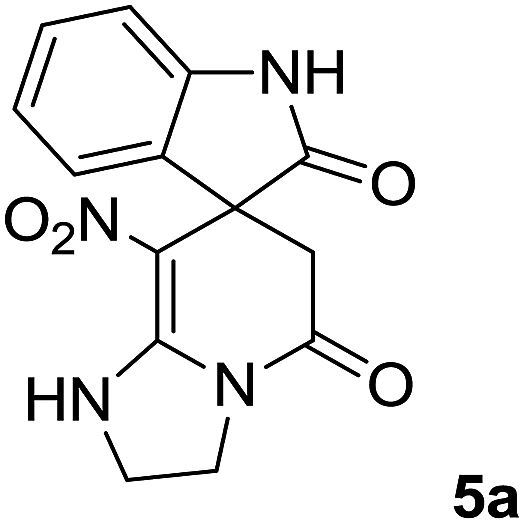
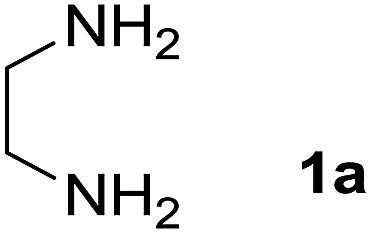
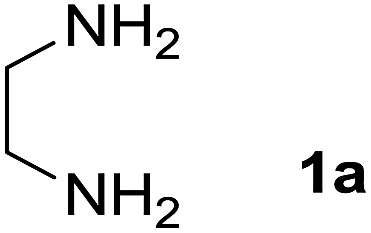
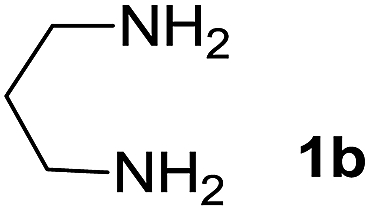
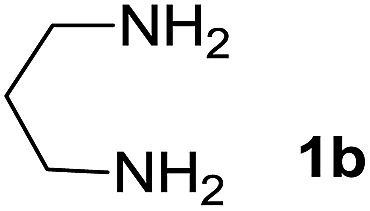
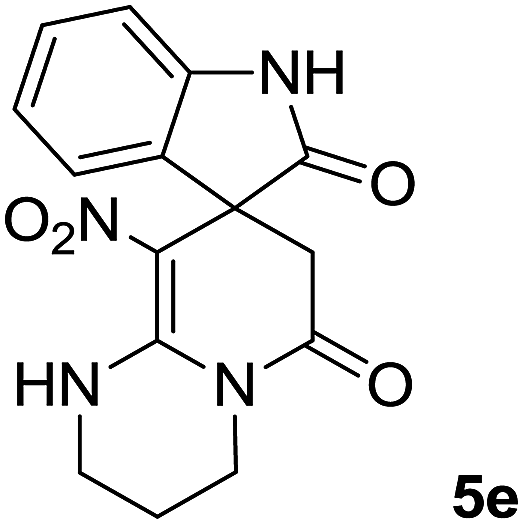

The 1H and 13C NMR spectra of the crude products clearly indicate the formation of the suggested products 5a–j. As a representative case, the key signals of the 1H and 13C NMR chemical shifts for 8-nitro-2,3-dihydro-1H-spiro[imidazo[1,2-a]pyridine-7,3′-indoline]-2′,5(6H)-dione 5a are shown in the ESI.†
The 1H NMR spectrum of 5a shows the two NH groups (δ 9.57, 10.55 ppm) that are exchangeable with D2O. The two protons of methylene appear at δ 2.36 and 3.04 ppm as two doublets. Four protons of the aromatic ring can be observed at δ 6.81–7.20 ppm as four doublets. The 1H-decoupled 13C NMR spectrum of 5a indicates 14 distinct resonances in accordance with the desired structure. The characteristic signal of the carbon of the CH2 group is seen at δ 41.8 ppm. The carbon of the spiro center appears at δ 49.5 ppm. The two signals at δ 105.6 and 153.6 ppm are related to C–NO2 and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NH, respectively. The two carbonyl groups appear at δ 165.6 and 177.6 ppm.
C–NH, respectively. The two carbonyl groups appear at δ 165.6 and 177.6 ppm.
The mass spectrum of 5a displays a molecular-ion peak at m/z 300, which is in agreement with the proposed structure. The IR spectrum of 5a shows broad absorption bands due to NH at 3327−1, stretching vibrations of the CH2 groups at 2920−1, strong absorption bands of carbonyl groups at 1725−1 and 1683−1 and absorption bands at 1480−1 and 1369 cm−1 related to NO2.
A plausible mechanism for the formation of spiropyridineoxindole 5 is depicted in Scheme 2. On the basis of the well-established chemistry of 1,1-bis(methylthio)-2-nitroethylene, initially, the addition of diamine 1 to 1,1-bis(methylthio)-2-nitroethylene 2 leads to the formation of ketene aminal 6. The second step involves the condensation of isatin 3 with Meldrum's acid 4 in the presence of p-toluenesulfonic acid to afford the Knoevenagel product 7. Then, the Michael addition of the ketene aminal 6 to adduct 7 affords the intermediate 8; it undergoes subsequent imine–enamine tautomerization, followed by intramolecular cyclization via the nucleophilic addition of –NH to the carbonyl group. Subsequently, one equivalent of CO2 and acetone are removed from intermediate 9 to give the corresponding product 5 (Scheme 2).
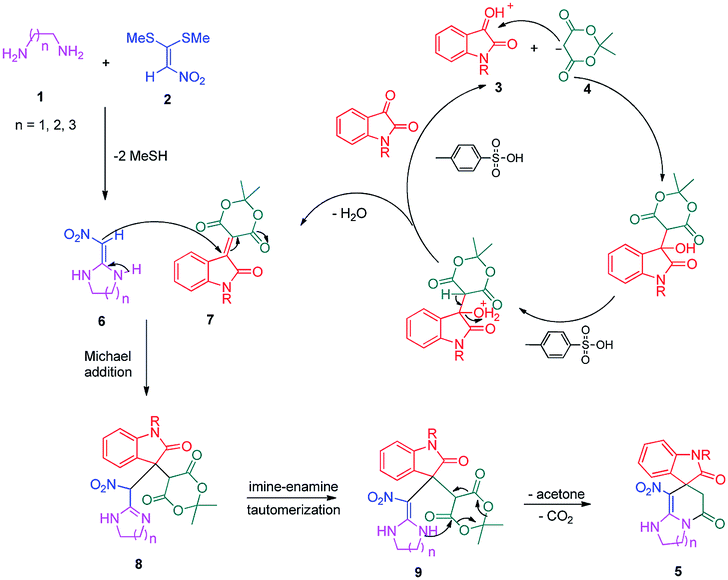 | ||
| Scheme 2 Proposed mechanism for the formation of the product 5a. | ||
Experimental
Materials
Meldrum's acid, 1,1-bis(methylthio)-2-nitro ethylene, diamines, various isatin derivatives, p-toluenesulfonic acid and solvents were purchased from Sigma-Aldrich and used as received without further purification. Melting points (mp) were determined with an electrothermal 9100 apparatus. Infrared (IR) spectra were recorded on a Bruker Tensor 27 spectrometer. Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker DRX-300 Avance instrument (300 MHz for 1H and 75.4 MHz for 13C) with CDCl3 and DMSO as solvents. Chemical shifts were expressed in parts per million (ppm) relative to internal TMS, and the coupling constant (J) was reported in hertz (Hz). All of the compounds were analyzed for mass data using Agilent 5975C VL MSD with a Triple-Axis Detector operating at an ionization potential of 70 eV.General procedure for the synthesis of 8-nitro-2,3-dihydro-1H-spiro[imidazo[1,2-a]pyridine-7,3′-indoline]-2′,5(6H)-dione (5a)
A mixture of ethylene diamine (66 mL, 1 mmol), 1,1-bis(methylthio)-2-nitroethylene (0.165 g, 1 mmol) and 10 mL EtOH in a 50 mL flask was refluxed for 6 h. After completion of the reaction (monitored by TLC, ethyl acetate/n-hexane, 1:1), isatin (0.147 g, 1 mmol), Meldrum's acid (0.144 g, 1 mmol) and p-toluenesulfonic acid (0.03 g, 0.2 mmol) were added to the reaction mixture, and it was stirred under reflux for 0.15 h. The progress of the reaction was monitored by TLC using ethyl acetate–n-hexane (1:1) as an eluent. After reaction completion, the precipitated product was collected by filtration and washed with EtOH to give the pure product 5a in 87% yield.
![[small upsilon, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0d5.gif) max/cm−1): 3327 (NH), 2920 (CH2), 1725, 1683 (2CO), 1632 (CC), 1480 and 1369 (NO2), 1277 (C–N); 1H NMR (300 MHz, DMSO): δ = 2.36 (1H, d, 2JHH = 16 Hz, CH2CO), 3.03 (1H, d, 2JHH = 16 Hz, CH2CO), 3.78–3.83 (2H, m, CH2NH), 3.90–4.06 (2H, m, CH2N), 6.82 (1H, d, 3JHH = 7.8 Hz, ArH), 6.88 (1H, d, 3JHH = 7.5 Hz, ArH), 7.11–7.20 (2H, m, ArH), 9.57 (1H, br, NHCH2), 10.55 (1H, s, NH–CO); 13C NMR (75 MHz, DMSO-d6); δ = 41.8 (CH2CO), 43.2 (CH2NH), 44.3 (CH2N), 49.5 (Cspiro), 105.6 (C–NO2), 110.1, 121.9, 122.7, 129.0 (4CH of Ar), 131.9, 142.1 (C–Ar), 153.6 (CC–NO2), 165.6 (NH–CO), 177.6 (CH2–CO); MS (EI, 70 eV): m/z (%) = 300 (56) [M]+, 254 (69), 239 (100), 225 (24), 198 (27), 155 (14), 69 (3), 44 (20). Anal. calcd for C14H12N4O4 (300.27): C, 56; H, 4.03; N, 18.66. Found C, 56.3; H, 4.2; N, 18.50.max/cm−1): 3334 (NH), 2886 (CH3), 1711, 1636 (2CO), 1488 and 1326 (NO2), 1157 (C–N); 1H NMR (300 MHz, DMSO): δ = 2.17 (1H, d, 2JHH = 20 Hz, CH2CO), 3.04 (1H, d, 2JHH = 20 Hz, CH2CO), 3.21 (3H, s, CH3), 3.67–3.78 (2H, m, CH2NH), 3.90–4.07 (2H, m, CH2N), 6.82–6.98 (2H, m, ArH), 7.11–7.30 (2H, m, ArH), 9.8 (1H, s, NH); 13C NMR (75 MHz, DMSO-d6); δ = 26.9 (N–CH3), 41.5 (CH2CO), 43.3 (CH2NH), 44.3 (CH2N), 49.1 (Cspiro), 105.5 (C–NO2), 109.0, 122.4, 122.6, 129.1 (4CH of Ar), 131.1, 143.6 (C–Ar), 153.6 (CC–NO2), 165.5 (NMe–CO), 175.9 (CH2–CO); MS (EI, 70 eV): m/z (%) = 314 (82) [M]+, 269 (12.5), 253 (100), 225 (17), 169 (12), 130 (13), 44 (14). Anal. calcd for C15H14N4O4 (314.30): C, 57.32; H, 4.4; N; 17.83. Found C, 57.6; H, 4.1; N, 18.60.max/cm−1): 3335 (NH), 2925, 2856 (C–H), 1711, 1636 (2CO), 1487 and 1369 (NO2), 1158 (C–N); 1H NMR (300 MHz, DMSO): δ = 1.11 (3H, t, 3JHH = 6.9 Hz, CH3), 2.30 (1H, d, 2JHH = 15.9 Hz, CH2CO), 3.05 (1H, d, 2JHH = 15.9 Hz, CH2CO), 3.62–3.71 (2H, m, CH2NH), 3.77 (2H, q, 3JHH = 6.9 Hz, CH3–CH2–N), 3.83–4.07 (2H, m, CH2N), 6.83 (1H, t, JHH = 7.8 Hz, ArH), 6.93 (1H, d, JHH = 7.5 Hz, ArH), 7.19 (1H, d, JHH = 7.8 Hz, ArH), 7.27 (1H, t, JHH = 7.5 Hz, ArH), 9.75 (1H, br, NH); 13C NMR (75 MHz, DMSO-d6): δ = 12.4 (CH3), 34.8 (CH3–CH2N), 41.5 (CH2CO), 43.2 (CH2NH), 44.3 (CH2N), 49.0 (Cspiro), 105.53 (C–NO2), 109.1, 122.4, 122.7, 129.1 (4CH of Ar), 131.2, 142.5 (C–Ar), 153.5 (CC–NO2), 165.5 (N–CO), 175.5 (CH2–CO); MS (EI, 70 eV): m/z (%) = 328 (100) [M]+, 282 (38), 267 (72), 239 (98), 224 (22), 197 (20), 158 (15), 44 (21). Anal. calcd for C16H16N4O4 (328.32): C, 50.53; H, 4.91; N, 17.06. Found C, 50.1; H, 4.6; N, 17.30.max/cm−1): 3412 (NH), 3032 (C–H of Ar), 2921 (CH3), 1716, 1619 (2CO), 1496 and 1323 (NO2), 1151 (C–N); 1H NMR (300 MHz, DMSO): δ = 1.95–2.09 (2H, m, CH2), 2.34 (1H, d, 2JHH = 15.3 Hz, CH2CO), 3.13 (1H, d, 2JHH = 15.3 Hz, CH2CO), 3.14 (3H, s, N–Me), 3.52–3.59 (2H, m, CH2NH), 3.72–3.78 (1H, m, CH2N), 3.88–3.95 (1H, m, CH2N), 6.94 (1H, t, JHH = 7.2 Hz, ArH), 7.01 (1H, d, JHH = 7.8 Hz, ArH), 7.09 (1H, d, JHH = 7.2 Hz, ArH), 7.28 (1H, t, JHH = 7.8 Hz, ArH), 11.50 (1H, s, NH); 13C NMR (75 MHz, CDCl3): δ = 19.0 (CH2), 26.9 (N–CH3), 39.2 (CH2–CO), 39.5 (CH2NH), 40.3 (CH2N), 48.0 (Cspiro), 107.8 (C–NO2), 108.6, 121.8, 122.6, 129.1 (4CH of Ar), 129.3, 143.4 (C–Ar), 152.7 (CC–NO2), 166.4 (NMe–CO), 175.9 (CH2–CO). Anal. calcd for C16H16N4O4 (328.33): C, 58.53; H, 4.91; N, 17.06. Found C, 58.9; H, 4.5; N, 17.20.C–NO2), 167.7 (N–CO), 174.1 (CH2–CO); MS (EI, 70 eV): m/z (%) = 314 (46) [M]+, 268 (100), 57 (41), 236 (31), 83 (30), 185 (20), 155 (19), 211 (14). Anal. calcd for C15H14N4O4 (314.30): C, 57.32; H, 4.49; N, 17.83. Found C, 57.5; H, 4.1; N, 17.50.C–NO2), 167.8 (N–CO), 175.5 (CH2–CO). Anal. calcd for C15H14N4O4 (324.35): C, 59.64; H, 5.30; N, 16.37. Found C, 59.4; H, 5.6; N, 16.10.C–NO2), 167.8 (N–CO), 175.1 (CH2–CO). Anal. calcd for C18H20N4O4 (356.38): C, 60.66; H, 5.66; N, 15.7. Found C, 60.9; H, 5.3; N, 15.50.
max/cm−1): 3327 (NH), 2920 (CH2), 1725, 1683 (2CO), 1632 (CC), 1480 and 1369 (NO2), 1277 (C–N); 1H NMR (300 MHz, DMSO): δ = 2.36 (1H, d, 2JHH = 16 Hz, CH2CO), 3.03 (1H, d, 2JHH = 16 Hz, CH2CO), 3.78–3.83 (2H, m, CH2NH), 3.90–4.06 (2H, m, CH2N), 6.82 (1H, d, 3JHH = 7.8 Hz, ArH), 6.88 (1H, d, 3JHH = 7.5 Hz, ArH), 7.11–7.20 (2H, m, ArH), 9.57 (1H, br, NHCH2), 10.55 (1H, s, NH–CO); 13C NMR (75 MHz, DMSO-d6); δ = 41.8 (CH2CO), 43.2 (CH2NH), 44.3 (CH2N), 49.5 (Cspiro), 105.6 (C–NO2), 110.1, 121.9, 122.7, 129.0 (4CH of Ar), 131.9, 142.1 (C–Ar), 153.6 (CC–NO2), 165.6 (NH–CO), 177.6 (CH2–CO); MS (EI, 70 eV): m/z (%) = 300 (56) [M]+, 254 (69), 239 (100), 225 (24), 198 (27), 155 (14), 69 (3), 44 (20). Anal. calcd for C14H12N4O4 (300.27): C, 56; H, 4.03; N, 18.66. Found C, 56.3; H, 4.2; N, 18.50.max/cm−1): 3334 (NH), 2886 (CH3), 1711, 1636 (2CO), 1488 and 1326 (NO2), 1157 (C–N); 1H NMR (300 MHz, DMSO): δ = 2.17 (1H, d, 2JHH = 20 Hz, CH2CO), 3.04 (1H, d, 2JHH = 20 Hz, CH2CO), 3.21 (3H, s, CH3), 3.67–3.78 (2H, m, CH2NH), 3.90–4.07 (2H, m, CH2N), 6.82–6.98 (2H, m, ArH), 7.11–7.30 (2H, m, ArH), 9.8 (1H, s, NH); 13C NMR (75 MHz, DMSO-d6); δ = 26.9 (N–CH3), 41.5 (CH2CO), 43.3 (CH2NH), 44.3 (CH2N), 49.1 (Cspiro), 105.5 (C–NO2), 109.0, 122.4, 122.6, 129.1 (4CH of Ar), 131.1, 143.6 (C–Ar), 153.6 (CC–NO2), 165.5 (NMe–CO), 175.9 (CH2–CO); MS (EI, 70 eV): m/z (%) = 314 (82) [M]+, 269 (12.5), 253 (100), 225 (17), 169 (12), 130 (13), 44 (14). Anal. calcd for C15H14N4O4 (314.30): C, 57.32; H, 4.4; N; 17.83. Found C, 57.6; H, 4.1; N, 18.60.max/cm−1): 3335 (NH), 2925, 2856 (C–H), 1711, 1636 (2CO), 1487 and 1369 (NO2), 1158 (C–N); 1H NMR (300 MHz, DMSO): δ = 1.11 (3H, t, 3JHH = 6.9 Hz, CH3), 2.30 (1H, d, 2JHH = 15.9 Hz, CH2CO), 3.05 (1H, d, 2JHH = 15.9 Hz, CH2CO), 3.62–3.71 (2H, m, CH2NH), 3.77 (2H, q, 3JHH = 6.9 Hz, CH3–CH2–N), 3.83–4.07 (2H, m, CH2N), 6.83 (1H, t, JHH = 7.8 Hz, ArH), 6.93 (1H, d, JHH = 7.5 Hz, ArH), 7.19 (1H, d, JHH = 7.8 Hz, ArH), 7.27 (1H, t, JHH = 7.5 Hz, ArH), 9.75 (1H, br, NH); 13C NMR (75 MHz, DMSO-d6): δ = 12.4 (CH3), 34.8 (CH3–CH2N), 41.5 (CH2CO), 43.2 (CH2NH), 44.3 (CH2N), 49.0 (Cspiro), 105.53 (C–NO2), 109.1, 122.4, 122.7, 129.1 (4CH of Ar), 131.2, 142.5 (C–Ar), 153.5 (CC–NO2), 165.5 (N–CO), 175.5 (CH2–CO); MS (EI, 70 eV): m/z (%) = 328 (100) [M]+, 282 (38), 267 (72), 239 (98), 224 (22), 197 (20), 158 (15), 44 (21). Anal. calcd for C16H16N4O4 (328.32): C, 50.53; H, 4.91; N, 17.06. Found C, 50.1; H, 4.6; N, 17.30.max/cm−1): 3412 (NH), 3032 (C–H of Ar), 2921 (CH3), 1716, 1619 (2CO), 1496 and 1323 (NO2), 1151 (C–N); 1H NMR (300 MHz, DMSO): δ = 1.95–2.09 (2H, m, CH2), 2.34 (1H, d, 2JHH = 15.3 Hz, CH2CO), 3.13 (1H, d, 2JHH = 15.3 Hz, CH2CO), 3.14 (3H, s, N–Me), 3.52–3.59 (2H, m, CH2NH), 3.72–3.78 (1H, m, CH2N), 3.88–3.95 (1H, m, CH2N), 6.94 (1H, t, JHH = 7.2 Hz, ArH), 7.01 (1H, d, JHH = 7.8 Hz, ArH), 7.09 (1H, d, JHH = 7.2 Hz, ArH), 7.28 (1H, t, JHH = 7.8 Hz, ArH), 11.50 (1H, s, NH); 13C NMR (75 MHz, CDCl3): δ = 19.0 (CH2), 26.9 (N–CH3), 39.2 (CH2–CO), 39.5 (CH2NH), 40.3 (CH2N), 48.0 (Cspiro), 107.8 (C–NO2), 108.6, 121.8, 122.6, 129.1 (4CH of Ar), 129.3, 143.4 (C–Ar), 152.7 (CC–NO2), 166.4 (NMe–CO), 175.9 (CH2–CO). Anal. calcd for C16H16N4O4 (328.33): C, 58.53; H, 4.91; N, 17.06. Found C, 58.9; H, 4.5; N, 17.20.C–NO2), 167.7 (N–CO), 174.1 (CH2–CO); MS (EI, 70 eV): m/z (%) = 314 (46) [M]+, 268 (100), 57 (41), 236 (31), 83 (30), 185 (20), 155 (19), 211 (14). Anal. calcd for C15H14N4O4 (314.30): C, 57.32; H, 4.49; N, 17.83. Found C, 57.5; H, 4.1; N, 17.50.C–NO2), 167.8 (N–CO), 175.5 (CH2–CO). Anal. calcd for C15H14N4O4 (324.35): C, 59.64; H, 5.30; N, 16.37. Found C, 59.4; H, 5.6; N, 16.10.C–NO2), 167.8 (N–CO), 175.1 (CH2–CO). Anal. calcd for C18H20N4O4 (356.38): C, 60.66; H, 5.66; N, 15.7. Found C, 60.9; H, 5.3; N, 15.50.The integrals in the 1H NMR spectrum of 5h and the particular carbon signals in the 13C NMR spectrum represent two structures that cannot be separated. The spectral information is given for one structure (A):
C–NO2), 167.6 (N–CO), 175.0 (CH2–CO).The presence of two chiral centers in 5i and 5j compounds led to four isomers: (3R,3′S), (3S,3′S) and their enantiomers. The 1H NMR and 13C NMR spectra also confirmed the existence of two types of diastereoisomers.
C–NO2), 165.6 (N–CO), 177.6 (CH2–CO); MS (EI, 70 eV): m/z (%) = 314 (82) [M]+, 314 (59), 269 (25), 253 (100), 240 (30), 212 (33), 155 (24), 41 (25). Anal. calcd for C15H14N4O4 (314.1): C, 57.32; H, 4.49; N, 17.83.C–NO2), 165.7 (N–CO), 175.9 (CH2–CO). Anal. calcd for C16H16N4O4 (328.32): C, 58.53; H, 4.91; N, 17.06. Found C, 58.2; H, 4.6; N, 17.30.Conclusion
We designed a novel and convenient procedure for the synthesis of three new classes of spiropyridineoxindoles with fused heterocyclic compounds (imidazole, pyrimidine and diazepine) in good yields via a four-component reaction among 1,1-bis(methylthio)-2-nitroethylene, various aliphatic diamines, Meldrum's acid and isatin derivatives using a catalytic amount of p-TSA. The present process has several important features including mild and facile reaction conditions, easy accessibility of reactants, a simple workup procedure, the use of ethanol as a solvent, short reaction times, and good-to-high yields. These structures having both indole and fused-pyridine moieties, which are some of the most typical privileged scaffolds, are completely new and there is no other report on their synthesis.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support of this research from Imam Khomeini International University, Iran is gratefully acknowledged.Notes and references
- J. F. M. Da-Silva, S. J. Garden and A. C. Pinto, J. Braz. Chem. Soc., 2001, 12, 273 CrossRef CAS.
- A. H. Abdel-Rahman, E. M. Keshk, M. A. Hanna and S. M. El-Bady, Bioorg. Med. Chem., 2006, 12, 2483 CrossRef PubMed.
- T. H. Kang, K. Matsumoto, Y. Murakami, H. Takayama, M. Kitajima, N. Aimi and H. Watanabe, Eur. J. Pharmacol., 2002, 444, 39 CrossRef CAS PubMed.
- T. Usui, M. Kondoh, C.-B. Cui, T. Mayumi and H. Osada, Biochem. J., 1998, 333, 543 CrossRef CAS PubMed.
- S. F. Thakor, M. Dinesh, M. P. Patel and R. G. Patel, Saudi Pharm. J., 2007, 15, 48 CAS.
- V. V. Vintonyak, K. Warburg, H. Kruse, S. Grimme, K. Hubel, D. Rauth and H. Waldmann, Angew. Chem., Int. Ed., 2010, 49, 5902 CrossRef CAS PubMed.
- F. Gatta, M. Pomponi and M. Marta, J. Heterocycl. Chem., 1991, 28, 1301 CrossRef CAS.
- G. Kumari, M. Nutan, M. Modi, S. K. Gupta and R. K. Singh, Eur. J. Med. Chem., 2011, 46, 1181 CrossRef CAS PubMed.
- A. Thangamani, Eur. J. Med. Chem., 2010, 45, 6120 CrossRef CAS PubMed.
- P. Rosenmond, M. Hosseini-Merescht and C. Bub, Liebigs Ann. Chem., 1994, 2, 151 CrossRef.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qui, Y. Ding, W. Gao, J. Stuckey and K. Krajewski, J. Am. Chem. Soc., 2004, 126, 16077 CrossRef PubMed.
- T. Okita and M. Isobe, Tetrahedron, 1994, 50, 11143 CrossRef CAS.
- B. K. S. Yeung, B. Zou, M. Rottmann, S. B. Lakshminarayana, S. H. Ang, S. Y. Leong, J. Tan, J. Wong, C. Fischli, E. A. Winzeler, F. Petersen, R. Brun, V. Dartois and T. T. Diagana, J. Med. Chem., 2010, 53, 5155 CrossRef CAS PubMed.
- T. Chen, X. P. Xu and S. J. Ji, J. Comb. Chem., 2010, 12, 659 CrossRef CAS PubMed.
- S. J. Yan, Y.-F. Niu, R. Huang and J. Lin, Synlett., 2009, 2821 CAS.
- (a) G. J. Meia and F. Shi, Chem. Commun., 2018, 54, 6607 RSC; (b) G. J. Meia, D. Li, G. X. Zhou, Q. Shi, Z. Cao and F. Shi, Chem. Commun., 2017, 53, 10030 RSC; (c) X. L. Jiang, S. J. Liu, Y. Q. Gu, G. J. Mei and F. Shi, Adv. Synth. Catal., 2017, 359, 3341 CrossRef CAS; (d) J. L. Wu, Y. C. Zhang, Y. Y. He, J. Y. Wang, P. Wu and F. Shi, Adv. Synth. Catal., 2016, 358, 2777 CrossRef CAS; (e) Y. Zhou, Y. Lu, X. Hu, H. Mei, L. Lin, X. Liu and X. Feng, Chem. Commun., 2017, 53, 2060 RSC; (f) Y. Wang, M. S. Tu, L. Yin, M. Sun and F. Shi, J. Org. Chem., 2015, 80, 3223 CrossRef CAS PubMed; (g) H. Zheng, X. Liu, C. Xu, Y. Xia, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2015, 54, 10958 CrossRef CAS PubMed.
- A. Rahmati and Z. Khalesi, Tetrahedron, 2012, 68, 8472 CrossRef CAS.
- (a) M. Stucchi, G. Lesma, F. Meneghetti, G. Rainoldi, A. Sacchetti and A. Silvani, J. Org. Chem., 2016, 81, 1877 CrossRef CAS PubMed; (b) M. Wu, W. W. He, X. Y. Liu and B. Tan, Angew. Chem., Int. Ed., 2015, 54, 9409 CrossRef CAS PubMed; (c) W. Dai, H. Lu, X. Li, F. Shi and S. J. Tu, Chem. Eur. J., 2014, 20, 11382 CrossRef CAS PubMed; (d) Q. N. Zhu, Y. C. Zhang, M. M. Xu, X. X. Sun, X. Yang and F. Shi, J. Org. Chem., 2016, 81, 7898 CrossRef CAS PubMed; (e) F. Shi, G. J. Xing, R. Y. Zhu, W. Tan and S. Tu, Org. Lett., 2013, 15, 128 CrossRef CAS PubMed; (f) C. S. Wang, R. Y. Zhu, J. Zheng, F. Shi and S. J. Tu, J. Org. Chem., 2015, 80, 512 CrossRef CAS PubMed; (g) W. Dai, X. L. Jiang, Q. Wu, F. Shi and S. J. Tu, J. Org. Chem., 2015, 80, 5737 CrossRef CAS PubMed.
- A. Alizadeh and L. Moafi, Helv. Chim. Acta, 2015, 98, 546 CrossRef CAS.
- R. A. Nagalakshmi, J. Suresh, S. Sivakumar, R. Ranjith Kumar and P. L. Nilantha Lakshman, Acta Crystallogr., 2014, 70, o971–o972 CAS.
- F. Yu, R. Huang, H. Ni, J. Fan, S. Yan and J. Lin, Green Chem., 2013, 15, 453 RSC.
- S. Ghadiri, M. Bayat and F. Hosseini, Monatsh. Chem., 2019, 150, 1 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ra10379h |
| This journal is © The Royal Society of Chemistry 2019 |