
Dispersive suspended-solidified floating organic droplet microextraction of nonsteroidal anti-inflammatory drugs: comparison of suspended droplet-based and dispersive-based liquid-phase microextraction methods
Behruz Barfi,
Alireza Asghari*,
Maryam Rajabi and
Nasim Mirkhani
Department of Chemistry, Semnan University, Semnan 35195-363, Iran. E-mail: aasghari@semnan.ac.ir
First published on 3rd December 2015
Abstract
Herein, a dispersive suspended-solidified floating organic droplet microextraction method was first developed to improve some limitations of droplet-based microextraction methods including long extraction times and uncertainties in the collection of low volume of extraction solvents coupled with high-performance liquid chromatography (HPLC). To the best of our knowledge, neither the extraction efficiency of droplet- and dispersive-based liquid-phase microextraction methods, under disperser solvent-free conditions, nor their ability to pre-concentrate nonsteroidal anti-inflammatory drugs (NSAIDs) from bio-fluid samples has been investigated so far. In this way, two droplet-based (directly suspended droplet and dispersive suspended), two solidified droplet-based (directly suspended-solidified floating organic droplet and dispersive suspended-solidified floating organic droplet), and two dispersive-based (air-assisted liquid–liquid and ultrasound-assisted emulsification) microextraction methods were studied and compared for the determination of three NSAIDs as model analytes. The influential parameters on the extraction efficiency of all methods were critically investigated and compared thermodynamically and kinetically. However, considering some advantages such as higher enrichment factors, shorter extraction time and simplicity in operation, the best results were obtained using the low density solvent-based air-assisted liquid–liquid microextraction (LDS-AALLME) method, which employed 65.0 μL of n-octanol as extraction solvent, 5 mL of sample at pH 2.5, without salt addition, and 10.0 extraction cycles (during 40 s). This method was validated with satisfactory results including low limits of detection (1.1 to 1.7 μg L−1), wide linear dynamic ranges (3.5 to 2448 μg L−1), acceptable recoveries (94 to 102%) and relative standard deviations (in terms of repeatability, <7.9%). Finally, the LDS-AALLME method coupled to HPLC was successfully applied for determination of ibuprofen, mefenamic acid and sodium diclofenac in human plasma and urine samples.
1. Introduction
In chemical analysis, sample preparation is frequently considered the bottleneck of the entire analytical method. Various sample preparation strategies have been developed based on exhaustive or non-exhaustive extraction of analytes from matrices. The main reason for extraction is to obtain a more concentrated sample, to eliminate interfering substances and to improve detection limits for specific compounds. There have been substantial efforts in the past two decades to adapt the existing extraction methods and develop new approaches to save time, labor, and materials.1–4 In this way, recent research activities have been oriented toward the development of miniaturized extraction methods such as solid phase microextraction (SPME)5 and liquid-phase microextraction (LPME),6 which are easy, fast and virtually-free or less organic solvent consumption. Although SPME has the advantages of portability and simplicity, the fiber is comparatively expensive, fragile, and has limited lifetime. In addition, sample carry-over is also a problem for SPME. Therefore, LPME was developed in order to overcome the shortcomings of SPME.7LPME has attracted increasing attention because it requires very little solvents and minimal exposure to toxic organic solvents, which make it a simple, quick, inexpensive and virtually solvent-free sample preparation method. Also, high enrichment factors are achievable because of the high ratio of sample volume to acceptor phase volume. Nowadays, LPME is widely used for the analysis of organic compounds8 and inorganic trace elements9 in environmental, biological, and food samples. Different configurations of LPME have recently emerged in three main categories including droplet-based LPME (D-LPME),10 hollow fiber-based LPME (HF-LPME)11 and dispersive-based LPME (Dis-LPME) methods.12,13
In the simplest form of D-LPME modes, which termed direct immersion single-drop microextraction (DI-SDME), an organic solvent or ionic-liquid droplet is held at the tip of a microsyringe needle and is directly immersed in the sample.6 The major drawback of this mode is that the microdrop suspended on the microsyringe needle is easily dislodged during stirring of the aqueous sample.14
To overcome this drawback, a novel D-LPME method named directly suspended droplet microextraction (DSDME) was first introduced by Lu and coworkers in 2006.15 Compared to DI-SDME, DSDME does not require special equipment, the organic drop is more stable, and the equilibrium is more quickly reached. In this method, a stir bar is placed at the bottom of a vial containing an aqueous sample and rotated at a speed required to cause a gentle vortex. If a small volume of an immiscible organic solvent with density lighter than water is added to the surface of the aqueous solution, the vortex results in the formation of a single droplet at or near the center of rotation. The droplet itself may also rotate on the surface of the aqueous phase, thereby increasing mass transfer. Other advantages of DSDME are simplicity, fastness and easy operation, because it requires only common laboratory equipment.16
However, despite its advantages, DSDME has two drawbacks as follow:
(i) Relatively small interfacial area between extraction solvent and aqueous sample lead to a long extraction time, and
(ii) Collection of extraction solvent can be accomplished with some uncertainties, especially, when the volume of extraction solvent is low.
To overcome the first drawback, a new version of DSDME i.e. dispersive suspended microextraction (DSME) was developed.17 In this technique, the extraction process is divided into two critical steps: (i) extraction, and (ii) restoration. During the extraction step, a continuous agitation at a high speed is provided and the extraction solvent dispersed into fine droplets, at which target analytes are extracted into the dispersed extraction solvent. This could significant enlarge the contact surface between immiscible phases and greatly reduce the equilibrium time.18 During the restoration step, two phases began to separate and the suspended extractant phase is formed, again. To overcome the second drawback, directly suspended-solidified floating organic droplet microextraction (DS-SFO) method was developed, at which the extractant is maintained as a micro-droplet throughout the extraction process and solidified after the extraction. This makes the extraction phase easy to collect.19
Regueiro et al. reported the application of ultrasonic irradiation as a substitution for the disperser solvent and named the procedure ultrasound-assisted emulsification microextraction (USA-EME).20 Ultrasound irradiation can lead to a process named cavitation. Cavitation is the creation and then immediate implosion of bubbles in a liquid. The physical process of cavitation is similar to boiling. The major difference between boiling and cavitation is the thermodynamic paths that precede the formation of the vapor. In cavitation process, bubble in a liquid rapidly collapses, producing a shock wave. Sufficient energy of this shock can break down the droplet of extraction phase and generate a smaller droplet size immediately after disruption, thus enhancing the emulsification.20–22 The consequence is a very efficient and relatively fast analyte extraction. After mass transfer, the two phases can be readily separated by centrifugation. In this way, USA-EME can be employed as a simple and efficient disperser solvent-free extraction and preconcentration method for organic and inorganic compounds in aqueous samples.13,23
Air-assisted liquid–liquid microextraction (AALLME) is one of the most recently used dispersive solvent-free LPME methods, which has been reported by Farajzadeh in 2012.24 In AALLME, a few microliters of a denser or lighter than water extraction solvent is transferred into an aqueous sample solution and then the mixture is repeatedly sucked into a glass syringe and then injected into the tube. After centrifugation of cloudy solution, the extractant is collected and used for further analysis.25,26 This method has been proved to be simple, rapid, efficient, and environmentally friendly.27,28
Most published analytical procedures focus on obtaining the very lowest possible limits of detection and limits of quantification. However, as a practical matter, it is just as important to focus on the time, precision, manual labor, and expense required for extraction. Hence, in the present study, the advantages of DSME and DS-SFO methods were emerged and a novel and efficient dispersive suspended-solidified floating organic droplet microextraction (Dis-S-SFO) method was first developed for the determination of non-steroidal anti-inflammatory drugs in bio-fluids by high performance liquid chromatography with ultra-violet detection (HPLC-UV). Then, it was compared with three suspended droplet-based LPME (including DSDME, DSME and DS-SFO) and two dispersive-based LPME (including USA-EME and low density solvent-based AALLME) methods. To the best of our knowledge, there is no report about the comparison of suspended droplet- and dispersive-based LPME methods basis on an identical term (i.e. enrichment factor) to evaluate their extraction and pre-concentrating abilities, under disperser solvent-free conditions.
To achieve this purpose, three nonsteroidal anti-inflammatory drugs (NSAIDs), i.e. diclofenac sodium (Dic), ibuprofen (Ibu), and mefenamic acid (Mef), were used as model analytes. NSAIDs form a group of analgesic, antipyretic and anti-inflammatory agents that are used with great frequency in both humans and animals since they do not induce sedation, respiratory depression or addiction.29 Because of their effectiveness in suppressing or preventing inflammation, NSAIDs are becoming the most commonly used medicines around the world. For the diagnosis or, more importantly, the differential diagnostic exclusion of cases of acute over-dosage or chronic abuse, a simple and efficient analytical procedure is necessary for the detection of these drugs in bio-fluid samples.30,31 After optimization, the results obtained showed that each of DSME, Dis-S-SFO, USA-EME and low density solvent-based AALLME (LDS-AALLME) methods has its unique capabilities, which could be applied as preferred method for extraction and determination of the analytes in human bio-fluid samples such as plasma and urine using high performance liquid chromatography with ultra-violet detection (HPLC-UV). However, the results showed that the LDS-AALLME is simpler, faster and more effective than the other methods, as it needed only 40 s to achieve the equilibrium with acceptable repeatabilities. Hence, it was selected as a preferred method for analyzing of ibuprofen, mefenamic acid and sodium diclofenac in human plasma and urine samples.
2. Experimental
2.1. Reagents and solutions
Standards of mefenamic acid (Mef), ibuprofen (IBP), and sodium diclofenac (DIC) were purchased from Sigma (Steinheim, Germany). 1-Octanol, toluene, n-heptane, cyclohexane, 2-dodecanol, 1-undecanol, n-hexadecane, acetone, methanol, sodium chloride, and ultra-pure water were all from Merck (Darmstadt, Germany). Trichloroacetic acid (TCAA) was obtained from Sigma. Sodium hydroxide and concentrated hydrochloric acid were bought from Merck, used to adjust the pH of the samples. Other reagents were of analytical grade and obtained from Merck.Stock standard solutions of each analyte were prepared separately by dissolving proper amounts of each drug in methanol at 1000 mg L−1 and stored at 4 °C. Mixtures of standard working solutions for extraction at different concentrations were prepared by dilution with ultra-pure water for optimization of parameters. The working solutions were freshly prepared by diluting the mixed standard solutions in ultra-pure water for the concentrations required. All the standard solutions were stored at 4 °C.
The optimum mobile phase consisted of water/acetonitrile/acetic acid (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :75:5, v/v/v) with a flow rate of 0.9 mL min−1. Prior to use, the mobile phase was filtered through a 0.45 μm membrane filter and degassed under vacuum. The analytes were monitored at 273 nm (at room temperature). The injection volume was 20 μL.
:75:5, v/v/v) with a flow rate of 0.9 mL min−1. Prior to use, the mobile phase was filtered through a 0.45 μm membrane filter and degassed under vacuum. The analytes were monitored at 273 nm (at room temperature). The injection volume was 20 μL.
2.2. Apparatus
A Knauer HPLC system (Berlin, Germany), equipped with a K-1001 HPLC pump, D-14163 degasser, and a K-2600 UV detector was used. Chromgate software (version 3.1) for HPLC system was employed to acquire and process chromatographic data. The chromatographic determinations were performed using an ODS III column (250 mm × ID 4.6 mm, 5 μm) from MZ-Analysentechnik (Mainz, Germany). The pH values for the solutions were measured using a PHS-3BW model pH-meter (Bell, Italy). Dispersion of the extraction solvent was enhanced using a 50/60 kHz (80 W) ultrasonic water bath (SW3, Switzerland). An EBA20 model centrifuge (Hettich, Germany) was used to accelerate phase separation.2.3. Sample preparation
Volunteers: the volunteers (between 25 to 35 years old) were recruited into the present study. The volunteers were all apparently healthy and none of them were taking medications. They were given oral instructions on the diet and also asked to restrain from using similar drugs or dietary supplements during three days before sampling. The experimentations in this study have absolutely served to maintaining, sampling, and analysis in accordance with ethical guidelines and recommendations for biomedical research and human laboratory of Declaration of Helsinki.32 Also, the research board of research & technology deputy of Semnan University has approved all results and the consent of all participants was obtained for research involving human subjects.000 rpm. After that, separated plasma was withdrawn into a Pyrex centrifuge tube and stored at −20 °C until analysis.Most of NSAIDs are extensively bounded to plasma proteins,33 and should be liberated prior to extraction. Blank plasma sample (2.5 mL) was spiked with particular level of the drug and sonicated for 5 min. The mixture was acidified with 200 μL hydrochloric acid (37%) to disturb the drug protein binding. Then, 250 μL TCAA (100%, w/v) was added to denature the proteins. These processes eventually led to the precipitation of proteins. Subsequently, the sample was centrifuged at 10000 rpm for 5 min. A volume of 2 mL of the supernatant was transferred to the sample vial and diluted with doubly distilled water to 5 mL.34 The resulting solutions were adjusted at pH 3.0, filtered and subjected to the examined methods.
Baseline plasma and urine samples were obtained 30 min before drugs administration.
2.4. Microextraction methods
In the extraction step (from opening the magnetic stirrer to turning down its speed at the restoration speed), the mixture was agitated for 1.0 min (extraction time) at 1200 rpm (extraction speed) and formed a cloudy solution. The analytes were extracted into the fine droplets of extractant.
In the restoration step (from ending the extraction step to the time when organic phase and aqueous phase were separated absolutely), the speed of the stirrer was turned down to 400 rpm (restoration speed) so that a steady and gentle vortex was formed. During this step, the dispersive droplets began to gather up in the top-center position of the vortex. After 5.0 min (restoration time), the organic phase was separated from aqueous phase and formed the final suspended phase. Then, 20.0 μL of the suspended phase was withdrawn injected into HPLC-UV system for further analysis.
3. Results and discussion
In two-phase droplet-based liquid-phase microextraction methods (such as single-drop microextraction and directly suspended-droplet microextraction), the microdrop can be thought of as essentially spherical and thus the extraction solvent has a minimum surface area to volume. This is one reason why many these methods may require long extraction times (usually higher than 10 min) for a satisfactory extraction. Increasing the volume of the aqueous sample may increase the amount of analyte that can be extracted, but will also increase the extraction time significantly. In contrary, dispersive-based LPME methods involve the dispersion of organic solvent as a “cloudy mixture” of tiny nanoliter-scale droplets within the aqueous phase. The extremely large interfacial area associated with these methods means that equilibrium can be reached rapidly have very large solvent to aqueous interfacial areas and reach equilibrium much faster. As a consequence, extraction equilibrium for USA-EME and AALLME (as instances of dispersive-based methods) is reached faster than droplet-based LPME extractions in part because the distances required for mass transfer are dramatically reduced in these methods, as well as larger accessible interfacial area of solvent.Although most published analytical procedures focus on obtaining the very lowest possible limits of detection, it is just as important to focus on the time, manual labor, and expense required for extraction method. In this way, six LPME methods were critically compared to consider all mentioned aspects, here. In order to simplify the experiments, the significant factors affecting the extraction efficiency of target analytes (in terms of EF) were divided into two categories as general and individual parameters. General parameters were first studied for all methods and individuals investigated for each method, afterwards.
3.1. General parameters
3.1.1.1. Type of extraction solvent in DSDME, DSME, LDS-AALLME, and USA-EME methods. The organic solvent used as the extraction solvent must have lower density than water (in the present work), a very low solubility in water and satisfactory extraction efficiency for analytes. Apart from these requirements, the organic solvent should also have a suitable viscosity to form a well-formed phase, especially in DSDME and DSME methods, and low volatility to prevent loss during extraction due to the low solvent consumption. Moreover, it should have good chromatographic behavior during the chromatographic separation. On the basis of these considerations, four organic solvents with different physicochemical properties including 1-octanol, toluene, n-heptane, and cyclohexane were tested. Among the studied solvents, toluene and n-heptane were not suitable solvents due to the instability and volatility of the extractant droplet in long times. 1-Octanol and cyclohexane were found to be appropriate extractant phases, as well as their good chromatographic behavior. However, for DSDME, DSME, LDS-AALLME, and USA-EME methods, 1-octanol was finally selected due to its relatively higher viscosity, good extractability, and lower solubility and volatility which allow a lower solvent consumption per analysis (Fig. 1a–d).
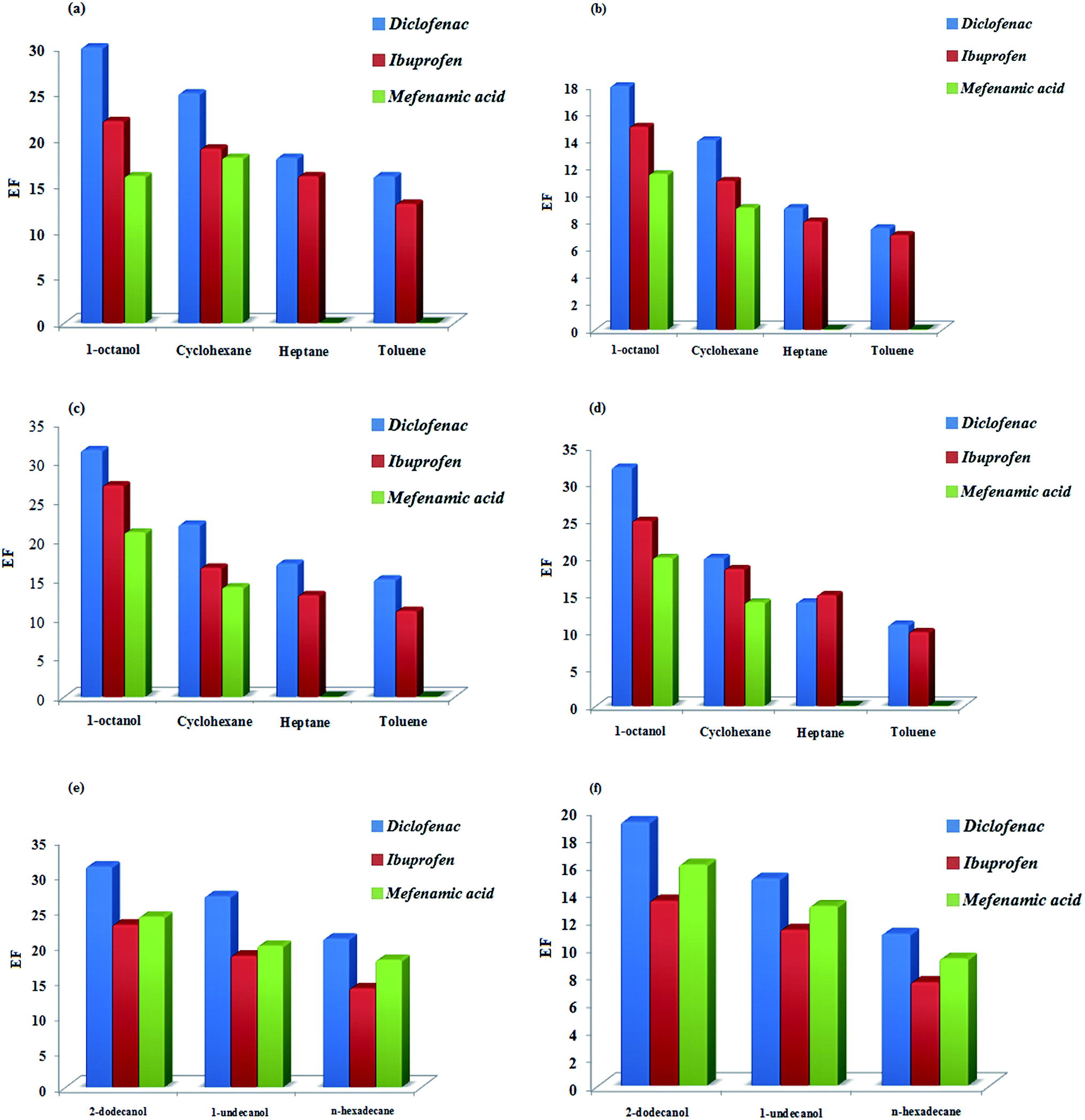 | ||
| Fig. 1 Effect of the type of extraction solvent on the analytes enrichment factors. (a) DSME, (b) DSDME, (c) USA-EME, (d) LDS-AALLME, (e) Dis-S-SFO, (f) DS-SFO. | ||
3.1.1.2. Type of extraction solvent in DS-SFO and Dis-S-SFO methods. Convenient collection of extractant phase is a crucial characteristic of microextraction methods. For suspended droplet-based microextraction methods, this convenience necessitates a droplet height large enough for needle insertion. Intuitively, a droplet with a greater volume will result in an increase in droplet height and be more convenient for collecting. However, droplet height depends on both droplet volume and shape. When the volume of extraction solvent is enough large, the shape of solvent droplet was nearly independent of collecting needle insertion. In contrast, at low volume of extractant, some uncertainties can be observed. In these cases, utilization of organic solvents that can be solidified at lower temperatures than ambient temperature can be a smart solution. Hence, compared with non-solidified suspended droplet-based microextraction methods, their solidified modes allow an increase in the enrichment factor as well as a decrease in the limit of detection in subsequent analysis steps.
As well as the criteria mentioned (in previous section) for suitable selection of solvents, they should have a melting point near room temperature in the range of 10 to 30 °C. In this way, three organic solvents including 1-undecanol (melting point (mp) = 13–15 °C), 2-dodecanol (mp = 17–18 °C) and n-hexadecane (mp = 18 °C) were examined. Because of its easy solidification, higher extraction efficiency, and better chromatographic behavior (better peak resolution) compared to other solvents tested, 2-dodecanol was found to be the best as extractant phase (Fig. 1e and f).
 | (1) |
Furthermore, the kinetics of extraction depends upon the Ai and Vo (eqn (2)). A larger Ai and lower Vo provide higher λ, which lead to faster equilibrium.
 | (2) |
![[small beta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c3.gif) o is the overall mass transfer coefficient for the organic phase in centimeters per second, k is the distribution ratio between the organic and aqueous phases, Caq is the analyte concentration in the aqueous phase at time t.36
o is the overall mass transfer coefficient for the organic phase in centimeters per second, k is the distribution ratio between the organic and aqueous phases, Caq is the analyte concentration in the aqueous phase at time t.36
Hence, in most cases, the lowest volume of the extraction solvent is the best choice to achieve the highest EF in a shorter time.
Different volumes of 1-octanol (25–70 μL for DSDME and DSME, 40–80 μL for LDS-AALLME, and 50–100 μL for USA-EME) and 2-dodecanol (20–50 μL for DS-SFO and Dis-S-SFO) were tested. Although the use of lower volumes of extraction solvent leads to higher extraction efficiency, the repeatability values are poor when the volumes are lower than selected amounts, due to the difficulty to uptake the extractant phase. High extraction efficiencies along with good repeatabilities were obtained when 50, 50, 65, and 80 μL of 1-octanol, and 40 and 40 μL of 2-dodecanol were used as extraction solvents in DSDME, DSME, LDS-AALLME, USA-EME, DS-SFO and Dis-S-SFO methods, respectively (Fig. 2a–f). Therefore, these volumes were selected as the optimal volume of extraction solvent.
 | ||
| Fig. 2 Effect of the volume of extraction solvent on the analytes enrichment factors. (a) DSME, (b) DSDME, (c) USA-EME, (d) LDS-AALLME, (e) Dis-S-SFO, (f) DS-SFO. | ||
 | ||
| Fig. 3 Effect of the pH on the analytes enrichment factors. (a) DSME, (b) DSDME, (c) USA-EME, (d) LDS-AALLME, (e) Dis-S-SFO, (f) DS-SFO. | ||
| K(salt) = K × 10+S[salt] | (3) |
| Caq(salt) = C × 10−S[salt] | (4) |
In some cases, there is no observed effect of salt addition or it can even suppress the extraction efficiencies. Relatively high concentrations of salts, as well as prohibition of phase separation, may modify the physical properties of the Nernst diffusion film and slow down the extraction kinetics which leads to decrease the extraction efficiency (salting-in effect). Also with increase in the viscosity and density of the medium due to the salt addition, ultrasound irradiation can be absorbed and dispersed as heat. This undesirable effect can prevent the extractant phase from being dispersed into fine droplets and, therefore, the efficiency of dispersion can be drastically reduced.
However, the outcome of salt addition is difficult to predict and only practical experiments can verify the effect of the addition of salts.
In this way, influence of ionic strength on the extraction efficiency was investigated by adding different amounts of NaCl (0–10% (w/v)) into the model sample. The salt addition had no significant effect on the extraction efficiency of DSDME, DSME, DS-SFO, Dis-S-SFO, slightly increased the efficiency of LDS-AALLME, and decreased the efficiency of USA-EME. Hence, salt addition was not used in the subsequent experiments.
If a solute introduces in a biphasic liquid system (including organic and aqueous phases), it distributes between the two phases. Assuming ideal mixtures, in the aqueous phase, the Gibbs free energy of analyte (A), or chemical potential, μaq A, is expressed by:
|
μaq A = μ0aq A + RTlnxaq A
| (5) |
|
μorg A = μ0org A + RTlnxorg A
| (6) |
If the chemical potential is not identical in the two phases, mass transfer of A occurs, the mole fractions x change so that the chemical potential of A becomes equal in both phases, i.e. the equilibrium is reached. Then:
 | (7) |
 is the distribution coefficient, k, which is usually expressed as molarity ratio and can be shown as:
is the distribution coefficient, k, which is usually expressed as molarity ratio and can be shown as:
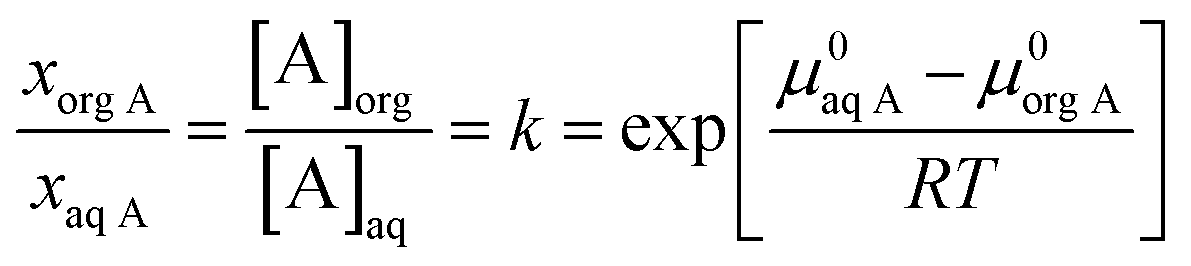 | (8) |
Eqn (7) and (8) show that the distribution coefficient is sensitive to temperature. Eqn (9) expresses the free energy of transfer, ΔG:
|
ΔG = RTlnk
| (9) |
Assuming the standard molar enthalpy is constant in a limited temperature range, the plot of lnk versus  (classical van't Hoff plots) should produce a straight line with slope
(classical van't Hoff plots) should produce a straight line with slope 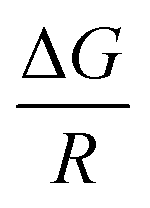 . As a general rule, it is possible to consider that the effect of temperature on the k value is not great if the solvents are not very miscible and the temperature change is not dramatic (an average change of 0.009logk unit per degree, either positive or negative).37 Meanwhile, increase in temperature also decreases the viscosity of solvent droplet which in turn facilitates the smooth and fast mass transfer of analytes from the aqueous phase into the organic droplet. It seems that the effect of temperature on the kinetics of analytes transfer between two phases is more significant. In other word, increase in the temperature can increase the mass transfer rate of the analytes. This increases o (eqn (2)), and as a consequence, extraction will be performed in a shorter period of time.
. As a general rule, it is possible to consider that the effect of temperature on the k value is not great if the solvents are not very miscible and the temperature change is not dramatic (an average change of 0.009logk unit per degree, either positive or negative).37 Meanwhile, increase in temperature also decreases the viscosity of solvent droplet which in turn facilitates the smooth and fast mass transfer of analytes from the aqueous phase into the organic droplet. It seems that the effect of temperature on the kinetics of analytes transfer between two phases is more significant. In other word, increase in the temperature can increase the mass transfer rate of the analytes. This increases o (eqn (2)), and as a consequence, extraction will be performed in a shorter period of time.
However, the mutual solubility of the two phases is also temperature dependent and, at high temperatures, the over-pressurization of the sample vial could also make the extraction system unstable. On the other hand, in LPME boiling point of the solvents is a limiting factor.
Considering the melting point of the extractant used in this method, the effect of extraction temperature on the extraction efficiency of target analytes was checked by varying the temperature within 25–45 °C. The results obtained illustrated that the extraction efficiency increased as the extraction temperature was increased up to ∼35 °C for DSDME, DSME, DS-SFO and Dis-S-SFO methods. After reaching a maximum at this temperature, the extraction efficiency was decreased. One possible reason is that the temperature of the extraction solvent also increases with temperature over time, resulting in less favorable distribution coefficients. With increasing the temperature, upper than 30 °C, the extraction efficiency of USA-EME was slightly decreased. However, further increase can cause to the loss in the volume of extractant phase and so, in extraction efficiency. The increase of temperature up to 45 °C had no significant effect on the LDS-AALLME efficiency. It can be due to high mass transfer rate of the analytes between two phases, at a short period of time.
3.2. Individual parameters
3.2.1.1. Extraction time in DSDME, DSME, DS-SFO, Dis-S-SFO methods. Mass transfer of the analytes between the two immiscible phases involved (sample solution and extraction solvent) is time dependent in droplet-based LPME methods. A reasonable extraction time is necessary to guarantee equilibrium between the samples and extractants and appropriate recovery of the analytes. Regarding eqn (10), the increase in the extraction time (t) leads to the decrease in the e−λt and as a result the increase in the (1 − e−λt). The maximum C0(t) is obtained when e−λt is the minimum and (1 − e−λt) is the maximum (preferably near unity). However, a long extraction time of microextraction to reach complete equilibrium may result in drop dissolution and a high rate of drop loss.
| C0(t) = Coeq (1 − e−λt) | (10) |
Bearing in mind that the whole analysis time depends directly on the time needed to perform all process (including extraction and restoration steps), 15, 6, 15, and 6 min were finally selected as suitable extraction times for DSDME, DSME, DS-SFO, Dis-S-SFO methods, respectively (Fig. 4a–d).
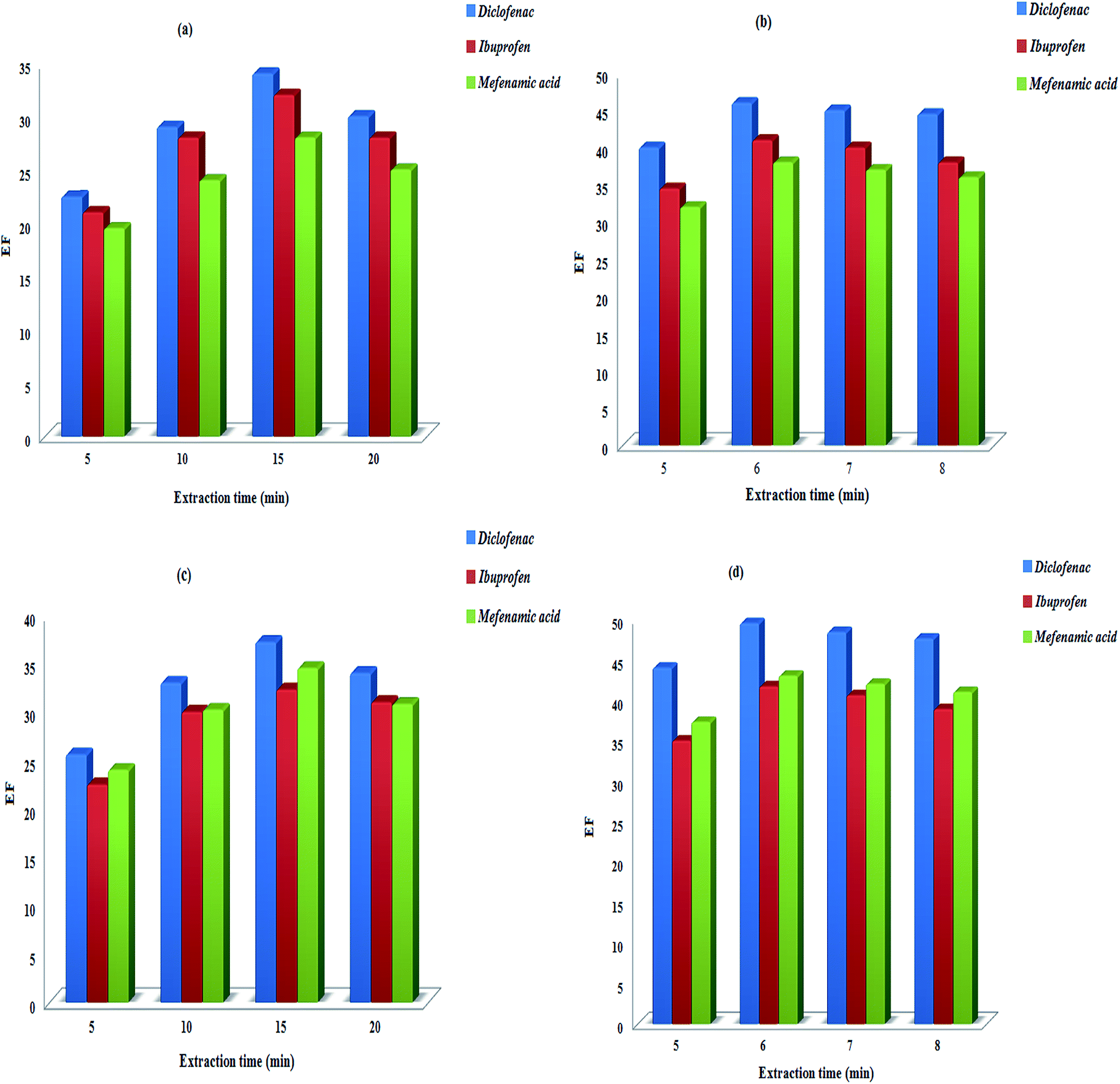 | ||
| Fig. 4 Effect of the extraction time on the analytes enrichment factors. (a) DSDME (b), DSME, (c) DS-SFO, (d) Dis-S-SFO. | ||
3.2.1.2. Sonication time in USA-EME method. Sonication plays an important role in the USA-EME method because it provokes the dispersion of extractant into the aqueous phase in the form of fine droplets that accelerate the transfer of analyte into the extraction phase. Hence, the effect of sonication time was evaluated in the range of 30–180 s (Fig. 5a). The results obtained showed that the extraction efficiency increased till 120 s of sonication and then decreased slightly.
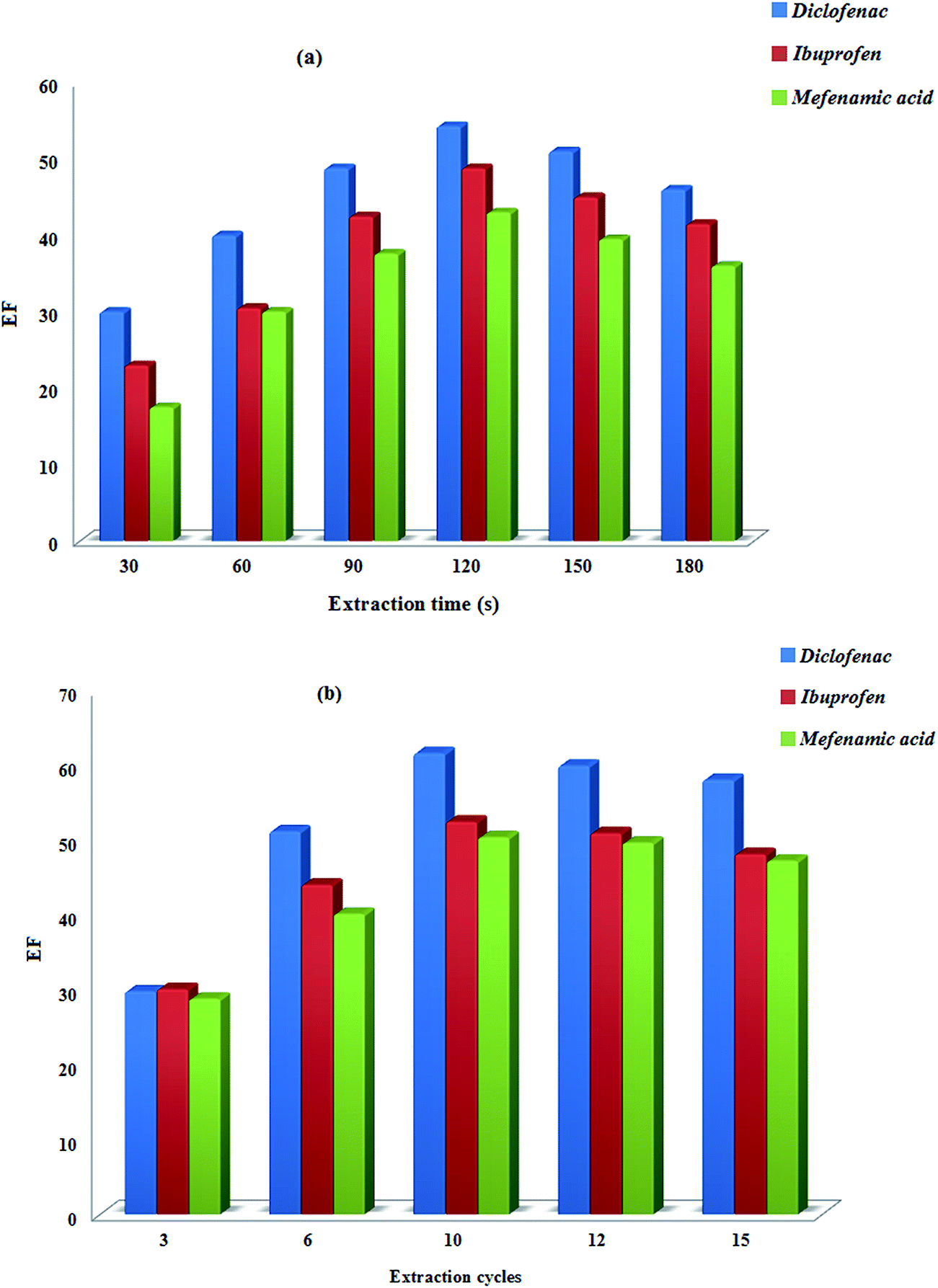 | ||
| Fig. 5 Effect of the extraction time (USA-EME) (a) and extraction cycles (LDS-AALLME) (b) on the analytes enrichment factors. | ||
3.2.3.1. Stirring rate in DSDME and DS-SFO methods. According to the film theory of convective-diffusive mass transfer for LPME system, high stirring rate can decrease the thickness of the diffusion film in the aqueous phase, so the aqueous phase mass-transfer coefficient will be increased with increased stirring rate (rpm). Furthermore, the rotation of the micro-droplet around a symmetrical axis may cause an internal recycling and intensify the mass transfer process inside the droplet. Since restoration of extractant phase is not considered for DSDME and DS-SFO methods, increasing stirring rate must be controlled, because it may be cause to sputtering of the solvent drops and influence the extraction efficiency.
Different stirring rates (500–800 rpm) were examined to achieve higher extraction efficiencies. The extraction efficiency increased and reached its maximum as the stirring rate was increased to 700 rpm, but declined obviously with greater agitation. It may be that a higher stirring rate (more than 800 rpm) generates a more unstable fluid field, thereby breaking the droplet, resulting in its dispersion in the aqueous phase. Therefore, the stirring rate was selected at 700 rpm for further analysis.
3.2.3.2. Stirring rate in DSME and Dis-S-SFO methods. In these methods, two stirring rates (extraction and restoration rates) were used. The first one was extraction rate under which a cloudy solution was formed and extraction solvent was dispersed as the fine droplets. The other was the restoration rate under which a vortex was obtained during the restoration step. In this step, the energy created by slow agitation is not enough for maintaining the fine droplets dispersed but can make the fine droplets gather up in the top-center position of the vortex.
The influence of the extraction rate was studied in the range of 900–1200 rpm. The results revealed that the extraction efficiency improved as the stirring rate increased. Hence, 1200 rpm (the maximum achievable stirring speed of the magnetic stirrer) was used for DSME and Dis-S-SFO methods.
The effect of restoration rate was examined in the range of 200–500 rpm in constant experimental conditions. Restoration speed below 200 rpm was not investigated, because it could not create a vortex which is easy to withdraw the suspended phase into the microsyringe. When the restoration speed was higher than 400 rpm, the suspended phase was not stable and easy for the dispersive droplets to gather up. The extraction efficiencies were seen to increase when the restoration rate was held at 400 rpm. Hence, this rate was used for further analysis.
3.3. Method validation
Based on the obtained results, DSME, Dis-S-SFO, USA-EME, and LDS-AALLME were shown to be faster and more efficient than DSDME and DS-SFO methods. To select the best method, limits of detection (LODs), limits of quantification (LOQs), linear dynamic ranges (LDRs), and relative standard deviations (in terms of repeatability) of four methods were calculated (Table 1). Sensitivity of the method was evaluated in terms of LOD and LOQ which were statistically calculated as 3 and 10 times of the standard deviations of seven replicate extractions of analyte minimum detectable concentrations divided on the calibration slope. Repeatabilities (calculated in terms of intra-day and inter-day precisions) were evaluated by analyzing five replicates of the model sample at three different concentration levels (low, middle, and high) in the same day and five different days. Enrichment factors (EFs) and relative recoveries (RR) of the analytes were used as the parameters to evaluate the method efficiency. The EF was calculated by eqn (11).
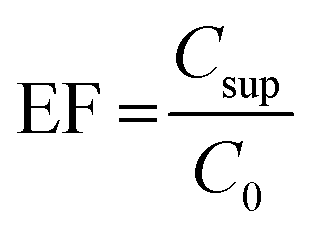 | (11) |
| Analytes | LODe (μg L−1) | LOQe (μg L−1) | LDRf (μg L−1) | Intra-day precision (%) | Inter-day precision (%) | EFg | CIh | Extraction time (min) |
|---|---|---|---|---|---|---|---|---|
| a Experimental conditions in DSME: “Extraction solvent: n-octanol, 50 μL; sample pH: 2.5; without salt addition; temperature of sample: 35 °C; total extraction time: 6 min; stirring rate of extraction step: 1200 rpm; stirring rate of restoration step: 400 rpm”.b Experimental conditions in Dis-S-SFO: “Extraction solvent: 2-dodecanol, 40 μL; sample pH: 2.5; without salt addition; temperature of sample: 35 °C; total extraction time: 6 min; stirring rate of extraction step: 1200 rpm; stirring rate of restoration step: 400 rpm; solidification time: 4 min”.c Experimental conditions in USE-EME: “Extraction solvent: n-octanol, 80 μL; sample pH: 2.5; without salt addition; temperature of sample: 30 °C; sonication time: 120 s; centrifugation time: 4 min”.d Experimental conditions in LDS-AALLME: “Extraction solvent: n-octanol, 65 μL; sample pH: 2.5; without salt addition; temperature of sample: 30 °C; numbers of extraction cycles: 10 cycles in 40 s; centrifugation time: 4 min”.e n = 7.f Linear dynamic range.g n = 3.h Consumptive index. | ||||||||
| DSMEa | ||||||||
| Diclofenac | 3.0 | 11.0 | 11.0–2200 | 4.5 | 5.3 | 50 ± 2 | ∼0.10 | 6 |
| Ibuprofen | 3.5 | 12.0 | 12.0–2727 | 4.4 | 4.9 | 44 ± 2 | ∼0.11 | |
| Mefenamic acid | 2.4 | 8.0 | 8.0–2093 | 4.8 | 5.7 | 43 ± 2 | ∼0.12 | |
|
||||||||
| Dis-S-SFOb | ||||||||
| Diclofenac | 2.0 | 7.0 | 7.0–2115 | 3.3 | 4.0 | 52 ± 1 | ∼0.10 | 10 |
| Ibuprofen | 3.0 | 10.0 | 10.0–2608 | 3.1 | 4.2 | 46 ± 1 | ∼0.11 | |
| Mefenamic acid | 1.9 | 4.0 | 4.0–1837 | 3.6 | 4.5 | 49 ± 1 | ∼0.10 | |
|
||||||||
| USA-EMEc | ||||||||
| Diclofenac | 2.3 | 7.5 | 7.5–2037 | 3.9 | 5.0 | 54 ± 2 | ∼0.09 | 6 |
| Ibuprofen | 2.0 | 7.0 | 7.0–2500 | 4.1 | 5.5 | 48 ± 2 | ∼0.10 | |
| Mefenamic acid | 3.0 | 10.0 | 5.0–2093 | 4.3 | 5.7 | 43 ± 2 | ∼0.12 | |
|
||||||||
| LDS-AALLMEd | ||||||||
| Diclofenac | 1.1 | 3.5 | 3.5–1864 | 6.2 | 7.3 | 61 ± 2 | ∼0.08 | 4 |
| Ibuprofen | 1.7 | 5.5 | 5.5–2448 | 6.6 | 7.9 | 52 ± 2 | ∼0.10 | |
| Mefenamic acid | 1.5 | 5.0 | 5.0–1875 | 6.3 | 7.8 | 50 ± 2 | ∼0.10 | |
The RR was calculated by eqn (12).
 | (12) |
However, in order to achieve this purpose, consumptive index (CI) was considered as a useful criterion and defined as:
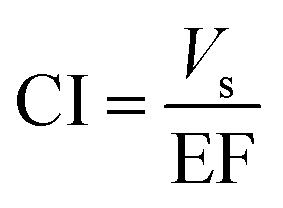 | (13) |
Three replicate extractions were performed in all calculations.
After optimization, the results showed that the DSME, Dis-S-SFO, LDS-AALLME and USA-EME methods have similar extraction efficiencies for the analytes. Although these methods are all simple, disperser solvent-free and convenient with organic solvent consumption at μL level, each of them has its unique capabilities and can be considered as a preferred microextraction method for the extraction of target analytes. The main advantages of DSME and Dis-S-SFO methods are; (i) the controlled stirrings for splitting and rejoining the organic droplets have avoided the use of centrifugation step, and (ii) the entire process involves only one step to extract target analytes as well as to separate and pre-concentrate the extracted phase. In contrary, they need more extraction times than USE-EME and LDS-AALLME methods.
Under the optimum conditions, the results showed that the repeatability and linearity of Dis-S-SFO were better than DSME and much better than that of USA-EME and LDS-AALLME methods. However, the sensitivity and extraction efficiency obtained by LDS-AALLME were higher than those obtained by other methods, reflecting that LDS-AALLME extracts the analytes much more efficiently as compared to examined methods. Besides, this method was faster and simpler than other examined methods. Altogether, the characteristics of LDS-AALLME were good enough for a practically reliable measurement, so that it was selected as a preferred method for extraction of target analytes (Table 1).
3.4. Application to real samples
After validation, the LDS-AALLME method was successfully applied to the analysis of plasma and urine samples taken from six healthy volunteers who were orally treated with 200, 250, and 250 mg of sodium diclofenac, ibuprofen, and mefenamic acid, respectively. The samples were collected 1, 2, 4, 8 and 12 h (after administration of tablets) and the maximum plasma and urinary excretion of the analytes were determined after 2 and 4 h, respectively. The quantification of the analytes was carried out using the standard addition method. Fig. 6 shows typical chromatograms obtained by analysis of standard mixture, plasma and urine samples extract from volunteers that was obtained 2 and 4 h after target analytes intake. | ||
| Fig. 6 Typical chromatograms of standards (20 μg mL−1) (a), spiked blank plasma (b), blank urine (c), plasma (d), and urine (e) after LDS-AALLME extraction, at optimum conditions (1) diclofenac, (2) ibuprofen, (3) mefenamic acid. | ||
Table 2 provides the results of three replicate plasma and urine analysis for all volunteers. To investigate accuracy of the method, the samples were spiked with certain amounts of under study drugs. The relative recoveries of the analytes were in the range of 94–102% (Table 2). The results showed that the LDS-AALLME can be useful for obtaining relevant clinical information related to bioactivity for these drugs. Also, this method can be used to determine the pharmacokinetic parameters of other NSAIDs analysed in these types of studies.
| Sample | Ibuprofen | Diclofenac | Mefenamic acid | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Added (μg L−1) | Found (found-real) (μg L−1) | RRb (%) | Added (μg L−1) | Found (found-real) (μg L−1) | RR (%) | A (μg L−1) | Found (found-real) (μg L−1) | RR (%) | |
| a Experimental conditions in USE-AALLME: “Extraction solvent: n-octanol, 30 μL; sample pH: 4; without salt addition; simultaneous sonication and numbers of extraction cycles: 5 cycles in 20 s; centrifugation time: 4 min”. brelative recovery, n = 3, cstandard deviation, *found, **(found-real). | |||||||||
| Plasma (after 12 h of ibuprofen administration) | 0.0 | 1029.3 ± 61.7c* | — | 0.0 | — | — | 0.0 | — | — |
| 200.0 | (202 ± 12.5)** | 101 | 10.0 | (9.7 ± 0.65) | 97 | 10.0 | (9.6 ± 0.61) | 96 | |
| Urine (after 12 h of ibuprofen administration) | 0.0 | 879.8 ± 56.3 | — | 0.0 | — | — | 0.0 | — | — |
| 200.0 | (198 ± 12.8) | 99 | 10.0 | (9.8 ± 0.61) | 98 | 10.0 | (10.2 ± 0.68) | 102 | |
| Plasma (after 12 h of diclofenac administration) | 0.0 | — | — | 0.0 | 487.9 ± 32.2 | — | 0.0 | — | — |
| 10.0 | (9.5 ± 0.60) | 95 | 100.0 | (98 ± 6.1) | 98 | 10.0 | (9.4 ± 0.62) | 94 | |
| Urine (after 12 h of diclofenac administration) | 0.0 | — | — | 0.0 | 325.3 ± 21.5 | — | 0.0 | — | — |
| 10.0 | (9.8 ± 0.64) | 98 | 100.0 | (101 ± 6.4) | 101 | 10.0 | (9.6 ± 0.64) | 96 | |
| Plasma (after 12 h of mefenamic acid administration) | 0.0 | — | — | 0.0 | — | — | 0.0 | 874.8 ± 58.6 | — |
| 10.0 | (9.6 ± 0.66) | 96 | 10.0 | (9.9 ± 0.63) | 99 | 200.0 | (190 ± 12.7) | 95 | |
| Urine (after 12 h of mefenamic acid administration) | 0.0 | — | — | 0.0 | — | — | 0.0 | 795.3 ± 51.7 | — |
| 10.0 | (10.1 ± 0.63) | 101 | 10.0 | (10.2 ± 0.67) | 102 | 200.0 | (194 ± 13.1) | 97 | |
4. Conclusions
In the present study, a dispersive suspended-solidified floating organic droplet microextraction method was developed to overcome long extraction times (associated with suspended droplet-based microextraction methods) and uncertainties in collection of low volume of extraction solvent (associated with dispersive suspended droplet-based microextraction methods) coupled to HPLC. Although the selected method showed higher extraction efficiencies and lower RSDs, total extraction time was higher than DSME due to necessary solidification step.Until now, no or very few studies have been published regarding comparison of droplet- and dispersive-based microextraction methods. In this way, two droplet-based (directly suspended droplet and dispersive suspended), two solidified droplet-based (directly suspended-solidified floating organic droplet and dispersive suspended-solidified floating organic droplet), and two disperser solvent-free dispersive-based (air-assisted liquid–liquid and ultrasound-assisted emulsification) microextraction methods were critically compared for the determination of three NSAIDs as model analytes. The results obtained showed that all DSME, Dis-S-SFO, LDS-AALLME and USA-EME methods are enough sensitive with low limits of detection that can be successfully applied to separation, preconcentration, and determination of NSAIDs in bio-fluid samples. However, USA-EME and LDS-AALLME methods showed higher recoveries and enrichment factors. However, the final results showed that LDS-AALLME is simpler, faster and more effective than the other methods, as it needed only 40 s to achieve the equilibrium with acceptable repeatabilities. Furthermore, it is more cost effective than the USA-EME, because a sonicator apparatus is not required. Hence, LDS-AALLME was selected as a preferred method for analyzing of ibuprofen, mefenamic acid and sodium diclofenac in human plasma and urine samples.
In comparison with other published methods for extraction of target analytes, the AALLME method has some advantages (Table 3) including (i) low amount of extraction solvent is consumed, (ii) it is simple and performed in a short period of time, (iii) the analytical merits are comparable to other extraction methods for the analytes, and (iv) no toxic disperser solvent – used in other LPME-based methods such as ethanol, acetone, acetonitrile and methanol – is used in this method. These characteristics are of key interest for laboratories doing routine analysis of this type of analytes in different real samples.
| Method | Matrix | Analyte | LOD | LDR | EF | Total volume of extraction solvent | Extraction time (min) | Reference |
|---|---|---|---|---|---|---|---|---|
| a Hollow-fiber liquid-phase microextraction.b Microextraction by packed sorbent.c Hollow-fiber liquid-phase microextraction.d Solid-phase extraction combined with supramolecular solvents.e Rotating disk sorptive extraction.f Liquid–liquid extraction.g Not reported. | ||||||||
| HF-LPMEa/UPLC-MS/MS | Real water, juice, soda, energy drinks | Salicylic acid, ibuprofen, naproxen, diclofenac | 0.5–1.25 μg L−1 | 1.0–5000 μg L−1 | 195–350 (For 5 mL of sample) | 15 μL | 30 | 38 |
| MEPSb/UHPLC | Human urine | Diclofenac, ibuprofen, acetylsalicylic acid, ketoprofen, naproxen | 1.07–16.2 μg L−1 | 10–20000 μg L−1 |
0.9–1.0 (for 20 μL of sample) | 20 μL | 5 | 39 |
| HF-LPMEc/HPLC-DAD and HPLC-FLD | Human urine | Diclofenac, salicylic acid, ibuprofen | 12.3–52.9 μg L−1 | 41.0–10000 μg L−1 |
70–1060 (for 50 mL of sample) | 50 μL | 15 | 40 |
| SPE-SUPRASFd/HPLC-UV | Human urine, water | Diclofenac, mefenamic acid | 0.4–7.0 μg L−1 | 1.0–300.0 μg L−1 | 431–489 (for 30 mL of sample) | 1500 μL | 25 | 28 |
| RDSEe/HPLC-UV | Human urine | Diclofenac, ibuprofen, ketoprofen, naproxen | 21.7–44.0 μg L−1 | 200.0–2000.0 μg L−1 | 15–18 (For 5 mL of sample) | 200 μL | 20 | 41 |
| LLEf/HPLC-UV | Human plasma | Ketoprofen, naproxen, fenoprofen, flurbiprofen, ibuprofen, diclofenac | 11.5–75.0 μg L−1 | 100.0–100000.0 μg L−1 | NRg | 600 μL | NRg | 42 |
| AALLME/HPLC-UV | Human plasma, human urine | Ibuprofen, diclofenac, mefenamic acid | 1.1–1.7 μg L−1 | 3.5–2448 μg L−1 | 50–61 (for 5 mL of sample) | 65 μL | ∼4 | This work |
References
- B. Barfi, A. Asghari, M. Rajabi, A. Barfi and I. Saeidi, J. Chromatogr. A, 2013, 1311, 30–40 CrossRef CAS PubMed.
- T. Ahmadi-Jouibari, N. Fattahi and M. Shamsipur, J. Pharm. Biomed. Anal., 2014, 94, 145–151 CrossRef CAS PubMed.
- F. Maya, B. Horstkotte, J. M. Estela and V. Cerdà, Trends Anal. Chem., 2014, 59, 1–8 CrossRef CAS.
- B. Barfi, A. Asghari, M. Rajabi, S. Sabzalian, F. Khanalipoor and M. Behzad, RSC Adv., 2015, 5, 31930–31941 RSC.
- J. Pawliszyn, Solid phase microextraction: theory and practice, John Wiley & Sons, 1997 Search PubMed.
- Y. He and H. K. Lee, Anal. Chem., 1997, 69, 4634–4640 CrossRef CAS.
- Y. Wang, Y. C. Kwok, Y. He and H. K. Lee, Anal. Chem., 1998, 70, 4610–4614 CrossRef CAS PubMed.
- Y. Yan, X. Chen, S. Hu and X. Bai, J. Chromatogr. A, 2014, 1368, 1–17 CrossRef CAS PubMed.
- E. M. Martinis, P. Berton and R. G. Wuilloud, Trends Anal. Chem., 2014, 60, 54–70 CrossRef CAS.
- L. Vidal, E. Psillakis, C. E. Domini, N. Grané, F. Marken and A. Canals, Anal. Chim. Acta, 2007, 584, 189–195 CrossRef CAS PubMed.
- A. V. de Bairros, R. M. de Almeida, L. Pantaleão, T. Barcellos, S. M. E. Silva and M. Yonamine, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 975, 24–33 CrossRef CAS PubMed.
- M. Parrilla Vázquez, P. Parrilla Vázquez, M. Martínez Galera, M. Gil García and A. Uclés, J. Chromatogr. A, 2013, 1291, 19–26 CrossRef PubMed.
- M. Rajabi, H. Ghanbari, B. Barfi, A. Asghari and S. Haji-Esfandiari, Food Res. Int., 2014, 62, 761–770 CrossRef CAS.
- E. Psillakis and N. Kalogerakis, Trends Anal. Chem., 2002, 21, 54–64 CrossRef.
- L. Yangcheng, L. Quan, L. Guangsheng and D. Youyuan, Anal. Chim. Acta, 2006, 566, 259–264 CrossRef.
- S. Wen, X. Zhu, X. Wu and X. Qin, Anal. Methods, 2014, 6, 9777–9782 RSC.
- Z.-H. Yang, Y. Liu, Y.-L. Lu, T. Wu, Z.-Q. Zhou and D.-H. Liu, Anal. Chim. Acta, 2011, 706, 268–274 CrossRef CAS PubMed.
- N. P. Petridis, V. A. Sakkas and T. A. Albanis, J. Chromatogr. A, 2014, 1355, 46–52 CrossRef CAS PubMed.
- S. Li, X. Yang, L. Hu, X. Cui, S. Zhang, R. Lu, W. Zhou and H. Gao, Anal. Methods, 2014, 6, 7510–7517 RSC.
- J. Regueiro, M. Llompart, C. Garcia-Jares, J. C. Garcia-Monteagudo and R. Cela, J. Chromatogr. A, 2008, 1190, 27–38 CrossRef CAS PubMed.
- O. Behrend, K. Ax and H. Schubert, Ultrason. Sonochem., 2000, 7, 77–85 CrossRef CAS PubMed.
- P. Walstra, Chem. Eng. Sci., 1993, 48, 333–349 CrossRef CAS.
- M. Rajabi, S. Asemipour, B. Barfi, M. R. Jamali and M. Behzad, J. Mol. Liq., 2014, 194, 166–171 CrossRef CAS.
- M. A. Farajzadeh and M. R. A. Mogaddam, Anal. Chim. Acta, 2012, 728, 31–38 CrossRef CAS PubMed.
- M. A. Farajzadeh and N. Nouri, Anal. Chim. Acta, 2013, 775, 50–57 CrossRef CAS PubMed.
- B. Barfi, A. Asghari, M. Rajabi, A. Goochani Moghadam, N. Mirkhani and F. Ahmadi, J. Pharm. Biomed. Anal., 2015, 111, 297–305 CrossRef CAS PubMed.
- X. You, Z. Xing, F. Liu and N. Jiang, J. Chromatogr. A, 2013, 1311, 41–47 CrossRef CAS PubMed.
- B. Barfi, A. Asghari, M. Rajabi and S. Sabzalian, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2015, 998, 15–25 CrossRef PubMed.
- G. G. Noche, M. E. F. Laespada, J. L. P. Pavón, B. M. Cordero and S. M. Lorenzo, J. Chromatogr. A, 2011, 1218, 9390–9396 CrossRef CAS PubMed.
- F. Rezaei, Y. Yamini, M. Moradi and B. Ebrahimpour, Talanta, 2013, 105, 173–178 CrossRef CAS PubMed.
- M. S. Beldean-Galea, V. Coman, D. Thiébaut and J. Vial, J. Sep. Sci., 2015, 38, 641–648 CrossRef CAS PubMed.
- Declaration of Helsinki, 59th WMA General Assembly, http://www.wma.net/en/30publications/10policies/b3/.
- E. G. C. Clarke, A. C. Moffat, M. D. Osselton and B. Widdop, Clarke's analysis of drugs and poisons: in pharmaceuticals, body fluids and postmortem material, Pharmaceutical Press, 2004 Search PubMed.
- H. Farahani, P. Norouzi, A. Beheshti, H. R. Sobhi, R. Dinarv and M. R. Ganjali, Talanta, 2009, 80, 1001–1006 CrossRef CAS PubMed.
- S. Magiera and Ş. Gülmez, J. Pharm. Biomed. Anal., 2014, 92, 193–202 CrossRef CAS PubMed.
- J. M. Kokosa, A. Przyjazny and M. Jeannot, Solvent microextraction: theory and practice, John Wiley & Sons, 2009 Search PubMed.
- J. Sangster, Octanol-water partition coefficients: fundamentals and physical chemistry, John Wiley & Sons, 1997 Search PubMed.
- H. Zhang, Z. Du, Y. Ji and M. Mei, Talanta, 2013, 109, 177–184 CrossRef CAS PubMed.
- S. Magiera, Ş. Gülmez, A. Michalik and I. Baranowska, J. Chromatogr. A, 2013, 1304, 1–9 CrossRef CAS PubMed.
- M. R. Payán, M. Á. B. López, R. Fernández-Torres, J. L. P. Bernal and M. C. Mochón, Anal. Chim. Acta, 2009, 653, 184–190 CrossRef PubMed.
- V. Manzo, M. Miró and P. Richter, J. Chromatogr. A, 2014, 1368, 64–69 CrossRef CAS PubMed.
- Y. Sun, K. Takaba, H. Kido, M. N. Nakashima and K. Nakashima, J. Pharm. Biomed. Anal., 2003, 30, 1611–1619 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |