
New highly brominated Mn-porphyrin: a good catalyst for activation of inert C–H bonds†
Abstract
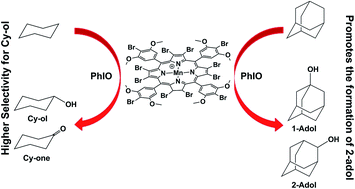
This work describes the synthesis and characterization of a novel third-generation catalyst 5,10,15,20-tetrakis-(4′-bromine-3′,5′-dimethoxyphenyl)-2,3,7,8,12,13,17,18-octabromoporphyrinatomanganese chloride [MnIIIBr12T3,5DMPP]Cl (Cat.2). The catalytic activity of Cat.2 in cyclohexane, adamantane, and n-hexane oxidation by iodosylbenzene (PhIO) or iodobenzene diacetate (PhI(OAc)2) was compared with the catalytic activity of [MnIIIT3,5DMPP]Cl (Cat.1), a second generation catalyst. The Cat.2/PhI(OAc)2 system led to higher yields of cyclohexane oxidation products (65%) with high selectivity for cyclohexanol (86%) as compared with Cat.1 (19% and 74%, respectively) and addition of water essentially did not alter total product yield. Addition of a small amount of imidazole to the Cat.1/PhIO system gave superior yields of cyclohexane oxidation products (64%) as compared with Cat.2 (52%). In all systems Cat.2 afforded significantly higher yields of 2-adamantanol, a product with great commercial value compared with 1-adamantanol. n-Hexane oxidation gave low total product yield; Cat.2 was more selective for alcohol products (2-hexanol and 3-hexanol).
 Please wait while we load your content...
Please wait while we load your content...

