The effect of the stacking fault on the diffusion of chemisorbed hydrogen atoms inside few-layered graphene†
Dong Hyen Chunga,
Hyein Guka,
Deajin Kimab,
Sang Soo Hanc,
Noejung Parkd,
Kihang Choib and
Seung-Hoon Choi*a
aInsilicotech Co., Ltd. C-602, Korea Bio Park, 694-1, Sampyeong-dong, Bundang-gu, Seongnam-si, Gyeonggi-do 463-400, Korea. E-mail: dhjung@insilicotech.co.kr; hinguk@insilicotech.co.kr; djkim@insilicotech.co.kr; shchoi@insilicotechc.co.kr; Fax: +82 31 628 0333; Tel: +82 31 628 0300
bDepartment of Chemistry, Korea University, 145, Anam-ro, Seongbuk-gu, Seoul 136-701, Korea. E-mail: kchoi@korea.ac.kr
cCenter for Computational Science, Korea Institute of Science and Technology, Hwarangno 14-gil 5, Seongbuk-gu, Seoul 136-791, Korea. E-mail: sangsoo@kist.re.kr
dInterdisciplinary School of Green Energy and Low Dimensional Carbon Materials Center, Ulsan National Institute of Science and Technology, UNIST-gil 50, Ulsan 689-798, Korea. E-mail: neojung@unist.ac.kr
First published on 11th December 2013
Abstract
We examined the diffusion of hydrogen atoms in mono-, bi- and tetralayer graphene with AB stacking and two bilayer graphene with stacking faults using density functional theory. The bi- and tetralayer graphene provide diffusion pathways with lower energy barriers inside the interlayer space. Inside the bi- and tetralayer graphene with AB stacking, the in-plane diffusion is more favorable than the inter-plane jumping. However, the stacking faults made by sliding layer planes lowers the energy barrier of the inter-plane jumping and the effective frequency of the inter-plane jump is larger than that of the in-plane diffusion inside the graphene layers with the stacking faults. This suggests that hydrogen atoms can diffuse over a long distance inside few-layered graphene with stacking faults jumping consecutively between adjacent layers.
1 Introduction
The hydrogenation of carbon-based nano materials has been attractive for various applications including electronic devices and hydrogen storage systems.1–5 As the hydrogenation patterns strongly affect the physical and/or chemical properties of the hydrogenated graphenes, many researchers have studied the thermodynamic, kinetic and structural characteristics of hydrogen atoms chemisorbed on the graphene surface.6–13 To finely control the graphene properties, understanding the atomic hydrogen kinetics on the graphene surface is indispensable.The kinetics of hydrogen atoms adsorbed on the graphene surface has raised particular attention since the spillover mechanism was proposed for hydrogen storage in carbon-based materials.5 The diffusion barrier of a hydrogen atom chemically adsorbed on the pristine graphene surface has been estimated to be around 1 eV,14–16 and a move from the edge of a stable hydrogenated island formed on the graphene surface needs to overcome an energy barrier of around 3 eV.17 In this regard, it was suggested that in order for the diffusion to take place with ease at room temperature, physisorbed hydrogen atoms18,19 or defect structures20,21 should be involved in the spillover process, but those arguments do not seem to be enough for an explanation on the spillover in carbonaceous materials.
Although the present interests are concentrated on monolayer graphene, recent researches revealed that bilayer graphene can show distinct properties such as gate-induced band gap opening22 and chiral quasiparticle.23 Similar with monolayer graphene, chemically modified bilayer graphene displays different electronic structures from the original bilayer.24–28 In half hydrogenated bilayer graphene (bilayer graphane) modeled by using the first principle calculations,25,26 interlayer C–C chemical bonds are formed resulting in a diamond-like structure capped by hydrogen atoms on both outer sides. It was shown by density functional theory calculations that external strain28 or electric bias25 allows the continuous tuning of the band gap of hydrogenated bilayer graphene.
The kinetics of hydrogen atoms adsorbed in multilayer graphene have rarely been investigated. Stable hydrogenation configurations29 and the energy barriers of in-plane diffusion and inter-plane jumping in bulk graphite16 have been investigated by density functional theory. However, the calculations have been carried out without proper corrections for van der Waals interactions. By tight binding molecular dynamics simulations on graphite, it was found that hydrogen atoms move to adjacent carbon atoms in the same or adjacent sheet with a diffusion barrier of 0.40 eV,30 but more detailed information about the hydrogen atom diffusion through interlayer space is still lacking. In this work, we calculated the diffusion barriers of chemisorbed hydrogen atoms in AB stacking bilayer and tetralayer graphene at the density functional theory level. In addition, we investigated the effect of the stacking fault (e.g. glide defects) on the hydrogen atom diffusion and found that the stacking fault can facilitate the diffusion by lowering the barrier of inter-plane jumping.
2 Computational method
All calculations were performed by a first principles quantum mechanical code, CASTEP program31 in the Materials Studio 5.5 package. The electron exchange-correlation functional was represented with the generalized gradient approximation of Perdew, Burke and Ernzerhof (PBE)32 augmented with the Tkatchenko–Scheffler dispersion correction (PBE-TS).33,34 The convergence tolerance for self-consistent field (SCF) iterations was set to 2.0 × 10−6 eV per atom and the values of the geometry optimization convergence thresholds for energy change, maximum force, maximum stress, and maximum displacement between optimization cycles are 2 × 10−5 eV per atom, 0.05 eV Å−1, 0.1 GPa, and 0.002 Å, respectively. Optimizing the unit cell of bulk graphite using PBE, PBE-TS and the local density approximation (LDA)35,36 the interlayer distance of graphite is 4.271, 3.320 and 3.307 Å, respectively. PBE underestimates the van der Waals interaction between the layer planes and LDA overestimates it, while the PBE-TS result shows better agreement with the experimental value of 3.336 Å.37 Therefore, PBE-TS was chosen as the exchange-correlation functional in all calculations. The core electrons were described by the ultrasoft pseudopotentials.38 To treat the metallic character of graphene, the Gaussian-like smearing scheme39 was adapted with the width of 0.1 eV. In the optimization of the structures, the kinetic energy cutoff of 400 eV was used and the 2 × 2 × 1 k-points were sampled in the first Brillouin zone by Monkhorst and Pack method.40 All calculations for hydrogenated states are spin polarized. The transition states were searched by the linear and quadratic synchronous transit method (LST/QST)41 implemented in CASTEP. The LST/QST method linearly interpolates a reaction pathway connecting reactants and products and finds the maximum energy state. Using that state, another conjugate gradient minimization is performed to locate a stationary point.When chemisorbed in the multilayer graphene, hydrogen atoms can bind to two types of carbon atoms: (i) carbon A having neighbors directly above and below in the adjacent layers; and (ii) carbon B facing the middle hollow sites of hexagons in adjacent layers (Fig. 1a). Thus, if a hydrogen atom is attached to carbon A (or B) from the upside (or from the downside) of layer 1, it is labeled as 1Au (or 1Bd). In order to investigate the hydrogen atom diffusion through interlayer space, monolayer (MNL), bilayer (BIL) and tetralayer (TEL) graphene with AB stacking were modeled.
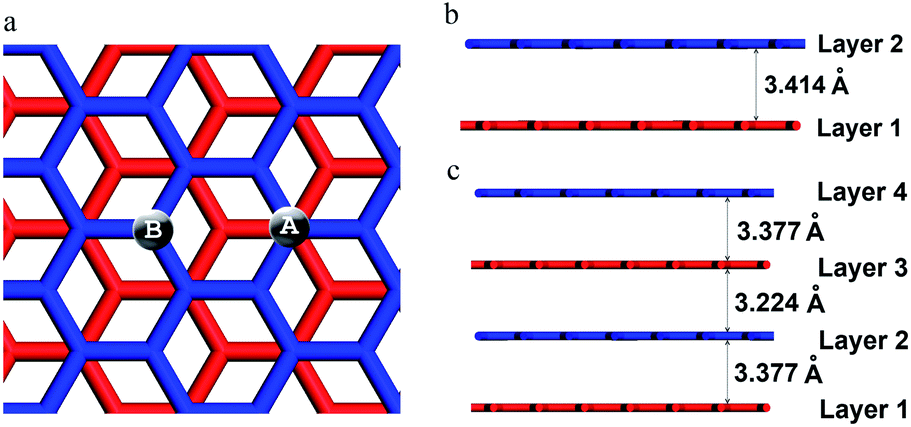 | ||
| Fig. 1 (a) A schematic description of carbon A and carbon B, (b) the bilayer graphene and (c) the tetralayer graphene with AB stacking. The layer planes are numbered sequentially from bottom to top. | ||
The dimensions of supercells 6 × 6 (MNL), 6 × 6 × 1 (BIL) and 6 × 6 × 2 (TEL) are 14.76 × 14.76 × 15.00, 14.73 × 14.73 × 18.32 and 14.73 × 14.73 × 24.96 Å, respectively. In all models ∼15 Å vacuum region was included in the direction perpendicular to the layer plane. The optimized structures show that the interlayer spacing of bulk graphite is well retained in the BIL and TEL structures (Fig. 1b and c).
3 Results and discussion
3.1 The diffusion of the H atom adsorbed in AB stacking graphenes
Before the kinetics study of the adsorbed hydrogen atom, it should be confirmed that hydrogen atom binding is thermodynamically favorable even in the interlayer space. To this end, we first calculated the binding energy, Ebind of a hydrogen atom using the following equation| Ebind = E(graphene) + E(H(g)) − E(graphene − H), | (1) |
The hydrogen binding occurring in the interlayer space (inside binding) is about 0.02–0.2 eV weaker than that occurring outside of the graphene (outside binding). This instability of the inside binding is caused by a strained structure of the hydrogenated carbon atom. Compared to the outside binding, the inside binding does not allow the hybridization of the hydrogenated carbon to be perfectly transformed into sp3 because the close contact between the adsorbed hydrogen atom and the opposite plane hinders the hydrogenated carbon from protruding (see Fig. S1 of the ESI†).
Next, we calculated the hydrogen diffusion energy barriers. Our calculation shows that the energy barriers are about 1.27–1.29 eV for the in-plane diffusion occurring outside the BIL and TEL (2Au ↔ 2Bu for BIL and 4Au ↔ 4Bu for TEL) and these barriers are very similar to that for the diffusion on MNL (Table 2). However, the barriers of the in-plane diffusion paths inside the BIL and TEL are lowered by 0.11–0.38 eV, which is attributed to the destabilized reactants and products as mentioned above and the more stable transition states inside the interlayer space. The stabilization of the transition states can be explained by considering the less deformed (more sp2-like) transition states formed between the layers. For the transition state of the in-plane path outside the BIL, two carbon atoms involved in the diffusion deviate from the plane by 0.189 Å, while inside the BIL they deviate by only 0.079 Å. The less deformed transition states inside the interlayer space are also observed in the TEL model (see Fig. S2 of the ESI†).
| Model | Diffusion path | Barrier (eV) | ||
|---|---|---|---|---|
| →a | ←a | |||
| a → is for the forward reaction and ← is for the backward reaction. | ||||
| MNL | In-plane | Ortho | 1.27 | 1.27 |
| BIL | In-plane | 2Au → 2Bu | 1.27 | 1.29 |
| 2Ad → 2Bd | 1.09 | 1.16 | ||
| Inter-plane | 2Ad → 1Au | 0.66 | 0.66 | |
| 2Ad → 1Bu | 1.32 | 1.38 | ||
| 2Bd → 1Bu | 1.33 | 1.32 | ||
| TEL | In-plane | 4Au → 4Bu | 1.28 | 1.29 |
| 4Ad → 4Bd | 1.02 | 1.09 | ||
| 3Au → 3Bu | 1.02 | 1.11 | ||
| 3Ad → 3Bd | 0.91 | 1.04 | ||
| Inter-plane | 4Ad → 3Au | 0.58 | 0.58 | |
| 4Ad → 3Bu | 1.23 | 1.32 | ||
| 4Bd → 3Bu | 1.27 | 1.29 | ||
| 3Ad → 2Au | 0.56 | 0.56 | ||
| 3Ad → 2Bu | 1.15 | 1.28 | ||
| 3Bd → 2Bu | 1.19 | 1.19 | ||
| ZIG | In-plane | 1 | 1.07 | 1.08 |
| 2 | 1.14 | 1.12 | ||
| Inter-plane | 3 | 0.95 | 0.93 | |
| 4 | 0.85 | 0.85 | ||
| 5 | 0.92 | 0.93 | ||
| 6 | 0.95 | 0.96 | ||
| ARM | In-plane | 1 | 1.08 | 1.08 |
| 2 | 1.14 | 1.14 | ||
| Inter-plane | 3 | 0.85 | 0.85 | |
| 4 | 1.18 | 1.19 | ||
The energy barrier of the vertical inter-plane jumping is 0.66 eV in the BIL (2Ad ↔ 1Au), and it reduces in TEL to 0.58 eV (4Ad ↔ 3Au) or 0.56 eV (3Ad ↔ 2Au). The barriers of other types of inter-plane jumping also decrease from 1.32–1.38 eV in the BIL to 1.15–1.32 eV in the TEL. In addition, the barriers are lower for the jumpings between layer 2 and 3 than for the corresponding jumpings between layer 3 and 4 of the TEL. The decrease in the energy barriers is closely related to two things: the destabilized reactants and products, and the travel distance of the hydrogen atom during the diffusion reaction. The shorter the travel distance is, the lower the energy barrier becomes. The relationship between the energy barrier and the travel distance will be more discussed later. Except for the vertical transfer, the inter-plane jumpings have larger barriers than the in-plane diffusion paths, and though the energy barrier for the vertical transfer is very low, it is not a useful pathway for the diffusion of the adsorbed hydrogen atom along the basal plane surface. This suggests that the diffusion of the hydrogen atom takes place by the in-plane paths and the inter-plane jumping hardly helps hydrogen atoms to move around inside the graphite layers with ABAB stacking.
3.2 The influence of the glide defects on the diffusion of chemisorbed H atoms
Gliding graphene sheets cause a bulk defect with alternative stacking structures. In the heat-treated vapor-grown carbon fibers the stacking fault with the graphene layers gliding in the “zigzag” direction have been found.49 The graphite epitaxially grown on the diamond (111) surface has been reported to have a new crystal structure with AA′ staking in which the layers shift in the “armchair” direction.50 We modeled two glide defect structures by modifying the BIL: one by shifting a layer in the “zigzag” direction (ZIG) and the other by shifting in the “armchair” direction (ARM) by a half the hexagon edge width (Fig. 2). The ZIG and ARM structures are less stable than the BIL with AB stacking by 1.17 × 10−3 and 2.15 × 10−4 eV per Catom, respectively. The interlayer spacing of the ZIG is 3.426 Å and that of the ARM is 3.418 Å which agrees well with the experimental value of 3.43 Å for the AA′ stacking structure.50 The hydrogen binding energies in the ZIG and ARM are very similar to those in the BIL structure (Table 1). | ||
| Fig. 2 The ZIG and ARM structures and the diffusion paths inside the interlayer space. In the magnified pictures, the dotted arrows indicate the inter-plane jump paths and the solid arrows indicate the in-plane diffusion paths. | ||
In the ARM structure, the energy barrier of the inter-plane path 3 is 0.85 eV, but that of path 4 is 1.18 eV that is larger than those of the in-plane paths. Therefore, hydrogen atoms are expected to diffuse through both the in-plane and inter-plane paths in the ARM structure. In the ZIG structure, two in-plane diffusion paths defined in Fig. 2 have almost the same barriers as those in the BIL structure. Large differences are found in the barriers of the inter-plane jumping. The energy barriers for the inter-plane jumping in the ZIG are less than 1 eV and are lower by about 0.4 eV than those in the BIL. Therefore, because all the possible inter-plane paths in the ZIG have low energy barriers, hydrogen atoms can diffuse along those pathways over a long distance by consecutive jumps between adjacent layers.
The diffusion rate Γ is usually given by the equation51
| Γ = v0e−ΔG/kBT = v0eΔS/kBe−ΔH/kBT = v*e−ΔH/kBT, | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 048.4 s−1 and that of the in-plane path inside the ZIG is 24477.9 s−1. The inter-plane jump has a larger effective frequency than the in-plane path. In addition to the lower energy barrier of the inter-plane jump between the layers of ZIG, the larger effective frequency of the inter-plane jump make the inter-plane jumping a more favorable diffusion way.
048.4 s−1 and that of the in-plane path inside the ZIG is 24477.9 s−1. The inter-plane jump has a larger effective frequency than the in-plane path. In addition to the lower energy barrier of the inter-plane jump between the layers of ZIG, the larger effective frequency of the inter-plane jump make the inter-plane jumping a more favorable diffusion way.
The lower energy barriers of the inter-plane jumpings in the ZIG structure arise from the short distance through which the hydrogen atom should travel as well as the less tightly bound hydrogen atom inside the interlayer space. In Fig. 3, the linear dependency of the energy barrier on the travel distance is shown. When the sheet-gliding takes place, new inter-plane diffusion paths with short travel distances are generated. This structural change makes the adsorbed hydrogen atoms more easily diffuse in the glide defect structures such as the ZIG than in the AB stacked bilayer graphene. Furthermore, as the glide defect is the bulk defect, it can affect the diffusion kinetics in the greater range compared to the point defects such as vacancies and Stone–Wales defects, and disorder introduced by functionalization, the effect of which is limited within a narrow range around them. Therefore, if chemisorbed hydrogen atoms are to be spread into the bulk, the glide defects would be more suitable for that purpose than the point defects. Especially, hydrogen storage by the spillover onto the graphitic surface could be accomplished more easily by the introduction of the glide defects because one of the main obstacles for understanding and realizing the spillover mechanism is the diffusion of spilled-over hydrogen atoms on graphitic surfaces over a long distance from the metal catalyst on which hydrogen molecules are decomposed to hydrogen atoms.17
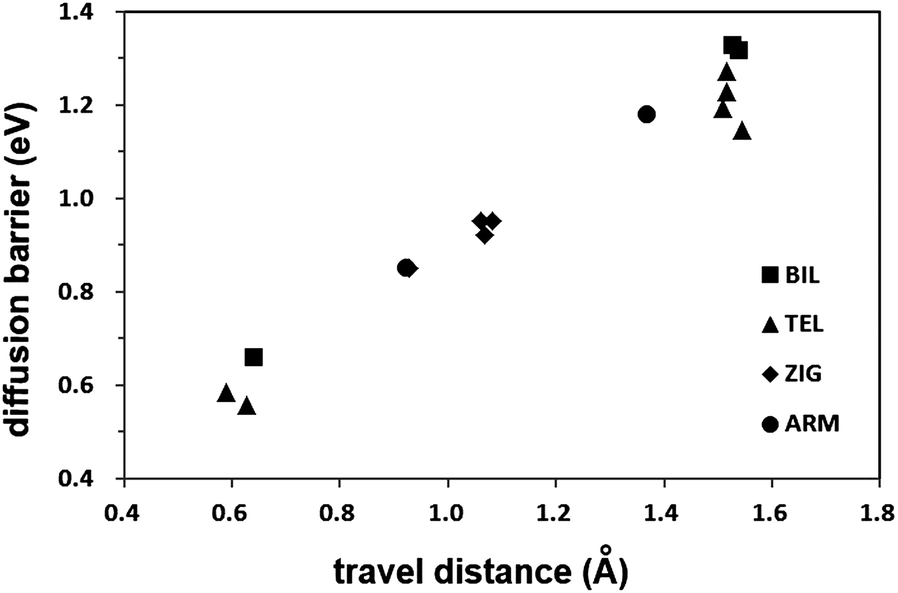 | ||
| Fig. 3 The energy barrier vs. travel distance of the inter-plane jump paths. The travel distance is defined as the distance from the hydrogen atom in the reactant to that in the product. | ||
4 Conclusions
In conclusion, we have calculated the diffusion energy barriers of hydrogen atoms chemisorbed in mono-, bi- and tetralayer graphene with AB stacking and bilayers with glide defect using density functional theory augmented with van der Waals interaction corrections. We found that the stacking fault such as glide defect can lower the diffusion barriers of the inter-plane paths and the inter-plane path has the larger effective frequency, and hence the stacking fault defect can generate new pathways through which hydrogen atoms can proceed jumping back and forth between adjacent layers. In this work, we do not take into account the quantum effects of nuclear motions such as phonon-assisted tunneling. For hydrogen, the quantum effects can reduce the barrier at room temperature by about 10%.13 Therefore, the barriers reported here can be renormalized as the 10% smaller value by the quantum effects.We expect that our results could provide a better understanding of the mechanisms of the hydrogenation processes and hydrogen storage in graphite and multilayer graphene materials. In the inelastic neutron scattering experiments on hydrogen spill-over by the Pt-doped activated carbon (Pt/AC) samples, it was indicated that the spilled-over hydrogen atom on the AC surface could diffuse more than 2.8 nm away from a Pt cluster.53 However, the long distance diffusion of the chemisorbed hydrogen atom has not been fully explained. The present work suggests that the bulk defects possibly existing in the AC carbon could contribute to the long diffusion length. The high mobility of hydrogen atoms inside the ball-milled graphite has been detected by nuclear-magnetic-resonance method.54–56 Although the latest paper assigned the highly mobile hydrogen motion to the rotation of methyl groups,56 given that the mechanical milling could make a lot of defects including the stacking faults in the graphite the fast diffusion inside the ball-milled graphite induced by the stacking faults could be one of the origins of the high mobility of the chemisorbed hydrogen atoms. Lastly, the reduction reaction of azoaromatic compounds separated by graphite foil from the catalytic iron powder57 might be explained by the hydrogen atom diffusion inside the graphite layers.
Acknowledgements
This research was supported by the Creative Research Program (2012–2014) of the Korea Atomic Energy Research Institute. We thank Accelrys Korea for the support of modeling software.References
- S. Niyogi, E. Bekyarova, J. Hong, S. Khizroev, C. Berger, W. de Heer and R. C. Haddon, J. Phys. Chem. Lett., 2011, 2, 2487 CrossRef CAS.
- Z. Sun, D. K. James and J. M. Tour, J. Phys. Chem. Lett., 2011, 2, 2425 CrossRef CAS.
- H. Gao, L. Wang, J. Zhao, F. Ding and J. Lu, J. Phys. Chem. C, 2011, 115, 3236 CAS.
- D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake, M. P. Halsall, A. C. Ferrari, D. W. Boukhalov, M. I. Katsnelson, A. K. Geim and K. S. Novoselov, Science, 2009, 323, 610 CrossRef CAS PubMed.
- J. A. J. Lachawiec, G. Qi and R. T. Yang, Langmuir, 2005, 21, 11418 CrossRef PubMed.
- L. F. Huang, M. Y. Ni, G. R. Zhang, W. H. Zhou, Y. G. Li, X. H. Zheng and Z. J. Zeng, J. Chem. Phys., 2011, 135, 064705 CrossRef PubMed.
- L. F. Huang, M. Y. Ni, Y. G. Li, W. H. Zhou, X. H. Zheng, L. J. Guo and Z. Zeng, Surf. Sci., 2011, 605, 1489 CrossRef CAS PubMed.
- A. A. Dzhurakhalov and F. M. Peeters, Carbon, 2011, 49, 3258 CrossRef CAS PubMed.
- A. R. Muniz and D. Maroudas, J. Appl. Phys., 2010, 108, 113532 CrossRef PubMed.
- V. V. Ivanovskaya, A. Zobelli, D. Teillet-Billy, N. Rougeau, V. Slidis and P. R. Briddon, Eur. Phys. J. B, 2010, 76, 481 CrossRef CAS.
- R. Balog, B. Jorgensen, J. Wells, E. Laegsgaard, P. Hofmann, F. Besenbacher and L. Hornekaer, J. Am. Chem. Soc., 2009, 131, 8744 CrossRef CAS PubMed.
- D. W. Boukhvalov, M. I. Katsnelson and A. I. Lichtenstein, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 035427 CrossRef.
- C. P. Herrero and R. Ramirez, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115429 CrossRef.
- L. F. Huang, M. Y. Ni, X. H. Zheng, W. H. Zhou, Y. G. Li and Z. Zeng, J. Phys. Chem. C, 2010, 114, 22636 CAS.
- D. W. Boukhvalov, Phys. Chem. Chem. Phys., 2010, 12, 15367 RSC.
- Y. Lei, S. A. Shevlin, W. Zhu and Z. X. Guo, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 134114 CrossRef.
- S. S. Han, H. Jung, D. H. Jung, S. H. Choi and N. Park, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 155408 CrossRef.
- G. M. Psofogiannakis and G. E. Froudakis, J. Phys. Chem. C, 2009, 113, 14908 CAS.
- X. Sha, M. T. Knippenberg, A. C. Cooper, G. P. Pez and H. Cheng, J. Phys. Chem. C, 2008, 112, 17465 CAS.
- H. Y. Wu, X. Fan, J. L. Kuo and W. Q. Deng, J. Phys. Chem. C, 2011, 115, 9241 CAS.
- L. F. Huang, M. Y. Ni and Z. Zeng, J. Phys.: Condens. Matter, 2011, 23, 435007 CrossRef PubMed.
- J. B. Oostinga, H. B. Heersche, X. Liu, A. F. Morpurgo and L. M. K. Vandersypen, Nat. Mater., 2008, 7, 151 CrossRef CAS PubMed.
- E. McCann, D. S. L. Abergel and V. I. Fal'ko, Solid State Commun., 2007, 143, 110 CrossRef CAS PubMed.
- F. M. Koehler, A. Jacobsen, K. Ensslin, C. Stampfer and W. J. Stark, Small, 2010, 6, 1125 CrossRef CAS PubMed.
- D. K. Samarakoon and X. Q. Wang, ACS Nano, 2010, 4, 4126 CrossRef CAS PubMed.
- O. Leenaerts, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 245422 CrossRef.
- J. M. B. LopesdosSantos, N. M. R. Peres and A. H. CastroNeto, Phys. Rev. Lett., 2007, 99, 256802 CrossRef CAS.
- Y. Zhang, C. H. Hu, Y. H. Wen, S. Q. Wu and Z. Z. Zhu, New J. Phys., 2011, 13, 063047 CrossRef.
- W. A. Dino, Y. Miura, H. Nakanishi, H. Kasai and T. Sugimoto, J. Phys. Soc. Jpn., 2003, 72, 1867 CrossRef CAS.
- C. P. Herrero and R. Ramirez, J. Phys. D: Appl. Phys., 2010, 43, 255402 CrossRef.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef.
- N. Marom, A. Tkatchenko, M. Scheffler and L. Kronik, J. Chem. Theory Comput., 2010, 6, 81 CrossRef CAS.
- J. P. Perdew and A. Zunger, Phys. Rev. B, 1981, 23, 5048 CrossRef CAS.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566 CrossRef CAS.
- V. Baskin and L. Meyer, Phys. Rev., 1955, 100, 544 CrossRef.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter, 1990, 41, 7892 CrossRef.
- A. D. Vita, Ph.D. thesis, Keele University, 1992.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- N. Govind, M. Petersen, G. Fitzgerald, D. King-Smith and J. Andzelm, Comput. Mater. Sci., 2003, 28, 250 CrossRef CAS.
- Z. Luo, T. Yu, Z. Ni, S. Lim, H. Hu, J. Shang, L. Liu, Z. Shen and J. Lin, J. Phys. Chem. C, 2011, 115, 1422 CAS.
- A. Nikitin, L. A. Naslund, Z. Zhang and A. Nilsson, Surf. Sci., 2008, 602, 2575 CrossRef CAS PubMed.
- T. Roman, W. A. Dino, H. Nakanishi, H. K. T. Sugimoto and K. Tange, Carbon, 2007, 45, 218 CrossRef CAS PubMed.
- M. Yang, A. Nurbawono, C. Zhang, R. Wu, Y. Feng and Ariando, AIP Adv., 2011, 1, 032109 CrossRef PubMed.
- S. Casolo, O. M. Lovvik, R. Martinazzo and G. F. Tantardini, J. Chem. Phys., 2009, 130, 054704 CrossRef PubMed.
- P. O. Lehtinen, A. S. Foster, Y. Ma, A. V. Krasheninnikov and R. M. Nieminen, Phys. Rev. Lett., 2004, 93, 187202 CrossRef CAS.
- P. W. Atkins, Physical Chemistry, Oxford University Press, 4th edn, 1990 Search PubMed.
- M. Endo, K. Oshida, K. Kobori, K. Takeuchi, K. Takahashi and M. S. Dresselhaus, J. Mater. Res., 1995, 10, 1461 CrossRef CAS.
- J. K. Lee, S. C. Lee, J. P. Ahn, S. C. Kim, J. I. B. Wilson and P. John, J. Chem. Phys., 2008, 129, 234709 CrossRef PubMed.
- G. H. Vineyard, J. Phys. Chem. Solids, 1957, 3, 121 CrossRef CAS.
- B. Montanari and N. M. Harrison, Chem. Phys. Lett., 2002, 364, 528 CrossRef CAS.
- C.-S. Tsao, Y. Liu, H.-Y. Chuang, H.-H. Tseng, T.-Y. Chen, C.-H. Chen, M.-S. Yu, Q. Li, A. Leuking and S.-H. Chen, J. Phys. Chem. Lett., 2011, 2, 2322 CrossRef CAS.
- G. Majer, E. Stanik and S. Orimo, J. Alloys Compd., 2003, 356–357, 617 CrossRef CAS.
- G. Majer, E. Stanik and S. Orimo, J. Alloys Compd., 2004, 377, 316 CrossRef CAS PubMed.
- E. Stanik, G. Majer, S. Orimo, T. Ichikawa and H. Fujii, J. Appl. Phys., 2005, 98, 044302 CrossRef PubMed.
- J. Ye and P. C. Chiu, Environ. Sci. Technol., 2006, 40, 3959 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: the structures of hydrogenated graphenes and transition states. See DOI: 10.1039/c3ra46626d |
| This journal is © The Royal Society of Chemistry 2014 |