DOI:
10.1039/C3RA47464J
(Paper)
RSC Adv., 2014,
4, 9213-9222
Synthesis of ordered mesoporous silica/ceria–silica composites and their high catalytic performance for solvent-free oxidation of benzyl alcohol at room temperature†
Received
10th December 2013
, Accepted 20th January 2014
First published on 22nd January 2014
Abstract
Highly ordered mesoporous Ce-incorporated MCM-41 and MCM-48 silica materials with Ce/Si ratio = 0.1–0.3 have been prepared in a surfactant-assisted hydrothermal method using hexadecyltrimethylammonium bromide (CTAB) as a structure-directing agent (SDA), cerium nitrate hexahydrate and tetraethyl orthosilicate (TEOS) as inorganic co-precursors under basic conditions. Thermal treatment at 823 K has allowed the removal of the organic template, resulting in the formation of long-range ordered 2D hexagonal (p6mm) MCM-41 and cubic (Ia3d) MCM-48, respectively, with ceria-rich particles. Highly ordered mesostructures for Ce-MCM-41 and Ce-MCM-48 materials were characterized with small angle X-ray scattering (SAXS) and wide-angle X-ray diffraction (XRD), nitrogen adsorption–desorption isotherms, UV-visible diffuse reflectance spectroscopy (UV-DRS), transmission electron microscopy (TEM). The elemental compositions were analyzed using elemental mapping from EDS, inductively coupled plasma atomic emission spectra (ICP-AES), X-ray photoelectron spectroscopy (XPS), and solid-state magic angle spinning (MAS) NMR. These high cerium containing mesoporous ceria–silica materials of ∼30 wt% showed an impressive performance in solvent-free highly selective liquid phase oxidation of benzyl alcohol to benzaldehyde at room temperature. This controlled oxidation of primary alcohol to aldehyde over highly stable, completely heterogeneous, non-air sensitive, and reusable Ce-incorporated silica composite can be an excellent gateway for industrial fine chemical synthesis.
1 Introduction
The hetero-atomic metal oxides with different oxidation state have been developed to explore innovative properties and applications as catalytic materials, especially, supported on another stable framework structure like zeolites,1 mesoporous silica,2 alumina,3 and titania4 and so on. Incorporation of metal atom onto the readily accessible framework of ordered mesoporous molecular sieves such as MCM-41 (hexagonal), MCM-48 (cubic), and SBA-15 (hexagonal), replacing a large amount of Si atom inside –O–Si–O– networks is one of main research topics for materials chemists.2,5,6 Two or three dimensional mesoporous silicas such as M41S family prepared through self-assembly of organic–inorganic precursors possess a variety of unique properties like high thermal stability, huge surface area, ordered pores, large pore size, and pore volume and so on.7,8 However, the aforementioned materials have limited applications due to structural inertness and absence of any acidic or redox species. Thus, the incorporation of heteroatoms within highly ordered silicate framework enhances the structural diversity of later leading to its versatile role in numerous advanced research fields.9
Heteroatom cerium (Ce), an early member of lanthanide series is a crucial element to be incorporated in silica matrices, because Ce-based materials have achieved enormous attention owing to their widespread promising applications in many industrial and economic grounds.10 Supported ceria and CeO2 based mixed oxides have imperative role in redox reactions,11 fluid catalytic cracking process (FCC),12 three-way catalysis (TWC),13 chemical mechanical planarization process (CMP),14 high temperature ceramics,15 solid oxide fuel cells (SOFC),16 CO2 activation,17 CO/NO removal,18 water purification,19 photocatalysis,20 and antibacterial activity21 and so on. However, a significant limitation encountered with pure CeO2 materials is low thermal stability and high cost for which they are rarely used as alone.22 Thus, catalytic amount of Ce when incorporated into the silicate building blocks of hexagonal MCM-41 or cubic MCM-48, leads to significant improvement of potential applications of supporting and catalytic material depending upon the structural stability supported metal oxides as well as may be cost effective.23 There are several examples of such Ce doping within mesoporous silica ordered framework via different methods.24 Dai et al. have prepared Ce-incorporated SBA-15 by a two-step direct synthesis method in acid media. However, the metal incorporation reported was up to 10%.25 Very recently, Dunne et al. reported a new ceria–silica composite material with high Ce content but the surface area of the material was quite low (i.e. ∼299 m2 g−1).26 Grafting method has also been employed to incorporate Ce within silica mesostructure, but about 18 wt% of Ce loaded in the material has been reported.23 Both the direct hydrothermal method and impregnation procedure fail to report a good amount of Ce doping into MCM-41 or SBA-15.27,28 Similarly, Ce-MCM-41 synthesized in aqueous hydrothermal method or Ce-MCM-48 obtained in presence of additional metal salt cannot fulfill the condition of high Ce loading within ordered silica structure.29 Hence, incorporation of an appreciable high amount (above 20 wt%) of Ce in silica along with the retention of ordered arrangement of support material is no doubt a challenging mission. Our recent publication reported the mesoporous silica/ceria–silica composites with high cerium content using a cerium hydroxide precursor.30 However, there was no further experimental example for any application. Also, we have tried to find further synthesis route for the Ce–O–Si mesopore wall with higher content of Ce chemical bond using a different well-soluble Ce precursor in aqueous media.
Here, we report a facile synthesis of highly ordered, stable mesoporous silica/ceria–silica composites using another cerium nitrate precursor well soluble in ammonium basic media. The mesoporous product also shows quite high Ce content up to 30 wt% of Ce/Si along with high specific surface area (∼376 m2 g−1). Not only that, our Ce-rich silica has been proved to be a highly efficient, heterogeneous, and recyclable catalyst for solvent-free oxidation of benzyl alcohol to benzaldehyde at room temperature in presence of tert-butyl hydroperoxide (TBHP) oxidant. Though there are several scientific reports on solvent-free oxidation of benzyl alcohol,31–33 but highly selective conversion of benzaldehyde at room temperature is rarely published. Other investigational approaches describing complete transformation of primary alcohol to aldehyde at normal temperature could not avoid the mediation of toxic solvent in reaction media.34,35 Selective methods employing liquid phase oxidation of benzyl alcohol over heterogeneous catalyst to produce chlorine-free benzaldehyde without formation of oxidation products like acid or ester is an important reaction in organic synthesis36 as well as in industrial catalytic applications.37 Hence, ceria–silica materials with good stability and reusability can function as a true alternative to more expensive Au, Ru, Pt or Pd catalysts31,32,38,39 for selective primary alcohol to aldehyde oxidation without using any additional solvent and high temperature, which deserves the priority to be environmental benign green process in industrial fine chemical production.
2 Experimental
2.1 Materials
Hexadecyltrimethylammonium bromide (CTAB, 98%) as a structure-directing agent, tetraethyl orthosilicate (TEOS, 98%) as a silica precursor, cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O) as a cerium source, ammonium hydroxide (NH4OH, 28.0–30.0% NH3) as a base and ethanol (EtOH) were purchased from Aldrich. Deionized water prepared in our laboratory was used as one of the solvents for all the samples preparation throughout our experiments. Benzyl alcohol and tert-butyl hydroperoxide (TBHP, 5.0–6.0 M in decane) were purchased from Aldrich for catalytic reactions. All the chemicals were used as received without further purification.
2.2 Synthesis of ceria-MCM-41 type mesoporous silica
In a typical synthesis of p6mm hexagonal framework ceria–silica material (HSCS-10), 1.5 g (4.1 mmol) of CTAB was dissolved in 30 mL of deionized water and 30 mL of EtOH in a glass bottle under vigorous stirring at room temperature with magnetic bar. This was followed by addition of 35 mL (0.5 mol) of NH4OH solution. After about 30 min, 3 mL (13.4 mmol) of TEOS was added to the surfactant solution and the mixture was stirred for another 30 min when heavy white precipitation formed gradually. Next, 0.58 g (1.34 mmol) of Ce(NO3)3·6H2O was dissolved in 15 mL of EtOH in another bottle and slowly added to the previous mixture. The final mixture was kept under stirring for another 24 h at room temperature, followed by ageing for 24 h at 373 K temperature in a convection oven. After that the mixture was cooled to room temperature, filtered and washed several times with deionized water and ethanol. The material was dried to get solid powder which was calcined at 823 K for 5 h under the constant flow of air. An analogous route was used for the synthesis of other MCM-41 type samples listed in Table 1, except the difference in amount of cerium salt. The final calcined samples with Ce/Si ratio 0.1 and 0.2 were named as HSCS-10 and HSCS-20, respectively.
Table 1 Physico-chemical data of various mesoporous silica/ceria–silica materialsa
| Sample |
fCe/Si |
Mesostructure |
SBET (m2 g−1) |
Vp (cm3 g−1) |
Dp (nm) |
d-spacing (nm) |
a (nm) |
w (nm) |
| Notation: fCe/Si = molar ratio of cerium nitrate to TEOS used in the synthesis gel; SBET = BET specific surface area determined in the range of relative pressures from 0.04 to 0.2; Vp = single-point pore volume at P/P0 = 0.99; Dp = mesoporous diameter at the maximum of the PSD curve obtained using BJH method; d-spacing = Bragg's spacing (= 2π/q*, q* is q value at maximum (100) peak for hexagonal p6mm and (211) peak for cubic Ia3d mesoporous structure); a = unit cell parameter (= 2d100/√3 for p6mm and √6d211 for Ia3d mesostructure), w = pore wall thickness (= a − Dp for p6mm and a/3.0919 − Dp/2 for Ia3d structure). |
| HSCS-10 |
0.10 |
p6mm |
499 |
0.92 |
2.15 |
3.95 |
4.56 |
2.41 |
| HSCS-20 |
0.20 |
p6mm |
349 |
0.47 |
2.10 |
3.77 |
4.35 |
2.25 |
| CSCS-10 |
0.10 |
Ia3d |
575 |
0.80 |
2.08 |
3.66 |
8.96 |
1.86 |
| CSCS-20 |
0.20 |
Ia3d |
376 |
0.70 |
2.00 |
3.63 |
8.89 |
1.87 |
2.3 Synthesis of ceria-MCM-48 type mesoporous silica
In a typical synthesis of Ia3d cubic framework ceria–silica material (CSCS-10), 1.2 g (3.3 mmol) of CTAB was dissolved in 50 mL of deionized water and 15 mL of EtOH in a glass bottle under vigorous stirring, followed by addition of 6 mL (0.09 mol) of NH4OH solution. After about 30 min, 1.8 mL (8.06 mmol) of TEOS was added to the surfactant solution and the mixture was stirred for another 30 min when heavy white precipitation formed gradually. Next, 0.35 g (0.81 mmol) of Ce(NO3)3·6H2O was dissolved in 10 mL of EtOH in another bottle and slowly added to the previous mixture. The final mixture was kept under stirring for another 24 h at room temperature, followed by ageing for 24 h at 373 K temperature in a convection oven. After that the mixture was cooled to room temperature, filtered and washed several times with deionized water, ethanol. The material was dried to get solid powder which was calcined at 823 K for 5 h under the constant flow of air. An analogous route was used for the synthesis of other MCM-41 type samples listed in Table 1, except the difference in amount of TEOS and cerium salt. The samples with Ce/Si ratio 0.1 and 0.2 were named as CSCS-10 and CSCS-20, respectively.
2.4 Characterizations
The small angle X-ray scattering (SAXS) data of the materials were recorded on a synchrotron (E = 10.5199 keV, λ = 1.1785 Å) of a 4C beam line in Pohang Accelerator Laboratory (PAL). Each sample was placed in a copper-alloy sample holder and secured on both sides using a Kapton tape. Wide angle X-ray diffraction (XRD) were performed in a PANanalytical Empyrean multipurpose diffractometer with Cu-Kα radiation (λavg = 1.5418 Å) at 40 kV and 30 mA in Korea Basic Science Institute (KBSI) Daegu center. The samples were prepared by grinding then putting on the microscope holder at room temperature. The spectra were recorded versus 2θ from 10 to 90 degrees with a scan step of 0.039391. SAXS experiments were also verified in Empyrean diffractometer.
Nitrogen sorption isotherms were obtained using a Micromeritics 2420 analyzer at 77 K. Prior to the measurement, all the samples were degassed at 393 K under vacuum below 30 μm Hg for at least 2 h. BET (Brunauer–Emmet–Teller) specific surface area were evaluated from the sorption data in P/P0 relative pressure range from 0.04 to 0.2. The total pore volume was calculated based on the adsorption amount at P/P0 of 0.99. PSD (pore size distribution) was obtained from the adsorption branches of the isotherms using a BJH method. The pore wall thickness (w) was estimated from pore size (Dp) obtained at the PSD maximum and the unit cell parameter (a) measured by SAXS (i.e. w = a − Dp for p6mm hexagonal and a/ξ0 − Dp/2 with ξ0 = 3.0919 for Ia3d cubic structure, respectively).
TEM images of the samples were recorded by a FEI TECNAI G2 F30 ST electron microscope operated at an accelerating voltage of 200 kV. Prior to the analysis, the samples were prepared by proper sonication (for 45 min) dispersing in ethanol and the solutions were dropped onto a porous carbon film on a copper grid then dried in air. Elemental mapping images for O, Si and Ce were recorded using a field emission SEM (JEOL JSM-4300F) equipped with embedded EDS system operated at an accelerating voltage of 15 kV. Electron microscopy was measured at the KBSI Seoul center.
UV-visible diffuse reflectance spectra (DRS) were obtained by using a Harrick Scientific diffuse reflectance attachment (DRP) with a Varian Cary 300 spectrophoto-meter at the KBSI Busan center. Samples were in dry state and the spectra were recorded at room temperature with 1–5 eV incident light and using MgO as background standard. Cerium weight percentage in the sample was determined by JobinYvon inductively coupled plasma (ICP) (ICP-OES, JY Ultima2C) analysis at the KBSI Seoul center adjusting the wavelength to 395.254 nm (for Ce atom).
X-ray photoelectron spectroscopic measurements were performed on a Theta Probe AR-XPS System (Thermo Fischer Scientific) by using monochromatic Al-Kα (1486.6 eV) radiation at the excitation source in KBSI Busan center. The charging of the samples was corrected by setting the binding energy of the adventitious carbon (C 1s) at 284.5 eV. The analysis was done at ambient temperature as well as pressure of typically less than 10−6 Pa and before recording the data all the samples were degassed in vacuum for overnight.
Solid state 29Si CP MAS NMR spectra were recorded with a Bruker ADVANCE II+ (400 MHz) spectrometer using a 4 mm magic angle (MAS) spinning probe at the KBSI Daegu center. The samples were spun at a spinning rate of 6 kHz, delay time of 3 s, contact time of 2 ms and Lamor frequency of 79.488 MHz. The chemical shift values were obtained with respect to the tetraethyl silane (TMS) reference peak.
2.5 Reduction of mesoporous silica/ceria–silica composite
The temperature-programmed reduction (TPR) of the samples were carried out from room temperature to 923 and 1123 K, respectively, with step-wise heating rate of average 5 °C min−1 followed by constant heating for 5 h at 923 and 1123 K, respectively. The TPR process was carried out in a flow of 7 vol% of hydrogen diluted in nitrogen with a total flow rate of gas mixture of 205 mL min−1.
2.6 Catalytic oxidation of benzyl alcohol
Liquid-phase oxidation of benzyl alcohol over various Ce catalysts was conducted in a magnetically stirred 25 mL capacity round bottom flask closed with stopper. In a typical oxidation reaction, 0.05 g of catalyst was added to a suspension containing 0.11 g of benzyl alcohol (1 mmol) and 1 g of TBHP (5–6 M in decane). The mixture was stirred at room temperature (298 K) for 24 h at a fixed stirring rate of 650 rpm. The reaction was also carried out at 343 K temperature in a 25 mL two necked round bottomed flask fitted with water condenser and placed in a magnetically stirred heating mantle. Aliquot of the reaction mixtures were withdrawn periodically for analysis. After the reaction, the catalyst was filtered and the reaction products with unconverted reactants were analyzed as well as identified using a 6890N GC/5975i MS (Agilent, USA) gas chromatography-mass spectrometry (GC/MS) system equipped with HP-5 capillary column. 1 μL sample was injected by a syringe with split ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to the injector at temperature 553 K and He gas was used as carrier gas at a flow rate of 1 mL min−1. For quantitative determination of the reactants and products the area% value of individual peak obtained from system software was used. Ion source of mass analysis was EI with analysis range of 50–800 m/z.
:1 to the injector at temperature 553 K and He gas was used as carrier gas at a flow rate of 1 mL min−1. For quantitative determination of the reactants and products the area% value of individual peak obtained from system software was used. Ion source of mass analysis was EI with analysis range of 50–800 m/z.
Percentage conversion and product selectivity were calculated as follows: conversion or total conversion (%) = [(peak area of all the products/summation of peak area of substrate and products) × 100], Product selectivity (%) = [(peak area of that particular product/summation of peak area of all the products) × 100].
3 Results and discussion
3.1 X-Ray analysis
2D hexagonal (p6mm) and 3D bicontinuous cubic (Ia3d) mesostructures of surfactant-free ceria–silica samples were determined using synchrotron small angle X-ray scattering (SAXS) as shown in Fig. 1. Each of the SAXS patterns of Fig. 1a and b representing ceria-MCM-41 type (HSCS-10 and HSCS-20) materials exhibits three peaks. One highly intense peak for (100) plane and two additional low intense peaks representing (110) and (200) planes are the characteristic of long range ordered 2D hexagonal (p6mm) mesostructure. Bragg (d100) spacing calculated from the respective q values (see Table 1) at the maximum peak (100) are 3.95 and 3.77 nm. Unit cell parameter (a) calculated from individual d-spacing is shown in Table 1. Fig. 1c and d shows SAXS patterns of Ce-MCM-48 (CSCS-10 and CSCS-20) materials. A high intense peak corresponding to (211) plane with small tail and other small peaks indicate the development of bicontinuous cubic mesostructure with Ia3d space group. Bragg spacing values of the maximum peaks are 3.66 and 3.63 nm for 10% and 20% cerium doped MCM-48, respectively.
 |
| | Fig. 1 Small angle X-ray scattering (SAXS) data of all the samples. SAXS data of hexagonal ceria–silica HSCS-10 (a) and HSCS-20 (b). SAXS data of cubic ceria–silica CSCS-10 (c) and CSCS-20 (d). | |
The unit cell parameter (a) calculated from the respective d-spacings and the wall thicknesses (w) of MCM-48 type samples are listed in Table 1. The conservation of ordered structure after high amount of Ce loading indicates the materials are highly stable in nature to act as a good host for adsorbing gas molecules. This type of huge cerium incorporation with retention of mesophase structure was also observed in case of ceria–silica synthesis with Ce(OH)4 precursor.30 Samples with Ce/Si ratio of 0.3 in the synthesis gel have also been prepared and tested by SAXS analysis. However, those do not retain ordered structure after calcination, so we do not discuss further about Ce samples containing Ce/Si higher than 0.2.
Wide angle powder X-ray diffraction (XRD) patterns of two calcined ceria–silica samples (i.e. HSCS-20 and CSCS-20 listed in Table 1) are shown in Fig. 2. Peaks observed at 2θ = 28.6, 32.8, 47.5, 56.3, and 76.7 degrees in both the samples (Fig. 2a and b) are attributed to the (111), (200), (220), (311) and (331) reflection planes of typical face-centered cubic CeO2 (JCPDS no. 034-0394).23,40 These diffraction peaks clearly indicate that Ce(III) ion incorporated in the silica framework has been oxidized to more stable Ce(IV) species during sol–gel synthesis and high temperature calcination process in presence of air.
 |
| | Fig. 2 Wide angle X-ray diffraction patterns of two mesoporous samples, HSCS-20 and CSCS-20. Vertical lines indicate the indexed peaks of CeO2 from the corresponding JCPDS data. | |
3.2 Surface area and pore size analysis
Textural properties like BET specific surface area, pore volume, and average pore diameter of these materials have been evaluated from N2 adsorption–desorption isotherms measurements. Fig. 3a displays the loops of adsorption/desorption isotherms of our all samples. All these physico-chemical data of Ce-MCM samples are summarized in Table 1. The calcined ceria–silica samples show typical type IV isotherms and an uptake of adsorbed volume observed in the adsorption branches at the relative pressure range P/P0 = 0.01–0.45, due to capillary condensation of nitrogen into mesopores, which is a characteristic of ordered mesoporosity present in the material.41 MCM-48 type samples exhibit higher volume uptake at this pressure range than the respective MCM-41 type which is reflected in the BET surface area values (Table 1). The hysteresis in the relative pressure range P/P0 = 0.5–0.9 is attributed to the presence of uniform mesopores and the sharp increase of the isotherm from P/P0 = 0.9–1.0 implies there is also some inter-particle porosity in the material.42
 |
| | Fig. 3 (a) Nitrogen adsorption (A)/desorption (D) isotherms of HSCS-20 ( ), CSCS-20 ( ), CSCS-20 ( ), HSCS-10 ( ), HSCS-10 ( ), CSCS-10 ( ), CSCS-10 ( ) measured at 77 K. (b) Respective pore size distribution (PSD) curves of all the samples, maximum representing individual mesopore diameter. ) measured at 77 K. (b) Respective pore size distribution (PSD) curves of all the samples, maximum representing individual mesopore diameter. | |
The corresponding pore size distribution (PSD) curves of Ce-MCM samples obtained by using BJH method are shown in Fig. 3b. Narrow PSD patterns are an indication of uniform mesoporous character of the material. The average pore diameters of the Ce samples observed corresponding to the maxima in PSD curves are also in the range of a typical mesopores (2–50 nm) nature. Respective pore diameters and pore volumes are given in Table 1. The decrease of specific surface area and pore diameter of the ceria–silica samples (Table 1) with increasing cerium content can be attributed to the relative ordering of the mesophase observed in SAXS patterns of the corresponding species.6
3.3 TEM images
Transmission electron microscopic images of Ce-MCM-41 and Ce-MCM-48 (Ce/Si = 0.2 in the synthesis gel) have been recorded to investigate the structural features at the atomic level as shown in Fig. 4a–d. In Fig. 4a, the spherical morphology of HSCS-20 particles is shown in μm scale and the average diameter of particles observed is 0.3–0.6 μm in size (also see ESI, Fig. S1†). Fig. 4b exhibits typical MCM-41 structure of HSCS-20 specimen consisting of a well-ordered hexagonal arrangement. The pore diameter estimated is about 2.0–2.5 nm which matches well with the average pore diameter obtained from PSD curve of N2 sorption analysis (Table 1). A closer inspection of this sample by HRTEM mainly reveals the existence of dark crystalline spots (Fig. 4c) due to ceria grains and the crystalline planes of CeO2 fluorite structure. Well dispersion of CeO2 crystallites in silica matrices is observed throughout the silica specimen as shown in Fig. 4c. The TEM image of the other Ce sample CSCS-20 presented in Fig. 4d demonstrates cubic mesostructure of the material with average pore diameter calculated in the range of 2.0–2.5 nm. This diameter is in reasonable agreement with Dp value obtained from PSD plot of the corresponding material.
 |
| | Fig. 4 TEM images of the morphology (a) hexagonal pattern of the sample (b) the lattice fringes corresponding to CeO2 incorporated in HSCS-20 (c) and cubic structure of CSCS-20 (d). | |
3.4 ICP-AES and elemental mapping
Cerium percentage in the material obtained from ICP-AES analysis is listed in Table 2. Sample with higher Ce content is our aim to synthesize for many potential applications and thus the data for HSCS-20 and CSCS-20 samples have been recorded. From the cerium wt% the content of remaining SiO4 can be easily evaluated and finally Ce/Si ratios are calculated as 0.26 and 0.29 for HSCS-20 and CSCS-20, respectively. These molar ratios of Ce/Si in the product are quite well matched with that used in synthesis gel.
Table 2 ICP quantification of Ce/Si molar ratio in the final mesoporous ceria–silica materials
| Sample |
Ce (ppm) |
Ce (wt%) |
Ce (mmol g−1) |
SiO4 (wt%) |
Si (mmol g−1) |
Ce/Si |
| HSCS-20 |
281400 |
28.140 |
2.008 |
71.860 |
7.804 |
0.26 |
| CSCS-20 |
308000 |
30.800 |
2.198 |
69.200 |
7.514 |
0.29 |
EDS elemental mapping for O, Si and Ce atoms for HSCS-20 and CSCS-20 samples are shown in Fig. 5a–f, respectively. All the images strongly indicate the presence of large amount of corresponding elements well distributed throughout the specimens.
 |
| | Fig. 5 EDS elemental mapping of O, Si, Ce of HSCS-20 (a–c) and O, Si, Ce of CSCS-20 (d–f), respectively. | |
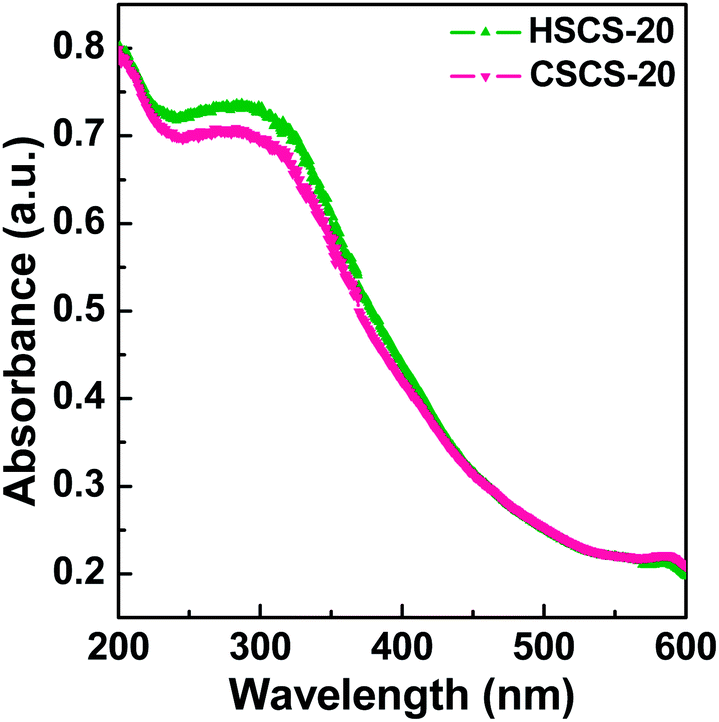
3.5 UV-visible spectral study
Diffuse reflectance UV-visible spectra is a very sensitive evidence to confirm the coordination environment and oxidation state of the transition metal ion embedded in the silica framework. UV-visible spectra of the Ce samples are shown in Fig. 6. Both the materials show a single broad maximum near ca. 300 nm which can be attributed to the charge transfer transition (ligand → metal) from O2− to Ce4+ species and this absorption band indicates one type of co-ordination environment of Ce(IV) ion dispersed uniformly throughout the specimen.43 Tetra-coordinated environment is most likely expected in both of the Ce samples as the lower energy electronic transition band (∼400 nm) responsible for hexa-coordinated Ce(IV) species is absent here.44
 |
| | Fig. 6 UV-visible diffuse reflectance spectra (DRS) of HSCS-20 and CSCS-20 samples recorded at room temperature. | |
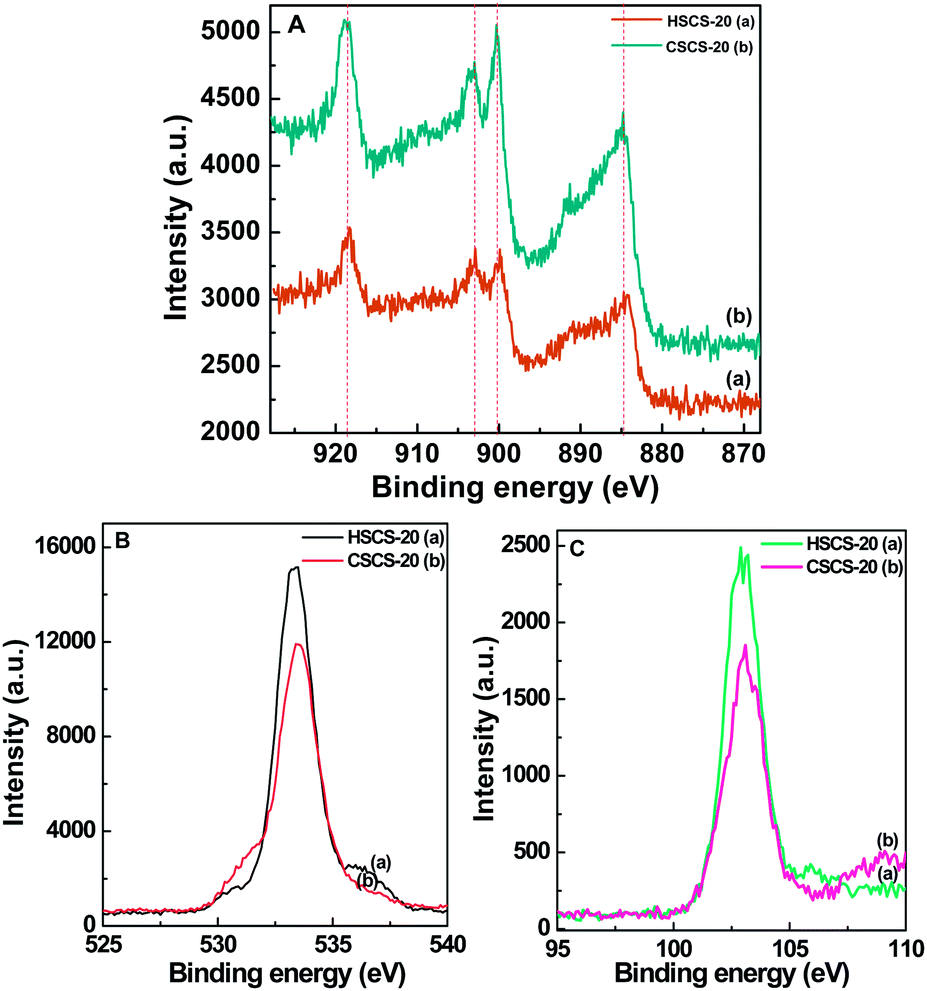
3.6 XPS analysis
In order to get further information about the chemical nature of Ce-incorporation in the mesoporous silica matrix and the interaction between supported CeO2 with silica framework, the samples are investigated by XPS technique. Fig. 7A–C show Ce 3d, O 1s, and Si 2p electron core level X-ray photoelectron spectra of calcined HSCS-20 and CSCS-20 samples, respectively, and the values of corresponding peaks are presented in Table 3. Ce 3d spectra of both the samples (Fig. 7A) consist of four peaks from binding energy range 918 to 884 eV. Binding energy in the range of 884–891 eV is associated with Ce 3d5/2 state,45 the two peaks in the range of 899–903 eV arise due to mixing of Ce 3d5/2 and Ce 3d3/2 spin-orbit coupling state and the last peak at 918 eV due to Ce 3d3/2 is exclusive for Ce(IV) state.46 Fig. 7B displays XPS spectra of O 1s electron from HSCS-20 and CSCS-20 samples and only one peak near ca. 533 eV can be attributed to the oxygen atom present in the form of SiO2. In addition to that a small hump near 531 eV indicates that few oxygen atoms in form of –O3SiOH are also available in the ceria–silica matrices.47 Si 2p XPS spectra of Ce samples in Fig. 7C show only one peak near BE value ca. 103 eV which corresponds to the presence of SiO2 species in the material.
 |
| | Fig. 7 XPS data showing A: Ce 3d, B: O 1s and C: Si 2p core level spectra recorded from (a) HSCS-20 and (b) CSCS-20 samples with respect to C 1s peak at 284.5 eV. Vertical lines indicate the corresponding peaks. | |
Table 3 XPS core level binding energies of HSCS-20 and CSCS-20 samples
| Sample name |
Binding energy (eV) |
| Ce 3d |
O 1s |
Si 2p |
| HSCS-20 |
918.56 |
533.34 |
103.03 |
| 903.25 |
| 900.14 |
| 884.81 |
| CSCS-20 |
918.28 |
533.47 |
103.07 |
| 902.75 |
| 899.84 |
| 884.22 |
3.7 Temperature-programmed reduction
Fig. 8 shows the wide angle X-ray diffraction patterns for the HSCS-10 (a) and HSCS-20 (b) samples heated at 923 K under flowing diluted H2. The two diffraction patterns show a suppressed cubic cerium dioxide crystalline phase similar to Fig. 2. However, the X-ray patterns for the samples heated up to 1123 K under flowing diluted H2 clearly show a mixed pattern with a new hexagonal structure Ce4.667Si3O13 (or Ce9.33(SiO4)6O2) (JCPDS no. 043-0441). The new crystalline structure is known to be an apatite-like structure with a hexagonal unit cell (a = 9.657 Å and c = 7.121 Å)30 which shows the oxidation state is changed above 1123 K from Ce4+ to Ce3+ inside mesoporous silica/ceria–silica composites.
 |
| | Fig. 8 Wide angle X-ray diffraction patterns of mesoporous silica/ceria–silica composites treated at 923 and 1123 K under flowing diluted hydrogen. Panel (a) is for HSCS-10 and panel (b) is for HSCS-20 sample treated at 923 K, respectively. Panel (c and d) are for HSCS-10 and HSCS-20 sample treated at 1123 K. | |
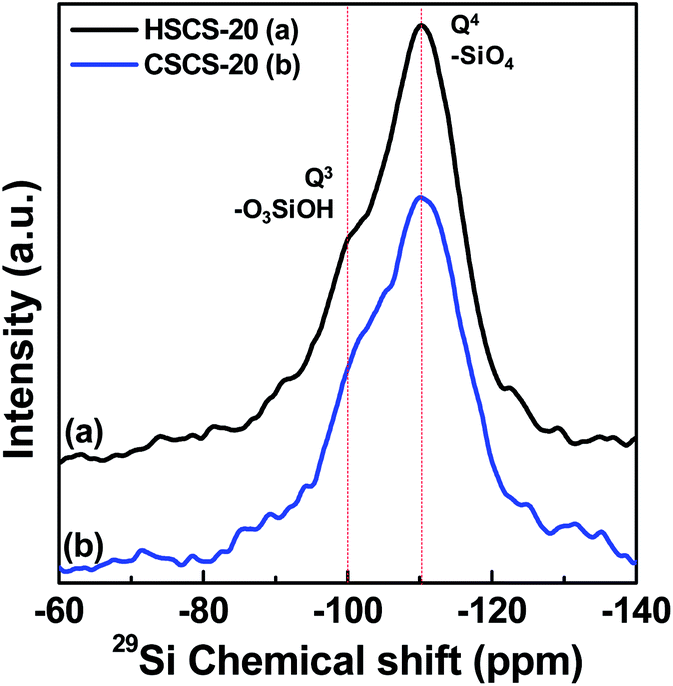
3.8 29Si MAS NMR study
Solid state 29Si CP MAS NMR spectra of two Ce samples are shown in Fig. 9 which provides significant information about the chemical environment of Si atom in ceria–silica materials. The figure shows one broad signal for Q4 (–SiO4) and another small hump for Q3 (–O3SiOH) with the chemical shift value at −110 and −100 ppm, respectively.27 The high intensity of Q4 signal indicates that large amount of Si atoms were fully condensed with the other Si or Ce atoms connected with oxygen.
 |
| | Fig. 9 29Si CP MAS NMR spectra of (a) HSCS-20 and (b) CSCS-20 samples showing Q4 and Q3 peaks by vertical lines. | |
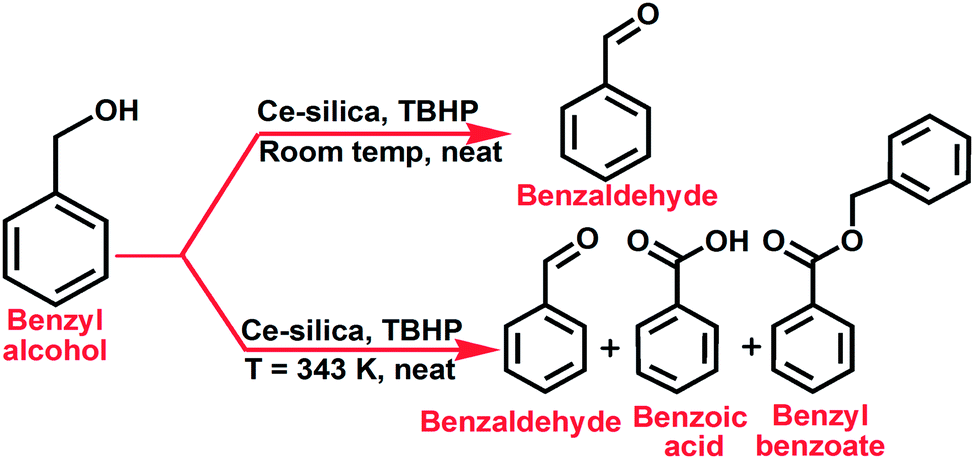
3.9 Catalytic performances
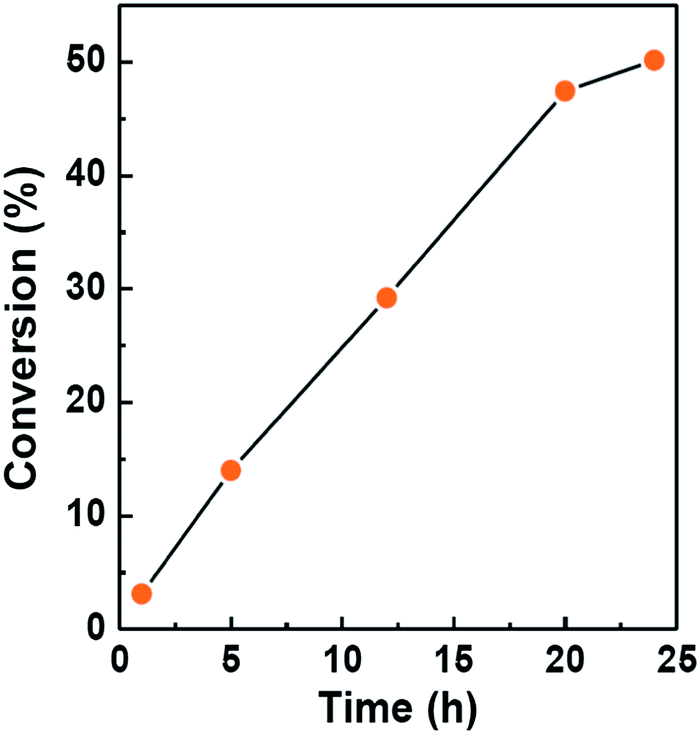
The catalytic activity of mesoporous Ce-silica solids have been examined for the oxidation of benzyl alcohol in presence of tert-butyl hydroperoxide (TBHP) oxidant under suitable reaction conditions (Scheme 1). Results of solvent free oxidation of benzyl alcohol at room temperature are presented in Table 4. Both the hexagonal (Entry 1) and cubic (Entry 2) samples appeared active in selective oxidation of primary alcohol to yield benzaldehyde as the only product, though cubic structured ceria–silica shows a little higher conversion than hexagonal one. Fig. 10 shows the conversion profile of benzyl alcohol with respect to reaction time over HSCS-20 catalyst. Additional solvent was not used in these reactions and 5.0–6.0 M solution of TBHP in decane form a complete homogeneous (organic) media with the reactant alcohol as well as product aldehyde to precede the overall reaction in forward direction. A blank reaction (Entry 3) has been carried out to understand the role of catalyst in this oxidation reaction. Strong oxidizing ability of Ce species present in catalyst support11 and its imperative role in catalytic oxidation27 is also confirmed from the low yield (i.e. ∼2%) of catalytic performance (Entry 4) using pure MCM-41.
 |
| | Scheme 1 Schematic for the oxidation of benzyl alcohol over ceria–silica catalyst. | |
Table 4 Result of catalytic solvent-free oxidation of benzyl alcohol over different catalystsa
| Entry |
Catalyst |
Conversion of benzyl alcohol (%) |
Selectivity (%) |
| Benz-aldehyde |
Benzoic acid |
Benzyl benzoate |
| Reaction conditions: benzyl alcohol = 1.0 mmol, TBHP = 1.0 g, catalyst = 0.05 g, T = 298 K, time = 24 h. Blank reaction carried out without catalyst. Reaction carried out with pure MCM-41. |
| 1 |
HSCS-20 |
50.17 |
100 |
— |
— |
| 2 |
CSCS-20 |
53.47 |
100 |
— |
— |
| 3b |
— |
0.67 |
— |
— |
— |
| 4c |
MCM-41 |
2.05 |
100 |
— |
— |
 |
| | Fig. 10 Catalytic activity test of HSCS-20 in solvent-free oxidation of benzyl alcohol at room temperature. | |
Activity of the catalysts has also been tested at elevated temperature (T = 343 K) and it is observed that the influence of temperature (Table 5) have accelerated the reaction rate and improved the alcohol conversion to almost 100%, but the selectively of product aldehyde is greatly reduced by the formation of benzoic acid and benzyl benzoate ester as over oxidation products of benzaldehyde. In fact, at higher temperature benzoic acid is produced as the major product with selectivity 85.02% and 84.21% for HSCS-20 and CSCS-20, respectively. Effect of Ce loading is clearly evident from the result of the reaction performed in presence of HSCS-10 (Ce/Si = 0.1) catalyst which is shown in Table 6. When Ce loading decreased from 28% to 16%, the conversion of benzyl alcohol also decreased from 50% to 23% under identical conditions.
Table 5 Effect of reaction temperature on oxidation of benzyl alcohola
| Entry |
Catalyst |
Temp. (K) |
Conversion of benzyl alcohol (%) |
Selectivity (%) |
| Benz-aldehyde |
Benzoic acid |
Benzyl benzoate |
| Reaction conditions: benzyl alcohol = 1.0 mmol, TBHP = 1.0 g, catalyst = 0.05 g, time = 24 h. |
| 1 |
HSCS-20 |
298 |
50.17 |
100 |
— |
— |
| 2 |
HSCS-20 |
343 |
100 |
12.69 |
85.02 |
1.38 |
| 3 |
CSCS-20 |
343 |
100 |
13.77 |
84.21 |
1.52 |
Table 6 Effect of Ce loading on oxidation of benzyl alcohola
| Entry |
Catalyst |
Ce loading in silica (wt%) |
Conversion of benzyl alcohol (%) |
Selectivity (%) |
| Benz-aldehyde |
Benzoic acid |
Benzyl benzoate |
| Reaction conditions: benzyl alcohol = 1.0 mmol, TBHP = 1.0 g, catalyst = 0.05 g, T = 298 K, time = 24 h. Cerium content was determined with ICP-AES method. |
| 1 |
HSCS-20 |
28.1 |
50.17 |
100 |
— |
— |
| 2 |
HSCS-10 |
16.6b |
22.95 |
100 |
— |
— |
The catalyst was recovered successfully after the reaction and easily regenerated by washing several times with ethanol and acetone, then finally heating at 373 K. The catalyst regenerated exhibits no change in its activity up to the third cycle of reusing (shown in Fig. 11) towards the alcohol oxidation (conversion and selectivity) which proves high reusability, good stability and true heterogeneity of the catalyst with minimum metal leaching.
 |
| | Fig. 11 Catalytic reusability test of HSCS-20 up to three cycles for alcohol oxidation at room temperature. | |
4 Conclusions
Mesoporous 2D hexagonal MCM-41 and 3D bicontinuous cubic MCM-48 with high Ce content up to 30 wt% have been prepared using CTAB surfactant, cerium(III) nitrate hexahydrate, and TEOS under ammonium basic conditions. It was found that ceria–silica composites showed high BET surface area over 349 m2 g−1 with narrow pore size distribution consisting 2.0–2.6 nm average pore diameter in spite of a high amount of Ce loading (up to 30 wt% as analyzed from ICP-AES). Most of the Ce present as Ce(IV), which was revealed from XPS 3d core-level spectra analysis and wide angle XRD data. XRD patterns showed a typical diffraction peak for FCC CeO2 nanostructure distributed throughout the silica matrices. Also, the cerium crystalline phase was changed to hexagonal structured Ce4.667Si3O13 when the sample was heated up to 1123 K under flowing 7 vol% of H2 diluted in N2. Besides, ceria–silica composite materials show a good efficiency in solvent-free oxidation of benzyl alcohol with a high selectivity of benzaldehyde at room temperature. At elevated temperature the materials also exhibit significant activity and good stability towards benzyl alcohol oxidation with benzoic acid as major product. Therefore, with proper selection of experimental conditions, mesoporous ceria–silica solids can demonstrate further applications as well as a recyclable, environmental friend, and economic route for oxidation of primary alcohol.
Acknowledgements
E.-B. Cho was supported partially by the New & Renewable Energy R&D program of the Korea Institute of Energy Technology Evaluation and Planning (KETEP) grant funded by the Korea government Ministry of Trade, Industry, and Energy (no. 20113020030040) and by the Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Education (NRF-2012R1A1A2000855). D. Kim was supported by the National Research Foundation (NRF-2012-R1A2A1A-05026313, NRF-2009-0093033, and NRF 2010-0027955) of the Korean Government. Experiments at PLS were supported in part by MSIP and POSTECH.
References
- A. Seidel, J. Loos and B. Boddenberg, J. Mater. Chem., 1999, 9, 2495 RSC.
- M. Nandi, P. Roy, H. Uyama and A. Bhaumik, Dalton Trans., 2011, 40, 12510 RSC.
- W. Cai, J. Yu, C. Anand, A. Vinu and M. Jaroniec, Chem. Mater., 2011, 23(5), 1147 CrossRef CAS.
- X. Li, Z. Zhuang, W. Li and H. Pan, Appl. Catal., A, 2012, 429–430, 31 CrossRef CAS PubMed.
- R. Peng, D. Zhao, N. M. Dimitrijevic, T. Rajh and R. T. Koodali, J. Phys. Chem. C, 2012, 116, 1605 CAS.
- N. Pal, M. Paul and A. Bhaumik, J. Solid State Chem., 2011, 184, 1805 CrossRef CAS PubMed.
- S. Inagaki, Y. Fukushima and K. Kuroda, J. Chem. Soc., Chem. Commun., 1993, 680 RSC.
- E.-B. Cho, M. Mandal and M. Jaroniec, Chem. Mater., 2011, 23, 1971 CrossRef CAS.
- A. Zukal, J. Mayerová and J. Čejka, Phys. Chem. Chem. Phys., 2010, 12, 5240 RSC.
- C. Sun, H. Liab and L. Chen, Energy Environ. Sci., 2012, 5, 8475 CAS.
- W. Yao, Y. Chen, L. Min, H. Fang, Z. Yan, H. Wang and J. Wang, J. Mol. Catal. A: Chem., 2006, 246, 162 CrossRef CAS PubMed.
- A. Trovarelli, C. de Leitenburg, M. Boaro and G. Dolcetti, Catal. Today, 1999, 50, 353 CrossRef CAS.
- J. G. Nunan, H. J. Robota, M. J. Cohn and S. A. Bradley, J. Catal., 1992, 133, 309 CrossRef CAS.
- X. Feng, D. C. Sayle, Z. L. Wang, M. S. Paras, B. Santora, A. C. Sutorik, T. X. T. Sayle, Y. Yang, Y. Ding and Y. S. Her, Science, 2006, 312, 1504 CrossRef CAS PubMed.
- F. A. Akopov and L. B. Borovkova, High Temp., 2011, 49, 862 CrossRef CAS.
- C. Périllat-Merceroz, G. Gauthier, P. Roussel, M. Huvé, P. Gélin and R.-N. Vannier, Chem. Mater., 2011, 23, 1539 CrossRef.
- A. Trovarelli, G. Dolcetti, C. de Leitenburg, J. Kaspar, P. Finetti and A. Santoni, J. Chem. Soc., Faraday Trans., 1992, 88, 1311 RSC.
- M. F. M. Zwinkels, S. G. Jaras and P. G. Menon, Catal. Rev. Sci. Eng., 1993, 35, 319 CAS.
- Y. I. Matatov-Meytal and M. Sheintuch, Ind. Eng. Chem. Res., 1998, 37, 309 CrossRef CAS.
- Y. Zhang, A. H. Yuwono, J. Wang and J. Li, J. Phys. Chem. C, 2009, 113, 21406 CAS.
- M. Negahdary, G. rezaMohseni, M. Fazilati, S. Parsania, G. Rahimi, S. Rad and S. R. Zarchi, Ann. Biol. Res., 2012, 3, 3671 CAS.
- A. Laachir, V. Perrichon, A. Badri, J. Lamotte, E. Catherine, J. C. Lavalley, J. El Fallah, L. Hilarie, F. Leonorm, E. Quemere, G. N. Sauvion and O. Touret, J. Chem. Soc., Faraday Trans., 1991, 87, 1601 RSC.
- J. Strunk, W. C. Vining and A. T. Bell, J. Phys. Chem. C, 2011, 115, 4114 CAS.
- A. R. Crozier, C. Schädle, C. Maichle-Mössmer, K. W. Törnroos and R. Anwander, Dalton Trans., 2013, 42, 5491 RSC.
- Q. Dai, X. Wang, G. Chen, Y. Zheng and G. Lu, Microporous Mesoporous Mater., 2007, 100, 268 CrossRef CAS PubMed.
- P. W. Dunne, A. M. Carnerup, A. Węgrzyn, S. Witkowski and R. I. Walton, J. Phys. Chem. C, 2012, 116, 13435 CAS.
- O. A. González Vargas, J. A. de los Reyes Heredia, A. Montesinos Castellanos, L. F. Chen and J. A. Wang, Mater. Chem. Phys., 2013, 139, 125 CrossRef PubMed.
- O. Aktas, S. Yasyerli, G. Dogu and T. Dogu, Ind. Eng. Chem. Res., 2010, 49, 6790 CrossRef CAS.
- Y. Shao, L. Wang, J. Zhang and M. Anpo, J. Phys. Chem. B, 2005, 109, 20835 CrossRef CAS PubMed.
- E.-B. Cho, S. Yim, D. Kim and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 12595 CAS.
- P. Zhang, Y. Gong, H. Li, Z. Chen and Y. Wang, Nat. Commun., 2013, 4, 1593 CrossRef PubMed.
- V. R. Choudhary, R. Jha and P. Jana, Green Chem., 2007, 9, 267 RSC.
- A. Villa, S. Campisi, C. Giordano, K. Otte and L. Prati, ACS Catal., 2012, 2, 1377 CrossRef CAS.
- S. Singha, M. Sahoo and K. M. Parida, Dalton Trans., 2011, 40, 11838 RSC.
- B. Karimi and F. K. Esfahani, Adv. Synth. Catal., 2012, 354, 1319 CrossRef CAS.
- M. Hundlucky, Oxidations in Organic Chemistry, American Chemical Society, Washington, DC, 1990 Search PubMed.
- M. Besson and P. Gallezot, Catal. Today, 2000, 57, 127 CrossRef CAS.
- A. Corma and M. E. Domine, Chem. Commun., 2005, 4042 RSC.
- A. Dijksman, A. Marino-González, A. MairataiPayeras, I. W. C. E. Arends and R. A. Sheldon, J. Am. Chem. Soc., 2001, 123, 6826 CrossRef CAS PubMed.
- Y. Jiang and N. Bahlawane, J. Alloys Compd., 2009, 485, L52 CrossRef CAS PubMed.
- A. Sayari, Y. Yang, M. Kruk and M. Jaroniec, J. Phys. Chem. B, 1999, 103, 3651 CrossRef CAS.
- C. J. Gommes, P. Ravikovitch and A. Neimark, J. Colloid Interface Sci., 2007, 314, 415 CrossRef CAS PubMed.
- S. C. Laha, P. Mukherjee, S. R. Sainkar and R. Kumar, J. Catal., 2002, 207, 213 CrossRef CAS.
- A. Bensalem, J. C. Muller and F. B. Verduraz, J. Chem. Soc., Faraday Trans., 1992, 88, 153 RSC.
- E. Cano, M. A. Garćia, M. A. Villegas, G. Battaglin, J. Llopis and J. M. Bastidas, J. Sol-Gel Sci. Technol., 2003, 27, 293 CrossRef CAS.
- A. K. Sinha and K. Suzuki, J. Phys. Chem. B, 2005, 109, 1708 CrossRef CAS PubMed.
- G. Zhang, Z. Shen, M. Liu, C. Guo, P. Sun, Z. Yuan, B. Li, D. Ding and T. Chen, J. Phys. Chem. B, 2006, 110, 25782 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ICP quantification and a SEM image of mesoporous ceria–silicas. See DOI: 10.1039/c3ra47464j |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 

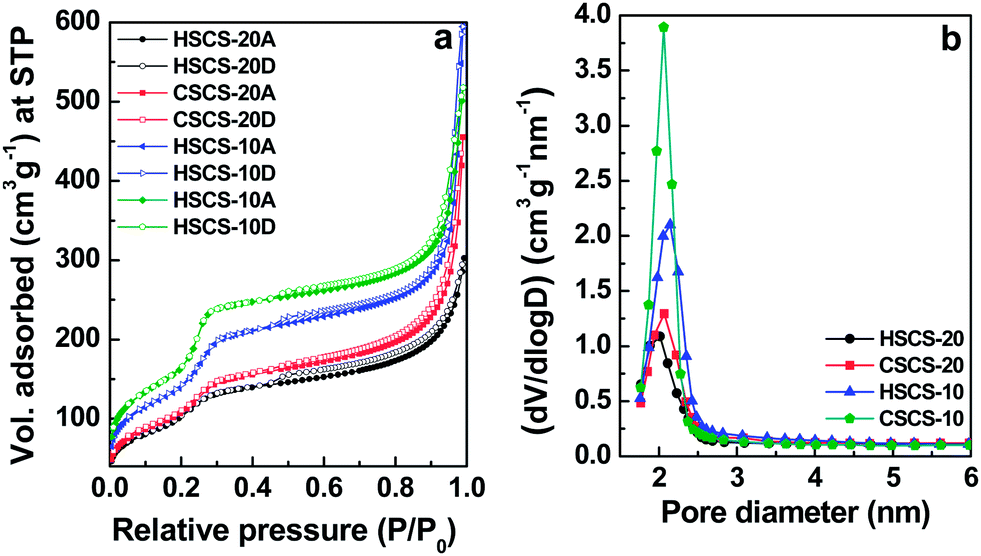
 ), CSCS-20 (
), CSCS-20 ( ), HSCS-10 (
), HSCS-10 ( ), CSCS-10 (
), CSCS-10 ( ) measured at 77 K. (b) Respective pore size distribution (PSD) curves of all the samples, maximum representing individual mesopore diameter.
) measured at 77 K. (b) Respective pore size distribution (PSD) curves of all the samples, maximum representing individual mesopore diameter.