
d-amino acid doping peptide hydrogel for the production of a cell colony†
Abstract
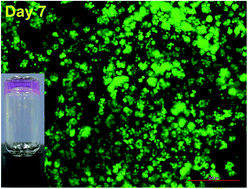
We report on a rationally designed D-amino acid doping peptide that can form hydrogels under neutral conditions and can be applied to form a cell colony of HeLa cells.
 Please wait while we load your content...
Please wait while we load your content...

