
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConsideration of the methanol-to-olefins (MTO) reaction over different zirconium species of the Zr-SAPO catalyst: a comprehensive periodic DFT investigation
Saeedeh Soheili and
Ali Nakhaei Pour*
and
Ali Nakhaei Pour*
Department of Chemistry, Faculty of Science, Ferdowsi University of Mashhad, Mashhad 9177948974, Iran. E-mail: a.nakhaei@um.ac.ir; nakhaeipoura@yahoo.com; Tel: +98-5138805539
First published on 15th May 2025
Abstract
Methanol-to-olefins (MTO) conversion is a crucial industrial process for producing valuable light olefins, but developing highly efficient and selective catalysts remains a significant challenge. The incorporation of zirconium has been shown to enhance the catalytic performance of MTO catalysts. In this study, periodic density functional theory (DFT) calculations were employed to investigate the stability and reactivity of zirconium species within the SAPO-18 framework. Results revealed that loading Zr3+ ions into 6-membered rings (6MRs) of the SAPO-18 framework (Zr-ZH) was the most favorable configuration for producing propylene, with a lower activation energy (0.46 eV) than that required for loading Zr4+ ions into 8-membered rings (8MRs) (Zr-ZOH). Analysis of Mulliken charges and partial density of states (DOS) suggested that the incorporation of zirconium into the SAPO-18 framework enhanced the electronic properties of the catalyst, leading to a significant increase in propylene selectivity. In summary, the DFT calculations provided valuable insights into the preferred coordination environments and electronic structures of zirconium species in the SAPO-18 catalyst. These results suggest that optimizing zirconium incorporation can lead to significant improvements in the catalytic performance of MTO processes, particularly with respect to propylene selectivity.
1. Introduction
Light olefins, such as ethylene and propylene, are crucial feedstocks in organic chemical processes.1,2 Microporous zeolites, with their diverse pore architectures and tunable acidity, have emerged as promising catalysts for the methanol-to-olefins (MTO) process.3–5 Among these, SAPO-18, a silicoaluminophosphate zeolite with the AEI framework, has garnered significant attention due to its small pore size and moderate acidity. These properties make it suitable not only for MTO but also for other applications, such as NH3-SCR.6 AEI, a small-pore zeolite framework, has also been adopted for vehicular applications. Recent studies have demonstrated enhanced hydrothermal stability in Al-rich AEI frameworks.7,8 Additionally, AEI has been observed to generally crystallize with fewer structural defects, which further enhances its hydrothermal stability.9The MTO reaction within SAPO-18 primarily occurs within its AEI cages, which are composed of 8-, 6-, and 4-membered rings (8MR, 6MR, and 4MR). However, coke formation, particularly from the accumulation of larger aromatic species, remains a major challenge, leading to catalyst deactivation.10 Incorporating metal species (Me) into the SAPO framework (MeSAPO) has emerged as a promising strategy to mitigate these issues and optimize catalytic performance. Metal incorporation can significantly influence the zeolite's physicochemical properties, including pore size, acidity, and electric field strength.11–17 MeSAPO can influence selectivity towards hydrocarbon species by altering the zeolite's physicochemical properties.15,18,19
The modified metal-incorporated SAPO enhances catalytic activity, lifetime, and light olefin yield.18 Bates et al. showed that the most stable site for isolated Cu2+ ions is the 6MR of SSZ-13.20 Peirong Niu et al. showed that the Co-Cr/SAPO catalyst exhibited stable performance in MTO over a time on stream of 600 min.21 Kang demonstrated that the selectivity of light olefins decreased more rapidly in CoSAPO than in other metal-incorporated SAPOs (FeSAPO-34 and NiSAPO-34) as the reaction time increased.15
One major obstacle to the widespread commercialization of Zr-containing zeolites, including their application in MTO, has been their limited hydrothermal stability. However, zirconium, an important transition metal that is finding increasing applications in catalysis, offers several potential advantages.11,22,23 Varzaneh et al. demonstrated that Zr-SAPO-34 with a low Brønsted acid strength had an improved catalyst lifetime.24 The incorporation of zirconium can improve the thermal stability of catalysts by providing enhanced thermal resistance.25 Metal incorporation can improve the acidic properties and reduce the diffusion limitations of the reactant molecules, which can positively affect the conversion performance of the MTO process.26 The incorporation of Zr in catalysts, particularly in zeolites like SAPO (silica–aluminum phosphate), leads to enhanced properties of the Lewis acid and catalytic activity.27,28 As already mentioned, the loading of Zr into the SAPO structure alters the physicochemical properties of these materials, which directly impacts their acidity and performance in catalytic reactions.29 This is due to Zr acting as a metal cation with unique characteristics, creating stronger centers for accepting electrons in acid–base reactions.30 The addition of Zr into SAPO can alter the Brønsted-to-Lewis acid site ratio (B/L), with the focus often being on enhancing the Lewis acid sites, which are commonly supplementary and suitable for reactions like hydrogenation and isomerization.31–33 Zr primarily increases the Lewis acidity of SAPO without significantly affecting the Brønsted acid properties,34 which is particularly beneficial in catalytic reactions that rely on Lewis's acid sites.35 These improvements help maintain the catalyst's activity under harsher conditions, thereby extending its lifespan.36 Zr can also modify the structure of SAPO's microspores, facilitating a better distribution of products and reactants throughout the catalyst structure, thereby improving the efficiency of catalytic reactions.37 In general, the addition of Zr to SAPO increases Lewis's acidity, making the catalyst more effective for Lewis-acid-driven reactions and enhancing its catalytic activity in processes such as cracking, alkylation, and isomerization.38
In the present work, metals were incorporated into SAPO in the present work to enhance its catalytic activity, selectivity, and lifetime. Due to its low acidic sites, Zr was incorporated into SAPO-18. Density functional theory (DFT) calculations were done to systematically study the MTO process on the Zr species. Mechanisms for the interaction of CH3OH with different Zr-SAPO-18 catalysts with Zr ions positioned in the 6MR and 8MR are proposed. The most favorable structure for producing propylene over a long lifetime is Zr-ZH. Additionally, the partial density of states (DOS) and Mullikan partial charges were calculated to confirm the oxidation state of Zr in the SAPO-18 structures.
2. Model
2.1. Structure
As a metal element, zirconium can significantly improve the performance of catalysts in complex processes, such as MTO conversion. In the MTO conversion process, it contributes to forming active sites that are crucial for olefin production, particularly propylene, and enhances the catalyst's resistance to thermal degradation, therefore extending its lifespan.29,39 SAPO is formed by substituting one Si atom for P in the CHA framework, which results in the creation of a Brønsted acid site.40,41 Table 1 summarizes the topological parameters of the SAPO-18 framework, along with the Zr-ZOH and Zr-ZH structures.| Structure | a (Å) | b (Å) | c (Å) | α (deg.) | β (deg.) | γ (deg.) | Cell formula |
|---|---|---|---|---|---|---|---|
| SAPO-18 | 9.47 | 9.47 | 18.63 | 90.25 | 90.25 | 85.27 | H2O48Al12Si2P10 |
| Zr-ZOH | 9.47 | 9.47 | 18.63 | 90.25 | 90.25 | 85.27 | H2O48Al12Si2P10 |
| Zr-ZH | 9.47 | 9.47 | 18.63 | 90.25 | 90.25 | 85.27 | H2O48Al12Si3P9 |
2.2. Computation
DFT calculations were performed using the CASTEP module within Materials Studio. This approach employs pseudopotentials to describe the core electrons and plane waves to represent the valence electrons. The Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) was utilized for the exchange–correlation interactions.42,43 A k-point grid with a spacing of 0.05 Å−1 was employed to sample the Brillouin zone. An energy cutoff of 450 eV was applied, and the self-consistent-field (SCF) tolerance was set at 1 × 10−6 eV per atom. For k-point separation, a value of 0.05 was considered. The values of the maximum force, and maximum displacement were taken to be 0.05 eV Å−1 and 0.002 Å, respectively. Spin polarization and van der Waals (vdW) interactions were included in all the computations. To determine the reaction barrier and transition state structures, a combination of the linear synchronous transit (LST) and quadratic synchronous transit (QST) methods were utilized. The LST method determines the location of the maximum energy along this path using a single interpolation. In contrast, the QST method increases the accuracy by combining maximum energy searches and constrained minimizations. The TS approximation obtained through this method is based on QST maximization. In this paper, a hybrid TS search method was used, which incorporates elements of both the LST and QST techniques, called LST/QST. The LST approach starts with interpolating between the reactant and product states to identify the maximum energy. This is followed by energy minimization in specific directions along the reaction path. Subsequently, the QST maximization step is modified, which is complemented by an additional double gradient minimization. This LST/QST cycle is repeated until the predefined convergence criteria are met.3. Result and discussion
3.1. Effects of Zr in the 6MR and 8MR
It is well-established that the MTO reaction proceeds through a complex mechanism involving aromatic intermediates that act as crucial co-catalysts in the formation of light olefins.44–46 Therefore, understanding the interactions between these intermediates and the zeolite framework, particularly within cavity-type structures, such as for SAPO-18, is critical. To effectively catalyze the MTO reaction for automotive applications, small-pore Zr-zeolites must exhibit unique and enhanced properties.47As shown in Fig. 1, Zr within the 8MR exhibits a five-fold coordination with oxygen atoms and creates [ZrIVOH]3+, while in the 6MR site, it forms bonds with four oxygen atoms and creates [Zr3+/H+]. These distinct coordination environments lead to the formation of two primary Zr species: Zr-ZH, located within the 6MR, and Zr-ZOH, located within the 8MR.48,49
 | ||
| Fig. 1 Local structures for (a) SAPO-18, (b) Zr4+ in 8MR, and (c) Zr3+ in 6MR. Mulliken partial charges are labeled in the figure. Zr, O, Si, Al, P, and H atoms are represented in blue, red, yellow, magenta, green, and white, respectively. | ||
There are theoretical and experimental evidence to support these structures with different Zr species and their distinct coordination environments.50–56
By increasing the acidity, Zr facilitates proton transfer, thereby promoting methylation reactions. Zr-ZH is formed through the simultaneous substitution of two Si atoms, directly creating Zr3+ on the 6MR. In contrast, Zr-ZOH is formed through the simultaneous substitution of a single Si atom, resulting in the formation of Zr4+ on the 8MR.
As shown in Fig. 2, the oxidation states of Zr according to the calculated DOS were +4 and +3 in the Zr-ZOH and Zr-ZH structures, respectively, which were consistent with the results in the 6MR and 8MR. For Zr-ZH and Zr-ZOH structures, one and two peaks with energies of 0.79 eV and (1.58 and 3.66 eV) related to d-orbitals appeared above the Fermi level in the DOS plots of the Zr 3d orbital, indicating a one-electron and two-electron loss in the Zr 3d orbital. A reduction in the number of valence electrons reduces the interactions, resulting in a decreased bandwidth.57,58 The greater the negativity of the d orbital energy, the more stable a structure becomes, reducing its tendency for electron transfer in the d orbital.
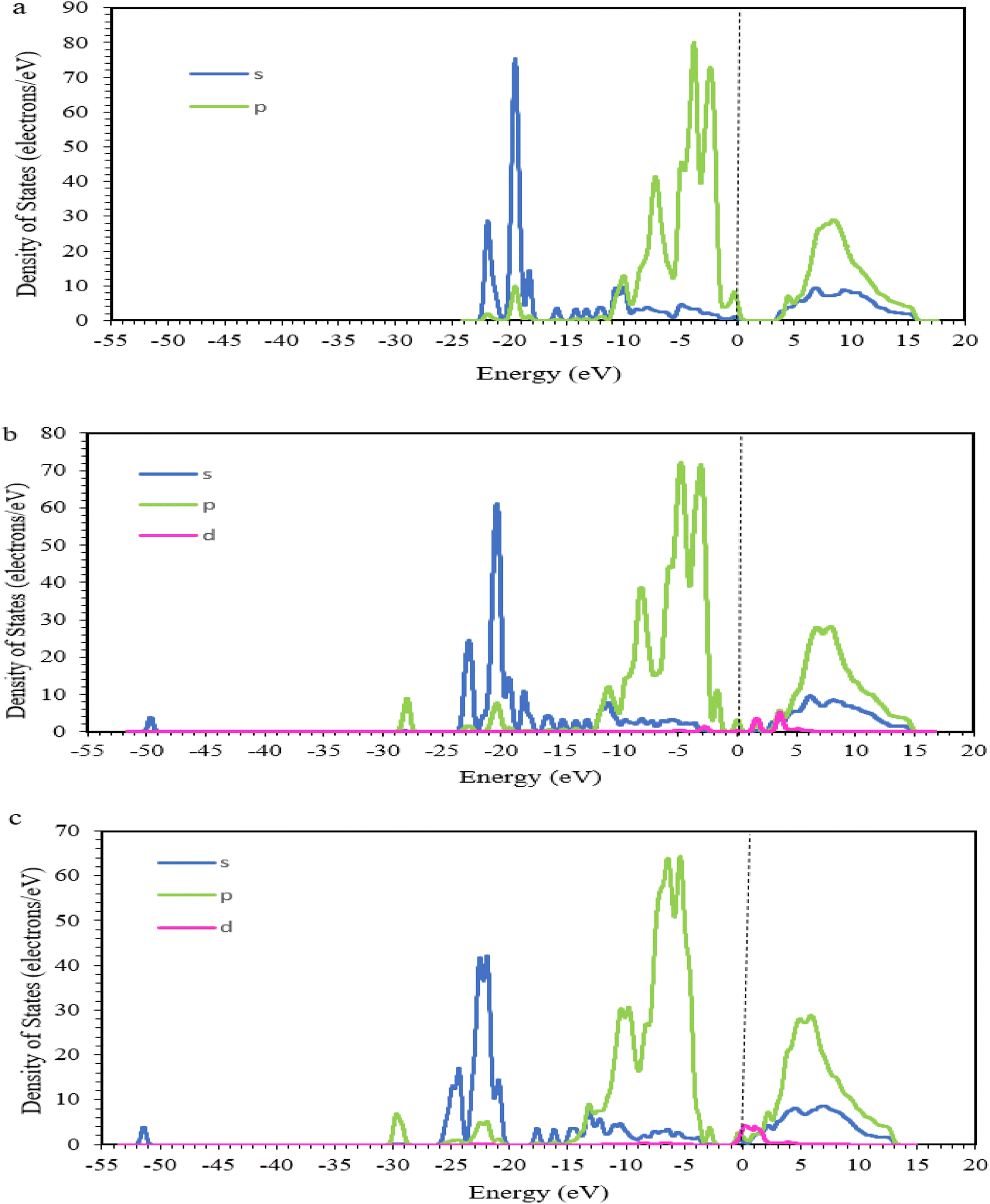 | ||
| Fig. 2 Density of states (DOS) of (a) SAPO-18, (b) Zr-ZOH (Zr in 8MR), and (c) Zr-ZH (Zr in 6MR), with the local structures inserted. The vertical lines represent the position of the Fermi level. | ||
In general, the DOS energy range is the same for all structures and no major changes occur in the s and p orbitals. However, the electron intensity decreases with Brønsted acid sites, such that the Zr-ZH structure has a lower electron density than the other structures.
The MTO reaction through a hydrocarbon pool mechanism involves a series of steps, including methylation, proton transfer, and olefin removal (Fig. 3). Zr can significantly influence these steps by modulating the acidity and electronic properties of the zeolite.
 | ||
| Fig. 3 Side-chain hydrocarbon pool mechanism for the MTO reaction catalyzed by PX in SAPO-18, Zr-ZOH, and Zr-ZH. | ||
Following the hydrocarbon pool mechanism (Fig. 3), this analysis focuses on the growth of the side chain through successive methylation of the intermediates containing exocyclic double bonds (e.g., P4/P8 → P5/P10) and deprotonation of the benzene cations (e.g., P2/P5 → P3/P6) for ethylene formation and (P2/P5/P10 → P3/P8/P11) for propylene formation. As shown in Fig. 1, Tables 2 and 3, the presence of Zr within the 6MR could enhance the reaction rate by increasing the electron density and influencing the electron affinity at the catalyst surface.
| Structure | P0 | P1 | TS1–2 | P2 | TS2–3 | P3 | P4 | TS4–5 | P5 | TS5–6 | P6 | TS6–7 | P7 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SAPO-18 | 0.00 | −1.00 | 0.40 | −0.63 | 0.32 | −0.25 | −0.30 | 0.73 | 0.12 | 1.16 | −0.51 | 0.52 | 0.12 |
| Zr-ZOH | 0.00 | −1.20 | 0.50 | −0.37 | 0.21 | −0.63 | −0.76 | 0.47 | 0.18 | 0.85 | −0.62 | 0.64 | −0.24 |
| Zr-ZH | 0.00 | −1.70 | 0.37 | −0.20 | 0.15 | −1.42 | −0.98 | 0.56 | 0.24 | 0.65 | −0.72 | 0.73 | −0.48 |
| Structure | P5* | TS5–8 | P8 | P9 | TS9–10 | P10 | TS10–11 | P11 | TS11–12 | P12 |
|---|---|---|---|---|---|---|---|---|---|---|
| SAPO-18 | 0.12 | 1.33 | −0.28 | −0.16 | 0.54 | −0.73 | 0.53 | −0.35 | 1.16 | 0.15 |
| Zr-ZOH | 0.18 | 0.96 | −0.56 | −0.43 | 0.49 | −0.29 | 0.33 | −0.23 | 0.80 | 0.28 |
| Zr-ZH | 0.24 | 0.76 | −0.71 | −0.65 | 0.37 | −0.15 | 0.20 | −0.19 | 0.71 | 0.35 |
3.2. Methylation
The catalytic behavior of Zr within the SAPO-18 framework was significantly influenced by its location within the 6MR or 8MR. As depicted in Fig. 3, the MTO reaction was initiated with the gem-methylation of p-xylene (PX), forming benzenium cations. This step involves the interaction between the OH group of methanol and the acid site of the zeolite, leading to the formation of the 3,3,6-trimethylcyclohexa-1,4-diene cation (methylPX+) (P1 → P2). The efficiency of the methylation step is directly correlated with the Brønsted acidity of the catalyst. The transition states (TS1-2) of SAPO-18, Zr-ZOH, and Zr-ZH structures are shown in Fig. 4. | ||
| Fig. 4 Transition state (TS1-2) structures of (a) SAPO-18, (b) Zr-ZOH, and (c) Zr-ZH. | ||
A stronger Brønsted acid facilitates proton transfer, lowering the activation energy for the formation of new bonds during methylation.59,60 This is because a strong Brønsted acid readily donates a proton, promoting the formation of catalytic intermediates and accelerating the reaction.59 The calculated activation energies for methanol in SAPO-18, Zr-ZOH, and Zr-ZH were 1.40, 1.70, and 2.07 eV, respectively. The stronger the Brønsted acid, the greater the decrease in the activation energy of the methylation step, as proton transfer can occur more easily, thereby reducing the energy required to cross the transition state and thus facilitating the reaction.59,61 The Brønsted acid property refers to a compound's ability to donate a proton (H+).62,63 As shown Fig. 4, the broken Z–H bond lengths in the SAPO-18, Zr-ZOH, and Zr-ZH structures are 1.61 Å, 1.74 Å, and 1.92 Å, respectively.
In the MTO process, protons play a crucial role in activating methanol molecules and forming intermediate compounds.64 As the Brønsted acidity increases, protons can more easily transfer to methanol molecules, facilitating the formation of methylation products and lowering the activation energy. The methylation step involves the interaction between the proton from the zeolite's acid site and the hydroxyl oxygen of methanol (Table 2). This interaction can be visualized as the formation of protonated methanol through an SN2-type transition state (TS1–2).
As shown Fig. 4, the methyl group of methanol attaches to PX structure. The broken O–C bond lengths of SAPO-18, Zr-ZOH, and Zr-ZH structures are 3.25, 2.94, and 2.53 Å and the formed C–C bond lengths are 1.53, 1.62, and 1.81 Å, respectively. As the Zr atom exhibits a greater covalent character, its Lewis properties are increased, while the partial positive charge on the hydrogen atom is decreased, leading to a reduction in its Brønsted properties.65,66 In the 6MR, the hydrogen atom displayed a stronger Lewis acidity than in the 8MR. As the Lewis acidity increased, the energy barrier for methylation also increased. The capture of methanol by the acid site of the zeolite was the first step in initiating the MTO reaction.67 The initial step in starting the MTO reaction involved the interaction of methanol with the acid site of the zeolite. Here, Zr enters the SAPO framework as a metal cation, creating Lewis acid centers.68 These centers can accept electrons from the reacting molecules and typically serve as catalytic sites for Lewis's acid reactions, such as hydrogenation, isomerization, and oxidation.69 Brønsted acids in zeolites are usually formed by the presence of protons (H+) at specific sites, often associated with oxygen-bonded hydrogen ions (Si–OH). Introducing Zr into the SAPO structure changes the balance between the Lewis acid and Brønsted sites by increasing the proportion of Lewis acids.31 As a result, the Lewis acidity increases, while the number or strength of the Brønsted acid sites remains largely unchanged. With an increase in the acidity of the zeolite, the extent of methanol physisorption also rises.70
It has been found that the activation energy for methylation decreases as the strength of the Brønsted acid sites increases, due to improved proton transfer and stabilization of the transition state.71 Consequently, the methylation activation energy here followed the trend: SAPO-18 < Zr-ZOH < Zr-ZH. Methanol (CH3OH) tends to adsorb on H+ sites in SAPO-18 more than on Zr-structures. As shown in Fig. 1, the partial charge on the hydrogen atom followed the trend: SAPO-18 > Zr-ZOH > Zr-ZH. As the partial charge of the H atom increased, the Brønsted acidic increased. Methylation in the 8MR is more facile than in the 6MR because of the higher partial charge of hydrogen. Zalazar et al. conducted an electron-density analysis of adsorbed methanol on acidic zeolite structures via a hydrocarbon pool mechanism.72 They found that the stronger OM–H⋯OZ interaction in H-beta, due to its larger cavity, led to higher ρ(r) values, ∇2ρ(r) < 0, and H(r) < 0, compared to in ZSM-5,72 suggesting the greater stabilization of the OM–H⋯OZ interaction in the H-beta cavity, which could be attributed to the higher electron density available within the larger cavity. Under reaction conditions, the adsorbed Zr-ZH becomes a stable and active site with a Zr3+ oxidation state. To evaluate the impact of the different Zr species on the MTO reaction, the transition state energies for the key steps in the hydrocarbon pool mechanism were calculated and are presented in Fig. 5–7.
 | ||
| Fig. 5 Energy profile of the PX pathway in the side-chain hydrocarbon pool mechanism on SAPO-18. | ||
 | ||
| Fig. 6 Energy profile of the PX pathway in the side-chain hydrocarbon pool mechanism on Zr-ZOH. | ||
 | ||
| Fig. 7 Energy profile of the PX pathway in the side-chain hydrocarbon pool mechanism on Zr-ZH. | ||
3.3. Deprotonation
In the hydrocarbon pool (HP) mechanism, the deprotonation step of the benzenium cations involves the transfer of a proton from the carbocation to a basic site within the zeolite framework. This process forms a Brønsted acid (HZ) and the corresponding conjugate base (Z−).73 The generated methylPX+ species can then undergo deprotonation, releasing a proton from its side-chain methyl group to the zeolite structure. This results in the formation of the intermediate P3 (3,3-dimethyl-6-methylenecyclohexa-1,4-diene) as shown in Fig. 3. The activation energies required for deprotonation in SAPO-18, Zr-ZOH, and Zr-ZH were determined to be 0.95, 0.58, and 0.35 eV, respectively (Table 2). This indicates that deprotonation is energetically more favorable in the presence of Zr, particularly within the 6MR (Zr-ZH). The presence of Zr3+ within the 6MR of SAPO-18 alters the electron-density distribution within the zeolite framework, leading to an increase in the basicity of the framework, which facilitates the deprotonation of the benzenium cations.The less Brønsted acidic in the ZrZ− structure cause to increase adsorption H atom from methylPX+. The activation energies for forming exocyclic double bonds (P2 → P3), (P5 → P8), and spiro features (P5 → P6) and (P10 → P11) in the deprotonation step increased with increasing the B/L ratio. The P3 structure is inherently unstable and tends to revert to PX+. However, the second methylation step, where methanol interacted with the exocyclic double bond of P4, leading to the formation of 6-ethyl-3,3-dimethylcyclohexa-1,4-diene (P5), could effectively prevent this back-reaction (Fig. 3).
3.4. P5 → P7: production of ethylene
To generate ethylene, a hydrogen atom from the ethyl side group needs to migrate to the ring carbon. The indirect hydrogen-transfer pathway begins with the deprotonation of P5 to the zeolite, and the transition from P5 to P6, and concludes with the proton returning from the zeolite to the ring carbon in the P6 → P7 step (Fig. 3). These steps are often facilitated by the presence of neighboring water molecules. Table 2 shows that the activation energies for the protonation step (P6 → P7) in SAPO-18, Zr-ZOH, and Zr-ZH were 1.03, 1.26, and 1.45 eV, respectively, indicating that ethylene production was energetically less favorable in the presence of Zr, particularly within the 6MR (Zr-ZH). The P6 intermediate was inherently unstable due to ring strain. The presence of Zr within the 6MR, with its lower Brønsted acidity, reduced the tendency for proton loss from the ethyl side chain, thereby inhibiting ethylene formation. Furthermore, the Zr-ZOH and Zr-ZH structures, with lower B/L ratios, generally exhibit higher selectivity towards propylene production compared to ethylene.3.5. P5 → P12: production of propylene
As shown in Fig. 3, propylene formation involved a different pathway compared to ethylene production. It began with the deprotonation of the CH2 group in the ethyl side chain of P5, forming an exocyclic double bond P8 (6-ethylidene-3,3-dimethylcyclohexa-1,4-diene) (P5 → P8). This process involved energy barriers of 1.21, 0.78, and 0.52 eV for the SAPO-18, Zr-ZOH, and Zr-ZH structures, respectively (Table 3). In the MTO process, a stronger Brønsted acidity generally favors ethylene production. This is because increased Brønsted acidity facilitates the protonation steps involved in the ethylene pathway, such as the P6 → P7 step. In contrast, the deprotonation steps involved in propylene formation (e.g., P5 → P8) are favored by a lower Brønsted acidity (Tables 2 and 3). The side chain can extend (P9 → P10) through the methylation of the exocyclic double bond in P9, facilitated by a third methanol molecule adsorbed on the surface. The production of propylene involved the deprotonation of P10 (6-isopropyl-3,3-dimethylcyclohexa-1,4-diene) to P11 (3,3-dimethyl-6-(2-methylcyclopropyl)cyclohexa-1,4-diene), followed by the protonation of P11. As shown in Table 3, the deprotonation step required overcoming energy barriers of 1.26, 0.62, and 0.35 eV. In contrast, the protonation step was comparatively straightforward, with energy barriers of 1.51, 1.36, and 0.90 eV for SAPO-18, Zr-ZOH, and Zr-ZH, respectively.Notably, the geometry of P11 closely resembles that of P6, characterized by a side spiro structure. The energy barriers for propylene formation (P11 → P12) were found to be significantly lower than those for ethylene formation (P6 → P7) in both Zr-ZOH and Zr-ZH, indicating a higher selectivity towards propylene production in these structures. This was attributed to the greater stability of the transition state involved in propylene formation, whereby a hydrocarbon cation was generated through protonation at the carbon of the side chain (Tables 2 and 3). Therefore, as the Brønsted acid strength increased, the reactions and pathways related to ethylene production proceeded more quickly and easily, while propylene production was less affected under the conditions that require the transfer of other groups. In conditions where the B/L ratio and oxidation number were lower, the tendency to produce propylene was greater.
The overall energy barriers for the rate-determining step in propylene formation were 0.80, 0.74, and 0.67 eV for SAPO-18, Zr-ZOH, and Zr-ZH, respectively. In contrast, the overall energy barriers for ethylene production were 0.45, 1.12, and 1.72 eV for SAPO-18, Zr-ZH, and Zr-ZOH, respectively. These results further support the higher selectivity towards propylene production in the presence of Zr, particularly within the 6MR (Zr-ZH). It can thus be concluded that the difference in the overall energy barrier of Zr-ZH showed its greater selectivity for propylene as the main olefin product compared to the other structures. Meanwhile, increasing the acid strength led to an increase in the rate of catalyst deactivation.73 Therefore, it can be concluded that the difference in the overall energy barrier indicated its higher selectivity for propylene as the main olefin product compared to the other structures.
4. Conclusion
This study demonstrated that the location of Zr within the SAPO-18 framework significantly influences its catalytic performance in the MTO process. The findings indicate that the 6MR site is thermodynamically more favorable for propylene production. It achieves this by strengthening the acidic properties, improving the catalytic efficiency, modulating the product selectivity, and stabilizing the catalyst, while lowering the activation energy. Notably, Zr's incorporation into the 6MR proved particularly effective in reactions demanding confined spaces and robust interactions with the reactants. The 6MR was better suited for reactions yielding propylene or similar products that involve more intricate structural requirements. Conversely, the 8MR, with its larger spatial arrangement, was more appropriate for ethylene production, as it facilitates the adsorption and processing of reactants and products. In the 6MR, Zr plays a more prominent role in enhancing propylene formation due to its ability to support reactions requiring significant conformational changes in a confined space. Additionally, a high propylene-to-ethylene (P/E) ratio was observed for Zr-ZH, even with low levels of Brønsted-to-Lewis (B/L) acidity. In conclusion, the variation in the overall energy barrier of Zr-ZH indicate its greater selectivity for propylene as the primary olefin product compared to the other configurations. The involvement of zirconium's d-orbitals contributed significantly to suppressing coke formation and enhancing catalyst stability.Data availability
Initial crystallographic data of SAPO can be obtained from https://doi.org/10.1039/c6cp02289h.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors of this work appreciate the financial support of the Ferdowsi University of Mashhad Research Council, Mashhad, Iran (grant no. 7849).References
- Z. Keyvanloo, A. N. Pour, F. Moosavi and S. M. K. Shahri, J. Mol. Graphics Modell., 2022, 110, 108048 Search PubMed.
- S. I. Jami, A. Nakhaei Pour, A. Mohammadi and S. M. Kamali Shahri, Chem. Eng. Technol., 2020, 43, 2100–2108 Search PubMed.
- S. Aghamohammadi, M. Haghighi and M. Charghand, Mater. Res. Bull., 2014, 50, 462–475 Search PubMed.
- M. Sedighi, H. Bahrami and J. Towfighi, J. Ind. Eng. Chem., 2014, 20, 3108–3114 Search PubMed.
- S. Soheili, A. N. Pour, A. Mohammadi and D. Y. Murzin, J. Mol. Graphics Modell., 2023, 124, 108555 Search PubMed.
- Y. Li, J. Deng, W. Song, J. Liu, Z. Zhao, M. Gao, Y. Wei and L. Zhao, J. Phys. Chem. C, 2016, 120, 14669–14680 Search PubMed.
- Y. Shan, W. Shan, X. Shi, J. Du, Y. Yu and H. He, Appl. Catal., B, 2020, 264, 118511 Search PubMed.
- Y. Shan, J. Du, Y. Zhang, W. Shan, X. Shi, Y. Yu, R. Zhang, X. Meng, F.-S. Xiao and H. He, Natl. Sci. Rev., 2021, 8, nwab010 Search PubMed.
- C.-R. Boruntea, L. F. Lundegaard, A. Corma and P. N. Vennestrøm, Microporous Mesoporous Mater., 2019, 278, 105–114 Search PubMed.
- U. Olsbye, S. Svelle, M. Bjørgen, P. Beato, T. V. Janssens, F. Joensen, S. Bordiga and K. P. Lillerud, Angew. Chem., Int. Ed., 2012, 51, 5810–5831 Search PubMed.
- J. Zhong, J. Han, Y. Wei, S. Xu, T. Sun, X. Guo, C. Song and Z. Liu, Chin. J. Catal., 2018, 39, 1821–1831 Search PubMed.
- M. Inoue, P. Dhupatemiya, S. Phatanasri and T. Inui, Microporous Mesoporous Mater., 1999, 28, 19–24 Search PubMed.
- S. Tian, S. Ji, D. Lü, B. Bai and Q. Sun, J. Energy Chem., 2013, 22, 605–609 Search PubMed.
- A. Z. Varzaneh, J. Towfighi and S. Sahebdelfar, Microporous Mesoporous Mater., 2016, 236, 1–12 Search PubMed.
- M. Kang, J. Mol. Catal. A: Chem., 2000, 160, 437–444 Search PubMed.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 Search PubMed.
- Y. Pu, X. Xie, W. Jiang, L. Yang, X. Jiang and L. Yao, Chin. Chem. Lett., 2020, 31, 2549–2555 Search PubMed.
- M. Sedighi, M. Ghasemi, M. Sadeqzadeh and M. Hadi, Powder Technol., 2016, 291, 131–139 Search PubMed.
- S. Hotevar and J. Levec, J. Catal., 1992, 135, 518–532 Search PubMed.
- S. A. Bates, A. A. Verma, C. Paolucci, A. A. Parekh, T. Anggara, A. Yezerets, W. F. Schneider, J. T. Miller, W. N. Delgass and F. H. Ribeiro, J. Catal., 2014, 312, 87–97 Search PubMed.
- P. Niu, X. Ren, D. Xiong, S. Ding, Y. Li, Z. Wei and X. Chen, Catalysts, 2020, 10, 785 Search PubMed.
- T. Yamaguchi, Catal. Today, 1994, 20, 199–217 Search PubMed.
- A. Amoozegar, M. Haghighi and S. Aghamohammadi, RSC Adv., 2016, 6, 51024–51036 Search PubMed.
- A. Z. Varzaneh, J. Towfighi and M. S. Moghaddam, Pet. Chem., 2020, 60, 204–211 Search PubMed.
- O. D. A. Salah Aldeen, M. Z. Mahmoud, H. S. Majdi, D. A. Mutlak, K. Fakhriddinovich Uktamov and E. Kianfar, Adv. Mater. Sci. Eng., 2022, 2022, 1–22 Search PubMed.
- J. Francis, E. Guillon, N. Bats, C. Pichon, A. Corma and L. Simon, Appl. Catal., A, 2011, 409, 140–147 Search PubMed.
- R. Zhang, S. He, F. Wang, B. Cai, J. Feng and H. Pan, Chem. Phys., 2024, 112344 Search PubMed.
- W. Xue, M. Ma, P. Hou, P. Zhang, W. Liu, Y. Liu, H. Yue, X. Wang, G. Tian and S. Feng, Fuel, 2024, 371, 132115 Search PubMed.
- S. H. Bakhtiar, A. Ismail, S. Ali, F. Raziq, S. A. Hussain, A. Zada, W. Dong, M. Zahid and Q. Fu, Inorg. Chem. Commun., 2024, 159, 111835 Search PubMed.
- S. Daliran, A. R. Oveisi, C.-W. Kung, U. Sen, A. Dhakshinamoorthy, C.-H. Chuang, M. Khajeh, M. Erkartal and J. T. Hupp, Chem. Soc. Rev., 2024, 53, 6244–6294 Search PubMed.
- W. Li, M. Li, H. Liu, W. Jia, X. Yu, S. Wang, X. Zeng, Y. Sun, J. Wei and X. Tang, Mol. Catal., 2021, 506, 111538 Search PubMed.
- R. Yadav and A. K. Singh, Catalysis for Clean Energy and Environmental Sustainability: Petrochemicals and Refining Processes, 2021, vol. 2, pp. 503–525 Search PubMed.
- J. H. Advani, A. Kumar and R. Srivastava, Catalysis in Confined Frameworks: Synthesis, Characterization, and Applications, 2024, pp. 363–396 Search PubMed.
- C. Liu, X. Wang, L. Xing, X. Cheng, X. Zhang, H. Li and M. Liu, Appl. Sci., 2023, 13, 4763 Search PubMed.
- S. Soheili, A. N. Pour and A. Mohammadi, Microporous Mesoporous Mater., 2021, 317, 111003 Search PubMed.
- I. B. Minova, N. S. Barrow, A. C. Sauerwein, A. B. Naden, D. B. Cordes, A. M. Slawin, S. J. Schuyten and P. A. Wright, J. Catal., 2021, 395, 425–444 Search PubMed.
- S. Rimaz, M. Kosari, M. Zarinejad and S. Ramakrishna, J. Mater. Sci., 2022, 1–39 Search PubMed.
- Q. Wang, M. Xing, L. Wang, Z. Gong, M. A. Nawaz, R. Blay-Roger, T. Ramirez-Reina, Z. Li and F. Meng, Mol. Catal., 2024, 569, 114567 Search PubMed.
- K. Ma, S. Zhao, M. Dou, X. Ma and C. Dai, ACS Catal., 2023, 14, 594–607 Search PubMed.
- M.-A. Djieugoue, A. Prakash and L. Kevan, J. Phys. Chem. B, 2000, 104, 6452–6461 Search PubMed.
- M. Fischer, Phys. Chem. Chem. Phys., 2016, 18, 15738–15750 Search PubMed.
- B. Delley, J. Chem. Phys., 1990, 92, 508–517 Search PubMed.
- C.-M. Wang, Y.-D. Wang, H.-X. Liu, Z.-K. Xie and Z.-P. Liu, J. Catal., 2010, 271, 386–391 Search PubMed.
- S. Xu, A. Zheng, Y. Wei, J. Chen, J. Li, Y. Chu, M. Zhang, Q. Wang, Y. Zhou and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 1190–1212 Search PubMed.
- M. Bjørgen, S. Svelle, F. Joensen, J. Nerlov, S. Kolboe, F. Bonino, L. Palumbo, S. Bordiga and U. Olsbye, J. Catal., 2007, 249, 195–207 Search PubMed.
- S. Svelle, F. Joensen, J. Nerlov, U. Olsbye, K.-P. Lillerud, S. Kolboe and M. Bjørgen, J. Am. Chem. Soc., 2006, 128, 14770–14771 Search PubMed.
- X. Liu and Z. Zhu, Adv. Sci., 2024, 11, 2306533 Search PubMed.
- T. Merle-Méjean, P. Barberis, S. B. Othmane, F. Nardou and P. Quintard, J. Eur. Ceram. Soc., 1998, 18, 1579–1586 Search PubMed.
- M. Zhang, M. Bosch, T. Gentle III and H.-C. Zhou, CrystEngComm, 2014, 16, 4069–4083 Search PubMed.
- C. Koschnick, M. W. Terban, R. Frison, M. Etter, F. A. Böhm, D. M. Proserpio, S. Krause, R. E. Dinnebier, S. Canossa and B. V. Lotsch, J. Am. Chem. Soc., 2023, 145, 10051–10060 Search PubMed.
- J. Zhang, B. An, Z. Li, Y. Cao, Y. Dai, W. Wang, L. Zeng, W. Lin and C. Wang, J. Am. Chem. Soc., 2021, 143, 8829–8837 Search PubMed.
- D. Yang and B. C. Gates, Acc. Chem. Res., 2021, 54, 1982–1991 Search PubMed.
- K. C. Park, J. Lim, G. C. Thaggard and N. B. Shustova, J. Am. Chem. Soc., 2024, 146, 18189–18204 Search PubMed.
- F. Farges, C. W. Ponader and G. E. Brown Jr, Geochim. Cosmochim. Acta, 1991, 55, 1563–1574 Search PubMed.
- S. Yuan, Y. P. Chen, J. Qin, W. Lu, X. Wang, Q. Zhang, M. Bosch, T. F. Liu, X. Lian and H. C. Zhou, Angew. Chem., Int. Ed., 2015, 54, 14696–14700 Search PubMed.
- D. Waroquiers, X. Gonze, G.-M. Rignanese, C. Welker-Nieuwoudt, F. Rosowski, M. Gobel, S. Schenk, P. Degelmann, R. André and R. Glaum, Chem. Mater., 2017, 29, 8346–8360 Search PubMed.
- S. Veiskarami, A. N. Pour, E. Saljoughi and A. Mohammadi, New J. Chem., 2024, 48, 9990–10001 Search PubMed.
- S. Veiskarami, E. Saljoughi and A. N. Pour, Int. J. Hydrogen Energy, 2024, 81, 225–236 Search PubMed.
- C. Chizallet, C. Bouchy, K. Larmier and G. Pirngruber, Chem. Rev., 2023, 123, 6107–6196 Search PubMed.
- P. Liu, J. Wang, L. Ling, X. Shen, X. Li, R. Zhang and B. Wang, Fuel, 2024, 357, 130000 Search PubMed.
- D. G. Barton, A. Bhan, P. Deshlahra, R. Gounder, D. Hibbitts, B. A. Kilos, G. Noh, J. M. Notestein, M. L. Sarazen and S. L. Soled, ACS Catal., 2024, 14, 10583–10613 Search PubMed.
- T. N. Dharmapriya, K.-Y. A. Lin and P.-J. Huang, Energy Fuels, 2024, 38, 16834–16852 Search PubMed.
- P. Thyssen, Found. Chem., 2024, 26, 225–253 Search PubMed.
- W. Cai, C. Wang, Y. Chu, M. Hu, Q. Wang, J. Xu and F. Deng, Nat. Commun., 2024, 15, 8736 Search PubMed.
- V. V. Butova, V. R. Zdravkova, O. A. Burachevskaia, I. E. Gorban, M. A. Soldatov and K. I. Hadjiivanov, Microporous Mesoporous Mater., 2025, 382, 113372 Search PubMed.
- L. Zhu, H. Yang, T. Xu, F. Shen and C. Si, Nano-Micro Lett., 2025, 17, 1–45 Search PubMed.
- M. Dehghani, M. Asghari, A. F. Ismail and A. H. Mohammadi, J. Mol. Liq., 2017, 242, 404–415 Search PubMed.
- S. L. Suib, J. Přech, E. Szaniawska and J. i. Čejka, Chem. Rev., 2022, 123, 877–917 Search PubMed.
- H. Y. Luo, J. D. Lewis and Y. Román-Leshkov, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 663–692 Search PubMed.
- G. J. Gomes, M. F. Zalazar, C. A. Lindino, F. R. Scremin, P. R. Bittencourt, M. B. Costa and N. M. Peruchena, Microporous Mesoporous Mater., 2017, 252, 17–28 Search PubMed.
- C.-M. Wang, Y.-D. Wang, Y.-J. Du, G. Yang and Z.-K. Xie, Catal. Sci. Technol., 2015, 5, 4354–4364 Search PubMed.
- M. F. Zalazar, E. N. Paredes, G. D. Romero Ojeda, N. D. Cabral and N. M. Peruchena, J. Phys. Chem. C, 2018, 122, 3350–3362 Search PubMed.
- S. Soheili and A. N. Pour, Phys. Chem. Chem. Phys., 2024, 26, 5226–5236 Search PubMed.
| This journal is © The Royal Society of Chemistry 2025 |