
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment and validation of an environmentally friendly fluorescence quenching method for linagliptin quantification using eosin Y: optimization via design of experiment and comparative greenness assessment†
Saud Alqahtania,
Ali Alqahtania,
Taha Alqahtania,
Adel Al Fateaseb and
Ahmed A. Almrasy *c
*c
aDepartment of Pharmacology, College of Pharmacy, King Khalid University, Abha, 62529, Saudi Arabia
bDepartment of Pharmaceutics, College of Pharmacy, King Khalid University, Abha, 62529, Saudi Arabia
cPharmaceutical Analytical Chemistry Department, Faculty of Pharmacy, Al-Azhar University, Cairo 11751, Egypt. E-mail: ahmedalialmrasy8@gmail.com
First published on 15th May 2025
Abstract
Diabetes management has increasingly relied on dipeptidyl peptidase-4 inhibitors like linagliptin, creating a need for environmentally sustainable analytical methods to replace conventional chromatographic techniques that often involve complex sample preparation, organic solvent usage, and expensive instrumentation. A sensitive and selective “turn-off” fluorescence quenching method was developed and validated for the determination of linagliptin using eosin Y as the fluorescent probe. The spectral characteristics and sensing mechanisms were investigated using Stern–Volmer analysis, Job's method, and thermodynamic studies, revealing a static quenching process driven by the formation of a non-fluorescent 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 linagliptin–eosin Y complex with a high Stern–Volmer constant (Ksv = 6.46 × 105 M−1). The influencing factors, including pH, buffer volume, eosin Y concentration, and incubation time, were optimized using a Box–Behnken experimental design. A significant reduced quadratic regression model was established, and the optimal conditions were found to be pH 5.25, buffer volume of 1 mL, eosin Y volume of 1.25 mL, and an incubation time of 5 min based on desirability function analysis that maximizes the quenching efficiency. The developed method demonstrated linearity in the range of 0.1–3.0 μg mL−1 with a correlation coefficient of 0.9999, a limit of detection of 0.03 μg mL−1, and accuracy of 99.59 ± 1.360%, in accordance with ICH guidelines. Selectivity was confirmed by the lack of interference from common pharmaceutical excipients and endogenous plasma components. The eosin Y-based fluorescence quenching method was successfully applied for the determination of linagliptin in pharmaceutical dosage forms and spiked human plasma samples. Statistical comparison of the proposed method with the reported HPLC-UV method revealed comparable analytical performance as evident by non-significant differences in the accuracy and precision profiles as well as interval equivalence testing. Furthermore, a comprehensive assessment of the environmental impact and analytical practicality of the proposed method was conducted, confirming its “green” and “blue” analytical profile. These findings establish the eosin Y-based fluorescence quenching method as a viable and environmentally friendly alternative for the routine analysis of linagliptin in various pharmaceutical and bioanalytical applications shedding light on the potential of spectrofluorometric techniques in green analytical chemistry and bioanalysis.
:1 linagliptin–eosin Y complex with a high Stern–Volmer constant (Ksv = 6.46 × 105 M−1). The influencing factors, including pH, buffer volume, eosin Y concentration, and incubation time, were optimized using a Box–Behnken experimental design. A significant reduced quadratic regression model was established, and the optimal conditions were found to be pH 5.25, buffer volume of 1 mL, eosin Y volume of 1.25 mL, and an incubation time of 5 min based on desirability function analysis that maximizes the quenching efficiency. The developed method demonstrated linearity in the range of 0.1–3.0 μg mL−1 with a correlation coefficient of 0.9999, a limit of detection of 0.03 μg mL−1, and accuracy of 99.59 ± 1.360%, in accordance with ICH guidelines. Selectivity was confirmed by the lack of interference from common pharmaceutical excipients and endogenous plasma components. The eosin Y-based fluorescence quenching method was successfully applied for the determination of linagliptin in pharmaceutical dosage forms and spiked human plasma samples. Statistical comparison of the proposed method with the reported HPLC-UV method revealed comparable analytical performance as evident by non-significant differences in the accuracy and precision profiles as well as interval equivalence testing. Furthermore, a comprehensive assessment of the environmental impact and analytical practicality of the proposed method was conducted, confirming its “green” and “blue” analytical profile. These findings establish the eosin Y-based fluorescence quenching method as a viable and environmentally friendly alternative for the routine analysis of linagliptin in various pharmaceutical and bioanalytical applications shedding light on the potential of spectrofluorometric techniques in green analytical chemistry and bioanalysis.
1. Introduction
Fluorescence spectroscopy has been widely recognized as a powerful analytical technique due to its high sensitivity, selectivity, and ease of use.1–3 Among the different fluorescent probes available, eosin Y has gained significant attention for its ability to act as an “on-off” sensitive probe, making it a versatile tool for the detection and quantification of various analytes, including pharmaceutical compounds.4–6 The probe is a xanthine dye with four bromide atoms attached to the fluorescein backbone, which gives it unique spectroscopic properties.7 In certain pH ranges, eosin Y exists as dianion which can interact with positively charged molecules through electrostatic interactions, resulting in fluorescence quenching.8 Such quenching phenomena can be exploited for the determination of many drugs particularly those with basic moieties maximizing the analytical potential of the technique.9 Another advantage of eosin Y is its high-water solubility, making it suitable for aqueous-based analytical methods making a significant contribution towards the development of green analytical chemistry.10 In fact, the greenness of this approach not only rises from the aqueous nature of the probe but also from the low energy requirements of the technique and the minimal waste generated during analysis.11Diabetes mellitus, a chronic metabolic disorder characterized by elevated blood glucose levels, can lead to serious complications including cardiovascular disease, kidney failure, and nerve damage.12 The International Diabetes Federation reported in 2021 approximately 537 million adults aged 20–79 years were living with diabetes, which is about 1 in 10 individuals.13 Linagliptin, a dipeptidyl peptidase-4 inhibitor, has become an important therapeutic option for diabetes management due to its ability to maintain normal blood glucose levels by preventing incretin hormone breakdown.14 With its favorable pharmacokinetic profile and low hypoglycemia risk,15 linagliptin's growing clinical use necessitates reliable analytical methods for its quantification. Developing sensitive, selective, and environmentally friendly techniques for linagliptin determination is therefore crucial for both pharmaceutical quality control and clinical applications, including therapeutic drug monitoring and pharmacokinetic studies.
A literature survey reveals that linagliptin determination methods are predominantly chromatographic techniques such as HPLC-UV16,17 and LC-MS/MS.18–20 These approaches, while effective, present several practical limitations. HPLC-UV methods typically require significant organic solvent consumption (often 5–10 mL per sample), with typical methods using acetonitrile-based mobile phases at flow rates of 1 mL min−1 for run times of 8–12 min. In addition, LC-MS/MS methods, while more sensitive, involve sophisticated instrumentation and substantial maintenance requirements, limiting their accessibility in resource-constrained laboratories. However, few fluorescent and spectrophotometric methods have been reported for linagliptin determination, either individually or in combination with other antidiabetic drugs. While spectrophotometric methods offer simplicity and cost-effectiveness, they frequently lack the sensitivity needed for accurate determination of linagliptin at low concentrations.21–23 The reported fluorescence-based techniques, on the other hand, primarily rely on derivatization step to produce fluorescent species such as o-phthalaldehyde,24 ninhydrin25 and NBD-Cl,26,27 making the analysis complex and potentially less environmentally friendly. For instance, Abu-hassan et al. developed a spectrofluorimetric method using o-phthalaldehyde as a derivatizing agent to form an isoindole fluorophore with strong fluorescence.24 Despite its sensitivity, this method requires derivatization steps, increasing sample preparation and analysis time. Additionally, the isoindole fluorophore emits in the blue region (λmax = 443 nm), potentially facing interference from endogenous components in biological samples. The NBD-Cl derivative developed by Elmasry et al. was reported to have better sensitivity compared to the OPA derivative, but it still involved a derivatization step and heating to 70 °C for 25 min further limiting the applicability and environmental friendliness of the procedure.26 These approaches present several environmental concerns that warrant consideration. First, derivatization procedures necessitate additional chemical reagents, which are typically dissolved in organic solvents, thereby increasing overall solvent consumption and waste generation. Second, multi-step derivatization processes inherently produce more chemical waste compared to direct detection methods. Besides, many derivatization reactions require elevated temperatures to proceed efficiently; for instance, NBD-Cl derivatization typically requires heating significantly increasing energy consumption. Moreover, certain derivatizing agents such as NBD-Cl possess intrinsic toxicity profiles that raise safety and environmental concerns. Finally, the extended sample preparation times associated with derivatization procedures increase the overall resource utilization per analysis.
Given these limitations, this study aims to develop a novel, sensitive, and environmentally friendly fluorescence-based analytical method for linagliptin determination using eosin Y as a fluorescence probe. The fluorescence quenching mechanism will be investigated using Stern–Volmer analysis, Job's method, and thermodynamic studies. Critical factors affecting the quenching process such as pH, buffer volume, eosin Y volume, and incubation time will be optimized using Box–Behnken experimental design to maximize analytical performance. The developed method will undergo validation according to International Council for Harmonization guidelines for linearity, sensitivity, accuracy, precision, robustness, and selectivity, ensuring reliability for practical applications. The method's real-world applicability will be demonstrated by analyzing linagliptin in pharmaceutical dosage forms and spiked human plasma samples. Finally, the method's greenness and blueness will be assessed compared to existing literature using the analytical greenness (AGREE),28 modified green analytical procedure index (MOGAPI),29 and blue applicability grade index (BAGI)30 tools, highlighting its environmental and analytical advantages.
2. Experimental
2.1. Materials and reagents
Linagliptin reference standard (98.75% purity) was procured from the Egyptian Drug Authority (EDA), Cairo, Egypt. The fluorescent probe eosin Y (2′,4′,5′,7′-tetrabromofluorescein disodium salt, ≥99% purity) was acquired from Sigma-Aldrich (St. Louis, MO, USA). To minimize potential fluorescent interference during analysis, HPLC-grade ethanol and acetonitrile were sourced from Sigma-Aldrich. The commercial pharmaceutical formulation Trajenta® tablets (containing 5 mg linagliptin per tablet) was purchased from a local pharmacy in Cairo, Egypt. Analytical-grade distilled water was utilized throughout all experimental procedures.Britton–Robinson universal buffer solutions were prepared fresh prior to each analytical session by combining precisely measured equimolar (0.04 M) solutions of orthophosphoric acid (H3PO3), boric acid (H3BO3), and glacial acetic acid (CH3COOH). The buffer pH was carefully adjusted to target values (pH range 3.0–7.0) using standardized 0.2 M sodium hydroxide solution, with pH confirmation performed before each analysis series. A 0.01% eosin Y working solution was freshly prepared daily in distilled water to maintain optimal fluorescent properties and solution stability.
Human blood samples were collected from healthy adult volunteers (n = 6, age 25–40 years, non-diabetic) who had not taken any medication for at least two weeks prior to collection. The study protocol was approved by the Ethics Committee of the Faculty of Medicine, Al-Azhar University, Damietta branch (approval no.: DFM-IRB00012367-25-02-056), and written informed consent was obtained from all donors. Blood samples were collected in EDTA-containing tubes, centrifuged at 2054 × g for 10 min at 4 °C to separate plasma, and stored at −20 °C until analysis.
2.2. Instrumentation
All fluorescence measurements were performed using a Jasco FP-6200 spectrofluorometer featuring a high-sensitivity 150 W xenon lamp and temperature-controlled sample compartment. Samples were analyzed in 1 cm path length quartz cells (four-sided polished) to ensure maximum optical clarity. Instrument parameters were optimized with excitation and emission slit widths set at 10 nm, scan speed at 4000 nm min−1, and photomultiplier tube voltage at medium sensitivity. The excitation wavelength was fixed at 305 nm (corresponding to eosin Y's maximum excitation), and emission spectra were collected from 475 to 650 nm to capture the complete fluorescence profile. Data acquisition and spectral processing were managed through Spectra Manager II software. Solution pH measurements were conducted using a calibrated Jenway 3510 pH meter equipped with a combination glass electrode (precision ± 0.01 pH units).2.3. Procedure
| Quenching efficiency (%) = [(F0 − F)/F0] × 100 | (1) |
The fluorescence emission spectra were recorded from 475 to 650 nm using an excitation wavelength of 305 nm. The fluorescence intensity at the emission maximum (544 nm) was measured for quantitative analysis. A blank solution containing all reagents except linagliptin was prepared simultaneously and measured under identical conditions to obtain the reference fluorescence intensity (F0) for quenching calculations.
:water, 50:50 v/v) was prepared and serially diluted to obtain working standards at seven concentration levels. Each concentration was analyzed in triplicate following the general procedure. The calibration curve was constructed by plotting the fluorescence quenching ratio (F0/F) against linagliptin concentration. Linearity was evaluated through calculation of the correlation coefficient (r2), y-intercept and slope.Accuracy was assessed through recovery studies at three concentration levels (0.3, 1.5, and 2.5 μg mL−1), analyzed in triplicate. The percentage recovery was calculated using the formula:
| Recovery (%) = [amount found/amount added] × 100 | (2) |
The robustness of the analytical method was investigated by deliberately introducing small variations (±0.1 pH units, ±0.1 mL buffer volume, ±0.05 mL eosin Y volume) in the optimized parameters. The effect of these variations on recovery and % RSD was evaluated, with acceptance criteria of recovery within 98–102% and RSD < 2%. The selectivity of the proposed method was investigated by analyzing different pharmaceutical excipients, and potential interfering species expected to be present in the analyzed samples at 10-fold excess concentration compared to the analyte. In addition, pooled plasma samples were analyzed to confirm the lack of interference from endogenous compounds.
2.4. Application to pharmaceutical dosage forms and spiked plasma
Ten Trajenta® tablets (each containing 5 mg linagliptin) were accurately weighed and finely powdered in a clean, dry mortar. An amount of powder equivalent to 5 mg linagliptin was precisely weighed, transferred to a 50 mL volumetric flask, and dissolved in 20 mL ethanol:water (50:50 v/v) solution. The mixture was sonicated for 15 min to ensure complete extraction of the active ingredient, then diluted to volume with the same solvent mixture. The resulting solution was filtered through a 0.45 μm membrane filter. Appropriate aliquots of the filtrate were further diluted to obtain final concentrations within the validated linearity range (0.1–3.0 μg mL−1) and analyzed following the general analytical procedure. Statistical comparison of the obtained results was performed against a reported HPLC-UV method using Student's t-test, F-test and interval hypothesis testing to assess for any significant differences in accuracy, precision and equivalence between the two methods.
For the preparation of spiked plasma samples, different aliquots of linagliptin standard solution were added to 2 mL drug-free plasma to obtain final concentrations of 2, 5, 15 and 25 μg mL−1. The spiked samples were vortexed for 30 s to ensure homogeneity. Protein precipitation was performed by adding 2 mL of acetonitrile to each 1 mL of spiked plasma. The mixtures were vortexed for 1 min and then centrifuged at 2827 × g for 15 min at 4 °C. The clear supernatants were carefully transferred to clean test tubes and evaporated to dryness under a gentle stream of nitrogen. The dried residues were reconstituted with 2 mL ethanol:water (50:50 v/v) solution, vortexed for 30 s, and filtered through 0.22 μm syringe filters. One mL of each of the filtered solutions was transferred to 10 mL volumetric flasks and analyzed according to the general analytical procedure. The plasma calibration curve was constructed by spiking drug-free plasma with known concentrations of linagliptin (0.1–3.0 μg mL−1), followed by the extraction procedure described above.
3. Results and discussion
3.1. Spectral properties of the eosin Y–linagliptin complex
Eosin Y, a tetrabromofluorescein derivative, demonstrates characteristic fluorescence properties influenced by solution pH. This xanthene dye exhibits two distinct pKa values (2.0 and 3.8) that govern its ionization state. At pH values above 3.8, eosin Y predominantly exists in its dianionic form, where the extended conjugated π-system, enhanced by the electron-withdrawing bromine substituents, creates a highly fluorescent molecular structure. Under the optimized conditions of our study, eosin Y displayed strong fluorescence emission at 544 nm when excited at 305 nm (Fig. 1A). This pronounced emission results from efficient radiative transitions facilitated by the rigid xanthene scaffold and the electronic influence of the bromine atoms, with additional contribution from the resonance effects of the carboxylate group.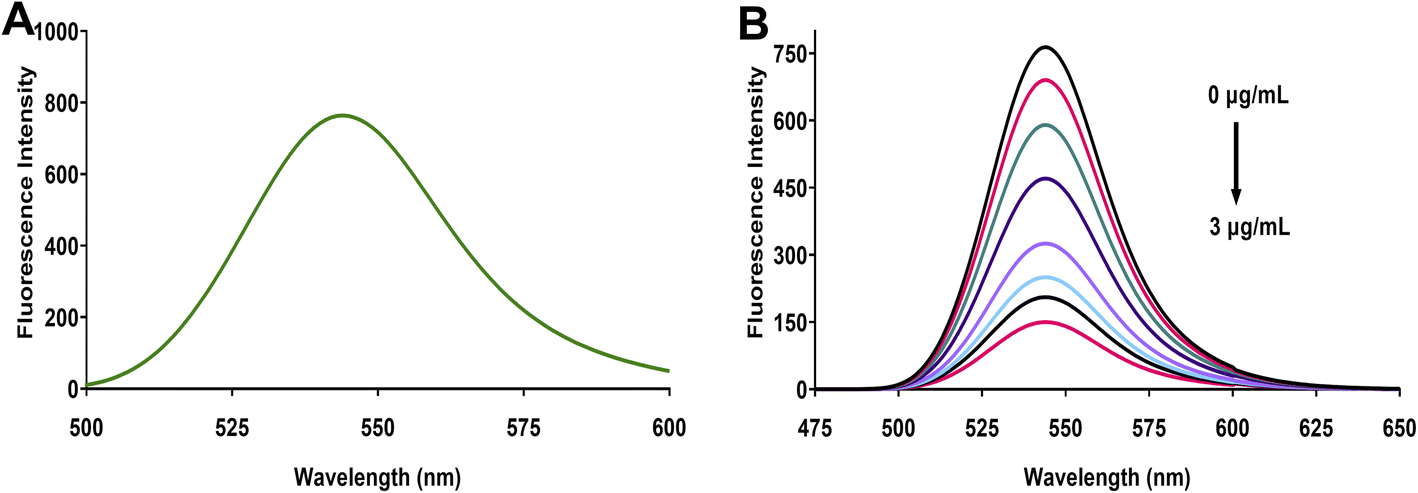 | ||
| Fig. 1 Spectrofluorimetric characteristics of the eosin Y–linagliptin system. (A) Emission spectrum of eosin Y (final concentration 0.00125% w/v, λex = 305 nm, λem = 544 nm) under optimized conditions (pH 5.25). (B) Concentration-dependent fluorescence quenching of eosin Y (final concentration 0.00125% w/v) upon addition of increasing concentrations of linagliptin (0–3 μg mL−1), demonstrating the “turn-off” sensing mechanism. | ||
When linagliptin is introduced to the eosin Y solution, a concentration-dependent fluorescence quenching is observed (Fig. 1B). The fluorescence intensity decreases progressively with increasing linagliptin concentration (from 0 to 3 μg mL−1), while the emission maximum remains unchanged at 544 nm. This quenching phenomenon can be explained by examining the molecular structures involved in the interaction (Fig. 2). At the optimized pH of 5.25, the protonated piperidine amino group of linagliptin interacts electrostatically with the negatively charged carboxylate group of eosin Y, forming an ion-pair complex. This interaction disrupts the electronic distribution within the xanthene chromophore of eosin Y, resulting in the deactivation of radiative transitions and consequent suppression of the fluorescence emission. The consistent relation between the degree of fluorescence quenching and linagliptin concentration provides the fundamental basis for developing a sensitive and selective spectrofluorimetric analytical method for linagliptin determination.
 | ||
| Fig. 2 Proposed mechanism of fluorescence quenching interaction between linagliptin and eosin Y. Based on Stern–Volmer analysis, Job's method, and thermodynamic studies, a hypothetical binding model is presented showing the formation of a 1:1 ion-pair complex at pH 5.25, where the protonated piperidine amino group of linagliptin potentially interacts electrostatically with the negatively charged carboxylate group of eosin Y, possibly disrupting the fluorophore's electronic distribution and causing fluorescence suppression. | ||
3.2. Fluorescence quenching mechanism studies
To comprehensively elucidate the interaction between eosin Y and linagliptin, multiple complementary analytical approaches have been employed including Stern–Volmer analysis, thermodynamic studies, and Job's method of continuous variation.Stern–Volmer analysis demonstrated a strong linear relationship between relative fluorescence intensity (F0/F) and linagliptin concentration, yielding a Stern–Volmer quenching constant (Ksv) of 6.46 × 105 M−1. This remarkably high constant suggested a static quenching mechanism involving ground-state complex formation rather than dynamic collisional processes. To confirm this hypothesis, the bimolecular quenching rate constant (kq) was calculated using the equation kq = Ksv/τ0, where τ0 represents the intrinsic fluorescence lifetime of eosin Y (1.43 ns).31 The calculated kq value of 4.52 × 1014 M−1 s−1 exceeds the diffusion-controlled limit (2 × 1010 M−1 s−1) by four orders of magnitude, definitively establishing static quenching as the predominant mechanism.
The strength of molecular interaction between eosin Y and linagliptin was quantified through the association constant (Ka), determined using the modified Stern–Volmer equation:
| F0/(F0 − F) = 1 + 1/(Ka[Q]) | (3) |
Linear regression analysis of F0/F versus 1/[Q] yielded an association constant of 6.34 × 105 M−1, indicating robust complex formation. This value aligned closely with our previously determined Stern–Volmer constant, providing internal consistency to our findings. The thermodynamic favorability of the interaction was assessed by calculating the Gibbs free energy change (ΔG°) using the relationship:
|
ΔG° = −RTlnKa
| (4) |
The calculated ΔG° value of −33.15 kJ mol−1 confirmed that complex formation occurs spontaneously under our experimental conditions.
To determine the binding stoichiometry, Job's method of continuous variation was employed where equal molar concentrations (6 × 10−5 M) of eosin Y and linagliptin were mixed in varying proportions while maintaining constant total volume. The difference in fluorescence intensity between the sample and the blank was plotted versus mole fraction of linagliptin, revealing a maximum at 0.5 mole fraction (Fig. 3). This observation indicates a 1:1 binding stoichiometry between eosin Y and linagliptin, consistent with the proposed ion-pair complex formation. This finding was independently verified using the double logarithmic modified Stern–Volmer equation:
|
log[(F0 − F)/F] = logKb + nlog[Q]
| (5) |
 | ||
| Fig. 3 Job's plot for the interaction between eosin Y and linagliptin showing the change in fluorescence intensity (ΔF) versus mole fraction of linagliptin. Equal molar concentrations (6 × 10−5 M) of eosin Y and linagliptin stock solutions were mixed in varying proportions while maintaining a constant total volume. Error bars represent standard deviations from triplicate independent determinations. The maximum at 0.5 mole fraction (indicated by green dashed line) confirms 1:1 binding stoichiometry between eosin Y and linagliptin. | ||
The slope of the linear plot (1.01) conclusively confirmed the 1:1 binding stoichiometry between eosin Y and linagliptin molecules. Collectively, these mechanistic investigations provide compelling evidence for the formation of a thermodynamically stable 1:1 ground-state complex between eosin Y and linagliptin, driven primarily by electrostatic interactions between the protonated amino group of linagliptin and the anionic carboxylate moiety of eosin Y, as illustrated in Fig. 2. However, the specific binding sites and exact nature of the molecular interactions illustrated represent the best hypothesis based on the molecular structures involved and the pH-dependence of the quenching effect. Further structural studies, such as NMR or computational modeling, would be required to definitively confirm these specific binding interactions.
3.3. Optimization of the sensing system
The sensing performance of the eosin Y–linagliptin system was systematically optimized using a Box–Behnken experimental design to evaluate the influence of four critical parameters: pH, buffer volume, eosin Y volume, and incubation time. A total of 27 experimental runs were performed (Table S1†), and the QE% (percent quenching efficiency) was chosen as the response variable to be maximized. The data was fitted to a second-order polynomial regression model, followed by backward elimination to identify the significant factors. The reduced quadratic equation was:| QE% = 63.344 + 3.4441A + 4.1158B + 9.65196C + 7.84096AC − 28.4205A2 − 6.68051B2 − 11.7978C2 | (6) |
ANOVA analysis confirmed the adequacy and statistical significance of the model, with an F value of 75.90 and a p-value less than 0.0001 (Table 1). Three main effects (pH, buffer volume, and eosin Y volume) as well as one interaction effect (pH × eosin Y volume) were found to be significant (Table 1). The positive coefficients for pH, buffer volume, and eosin Y volume and the interaction effect between pH and eosin Y volume indicate that increasing their levels favors higher QE% values. On the other hand, the negative quadratic coefficients suggest the existence of an optimum level for each parameter beyond which the response diminishes. These results comes in line with main effect plots which showed that the QE% reaches a maximal response at certain optimum values of each significant factor followed by a decline upon further increase or decrease of the parameter levels (Fig. 4). The quadratic effect of pH could be attributed to the pH dependence of the ionization state of eosin Y, which significantly impacts its fluorescence properties and interaction with linagliptin (Fig. 4A). The buffer volume also shows quadratic effect as the QE% decreased at low buffer volumes, likely due to insufficient buffering capacity where at high buffer volumes, increasing the ionic strength may interfere with the ion-pair formation via competitive effects (Fig. 4B). Eosin Y volume was the most significant factor affecting the QE%, illustrating the critical role of the probe concentration in determining the sensing efficiency. At lower eosin Y volumes, the available probe molecules may be insufficient to effectively interact with linagliptin, while at higher volumes, self-quenching and interference from the excess probe can lead to lower fluorescence emission (Fig. 4C). The incubation shows no significant effect suggesting that the complexation between eosin Y and linagliptin reaches equilibrium rapidly (Fig. 4D).
| Source | Sum of squares | df | Mean square | F-Value | p-Value | |
|---|---|---|---|---|---|---|
| Model | 6709.15 | 7 | 958.45 | 75.90 | <0.0001 | Significant |
| A-pH | 142.34 | 1 | 142.34 | 11.27 | 0.0033 | |
| B-buffer volume | 203.28 | 1 | 203.28 | 16.10 | 0.0007 | |
| C-eosin Y volume | 1117.92 | 1 | 1117.92 | 88.53 | <0.0001 | |
| AC | 245.92 | 1 | 245.92 | 19.47 | 0.0003 | |
| A2 | 4846.37 | 1 | 4846.37 | 383.77 | <0.0001 | |
| B2 | 267.78 | 1 | 267.78 | 21.20 | 0.0002 | |
| C2 | 835.12 | 1 | 835.12 | 66.13 | <0.0001 | |
| Residual | 239.94 | 19 | 12.63 | |||
| Lack of fit | 228.29 | 17 | 13.43 | 2.31 | 0.3448 | Not significant |
| Pure error | 11.65 | 2 | 5.82 | |||
| Cor total | 6949.09 | 26 |
 | ||
| Fig. 4 Main effect plots showing the influence of individual parameters on quenching efficiency (QE%). (A) Effect of pH showing optimal response at approximately pH 5.25 with significant quadratic effect. (B) Effect of buffer volume with moderate quadratic relationship and optimal response around 1.0 mL. (C) Effect of eosin Y volume on quenching efficiency (QE%). Different volumes (0.5–1.5 mL) of 0.01% eosin Y solution were added to a final volume of 10 mL, showing optimal response at approximately 1.25 mL. (D) Effect of incubation time showing non-significant influence across the studied range (5–20 min). A fixed linagliptin concentration of 1.5 μg mL−1 was used for all optimization experiments. | ||
The validity of the model was confirmed by inspecting different statistical parameters (Table S2†) such as the coefficient of determination (R2 = 0.9655) and the adjusted R2 (0.9528), both indicating a high goodness-of-fit. The predicted R2 value of 0.9105 was in reasonable agreement with the adjusted R2 of 0.9528, supporting the model's predictive ability. The adequate precision ratio of 28.05 exceeded the minimum threshold of 4, further verifying the model's suitability for navigating the design space. Several diagnostic plots were generated to ensure the model assumptions were met, including normal probability plot, actual versus predicted plot, and residuals versus predicted plot, all of which confirmed the satisfactory fit of the model (Fig. S1–S3†). Inspecting the residual vs. run plot showed a random scattering of the points, indicating the absence of any systematic error or unusual observations (Fig. S4†). Also, the leverage vs. run plot showed all the data points within the acceptable range, ruling out any influential or outlier observations (Fig. S5†).
A major advantage of the Box–Behnken design is its ability to study the interaction effects between the factors using the 2D interaction (Fig. 5A) and the 3D response surface plots (Fig. 5B). The 3D response surface plot shows the combined effect of pH and eosin Y volume on the QE%, revealing a pronounced synergistic interaction. At low pH and low eosin Y volume, the QE% is suboptimal, but as both factors increase, the response shows a sharp escalation, reaching a maximum at around pH 5.25 and 1.25 of eosin Y. The curvature of the surface clearly indicates the quadratic nature of the individual effects. Desirability function optimization was employed to maximize the QE% by simultaneously optimizing the three significant factors and keeping the incubation time minimized to allow for rapid and cost-effective analysis (Fig. 5C). The optimal conditions were determined to be: pH 5.25, buffer volume 1 mL, and eosin Y volume 1.25 mL, with a predicted maximum QE% of 66.35% using 1.5 μg per mL linagliptin. These optimal conditions were subsequently employed for all further analytical validation and application studies.
 | ||
| Fig. 5 (A) 2D interaction plot demonstrating the synergistic effect between pH and eosin Y volume on quenching efficiency. (B) 3D response surface plot visualizing the combined quadratic effects of pH and eosin Y volume on QE%. (C) Desirability function analysis showing the optimal experimental conditions, yielding a predicted maximum QE% of 66.36%. | ||
3.4. Analytical method validation
Following optimization and mechanistic investigation, the developed fluorescence quenching method underwent comprehensive validation according to ICH Q2(R1) guidelines. The method demonstrated acceptable analytical performance across all evaluated parameters (Table 2).| Parameters | Linagliptin | |
|---|---|---|
| a Average of 9 determinations (3 concentrations repeated 3 times).b % RSD of 9 determinations (3 concentrations repeated 3 times) measured on the same day.c % RSD of 9 determinations (3 concentrations repeated 3 times) measured in the three consecutive days. | ||
| Excitation wavelength (nm) | 305 | |
| Emission wavelength (nm) | 544 | |
| Linearity range (μg mL−1) | 0.1–3.0 | |
| Slope | 1.3803 | |
| Intercept | 0.9575 | |
| Correlation coefficient (r2) | 0.9999 | |
| LOD (μg mL−1) | 0.0315 | |
| LOQ (μg mL−1) | 0.0946 | |
| Accuracy (% R)a | 99.59 ± 1.360 | |
| Repeatability precision (% RSD)b | 1.365 | |
| Intermediate precision (% RSD)c | 1.765 | |
| Robustness (% R) | Buffer (pH) | 98.41 ± 1.076 |
| Buffer volume (mL) | 100.52 ± 1.242 | |
| Reagent volume (mL) | 99.56 ± 1.022 | |
Selectivity assessments were carried out by analyzing the fluorescence signal of common pharmaceutical excipients and potential interferents typically encountered in linagliptin formulations and biological matrices. As shown in (Fig. 6), potential interferents, including common tablet excipients, (starch, lactose, magnesium stearate, cellulose, sodium lauryl sulfate) and biological matrix components (Na+, K+, Ca2+, Mg2+, Ni2+, SO42−, PO43−) produced negligible fluorescence quenching (<2%) compared to linagliptin (67%). Selectivity investigations were extended to clinically relevant interferents. Primary amines (glycine, alanine) showed minimal interference (<3%), while ethanolamine produced slightly higher quenching (<5%). Co-administered antidiabetics demonstrated moderate interference (empagliflozin: <9%, metformin: <8%), while structurally similar DPP-4 inhibitors exhibited the highest interference (sitagliptin: <13%, saxagliptin: <14%) as expected due to their similar molecular structures. This level of interference remains acceptable for accurate analysis. Antibiotics (ciprofloxacin: <8%, amoxicillin: <6%) and pooled plasma (<3%) showed sufficiently low interference to permit reliable determination in biological matrices. These results validate the method's selectivity in pharmaceutical formulations and biological samples containing multiple medications.
 | ||
| Fig. 6 Selectivity of the eosin Y-based fluorescence quenching method. Bar chart demonstrating the significant quenching efficiency of linagliptin (1.5 μg mL−1, 67%) compared to minimal interference from pharmaceutical excipients and ions (<2%), primary amines (<5%), antibiotics (<8%), co-administered antidiabetics (<9%), and other DPP-4 inhibitors (<14%), confirming the method's selectivity for pharmaceutical and biological analysis. All potential interferents were tested at 10-fold excess concentration (15 μg mL−1) relative to linagliptin. | ||
The validation results collectively demonstrate that the developed fluorescence quenching method using eosin Y provides a reliable, sensitive, and selective analytical tool for linagliptin determination, fulfilling all requirements for pharmaceutical quality control and bioanalytical applications.
3.5. Application to pharmaceutical formulations and spiked plasma samples
The optimized and validated fluorescence quenching method was successfully applied for the determination of linagliptin in commercial pharmaceutical preparations (Trajenta® tablets) and in spiked human plasma samples, demonstrating its practical utility for both quality control and bioanalytical applications.| Method | Meanb | SD | t-Test (2.306) c | P value | F value (6.338) b | P value | θLd | θUd |
|---|---|---|---|---|---|---|---|---|
| a The HPLC method was reproduced according to the procedure reported in ref. 16.b Average of five determinations.c The values in parenthesis are tabulated values of “t“and “F” at (P = 0.05).d Bias of ±2% is acceptable. | ||||||||
| Developed method | 100.30 | 1.344 | 0.311 | 0.764 | 1.775 | 0.592 | −1.499 | 1.967 |
| Reported method a | 100.07 | 1.009 | ||||||
Further validation of method equivalence was conducted through interval hypothesis testing as shown in (Table 3). The calculated lower (θL = −1.499) and upper (θU = 1.967) confidence limits fell within the predetermined acceptance range of ±2%, demonstrating that any differences between the methods were analytically insignificant. These comprehensive statistical analyses confirm the reliability of the proposed fluorescence quenching method for the routine quality control analysis of linagliptin in pharmaceutical dosage forms.
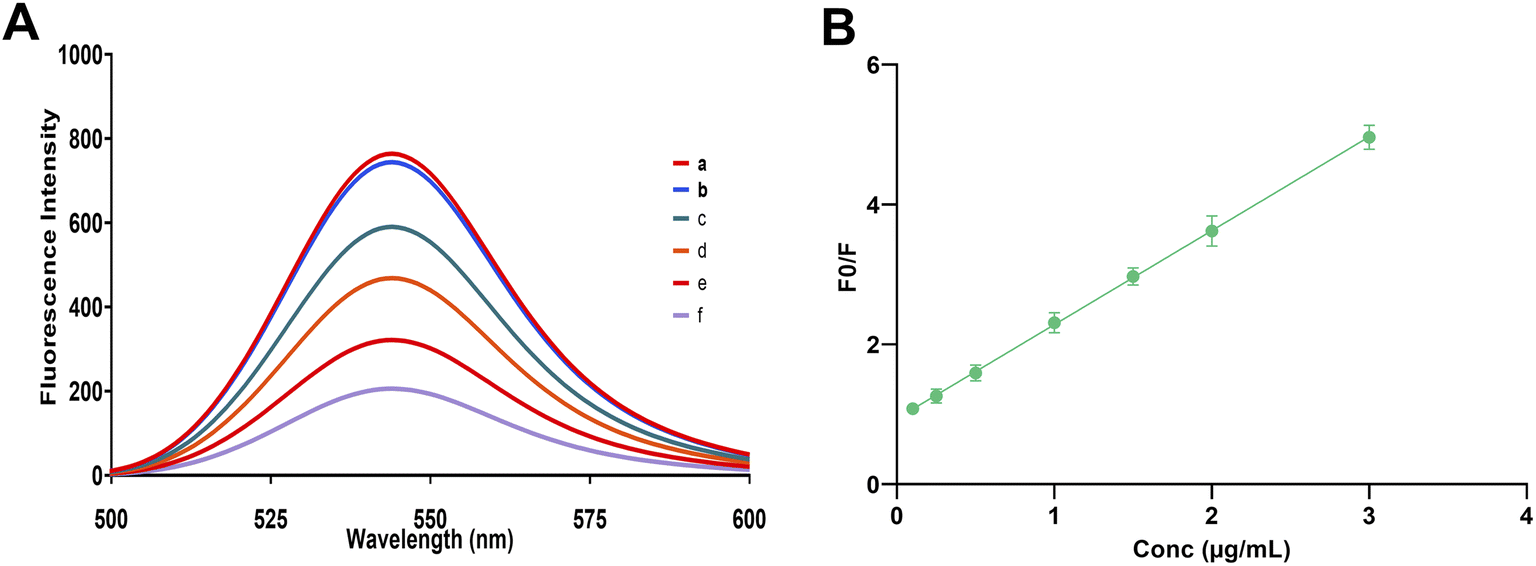 | ||
| Fig. 7 (A) Representative fluorescence emission spectra (λex = 305 nm) showing: (a) eosin Y alone, (b) eosin Y with blank plasma extract, and (c–f) eosin Y with plasma samples spiked with linagliptin at final concentrations of 0.25, 0.5, 1.0, and 2.0 μg mL−1, respectively. (B) Calibration curve showing the linear relationship between F0/F and linagliptin concentration (0.1–3.0 μg mL−1). Each point represents the mean of three independent determinations (error bars represent ±SD). | ||
The method achieved satisfactory recoveries across the concentration range of 0.20–2.00 μg mL−1 in spiked plasma samples. The mean recoveries ranged from 95.51% to 104.75%, with RSD values between 0.661% and 2.340%, indicating excellent reproducibility and accuracy in complex biological matrices (Table 4). All recovery values were within the acceptable range for bioanalytical methods (85–115%), confirming the method's reliability for potential clinical and pharmacokinetic studies.
| Spiked (μg mL−1) | Found (μg mL−1) | Recovery (%) | RSD (n = 3, %) |
|---|---|---|---|
| 0.10 | 0.096 | 95.51 | 2.136 |
| 0.20 | 0.193 | 96.55 | 1.856 |
| 0.30 | 0.312 | 104.05 | 1.862 |
| 0.50 | 0.481 | 96.25 | 0.661 |
| 1.00 | 1.048 | 104.75 | 2.340 |
| 2.00 | 1.987 | 99.36 | 1.083 |
While the method demonstrated satisfactory recovery from pooled plasma samples, it is important to acknowledge that biological matrices exhibit considerable inter-individual variability. Endogenous reductants (ascorbic acid, uric acid), amino acids (particularly cysteine, tryptophan, and tyrosine), and other plasma constituents have been reported to interact with eosin Y through various quenching mechanisms.32 The concentrations of these potential interferents fluctuate significantly between patients and can be influenced by factors such as diet, medication use, and pathophysiological conditions. This biological variability represents a limitation that should be considered when extending the method to clinical applications. For therapeutic drug monitoring or pharmacokinetic studies in diverse patient populations, matrix-matched calibration approaches or standard addition techniques may be necessary to mitigate these matrix-specific effects.
3.6. Environmental impact and analytical practicality assessment
The environmental sustainability of the developed eosin Y-based fluorescence quenching method for linagliptin determination was comprehensively evaluated using three complementary assessment tools: AGREE, MOGAPI, and BAGI (Table 5). An AGREE greenness score of 0.73 was achieved by the proposed method, which was found to be substantially higher than those obtained for conventional HPLC-UV (0.61),16 LC-MS/MS (0.60)18 and NBD-Cl derivatization fluorescence method26 (0.5) techniques (Table 5). This superior environmental performance can be attributed to several key factors: the predominant utilization of water as the primary solvent system, limited organic solvent consumption (acetonitrile being used solely for plasma protein precipitation), decreased energy requirements due to room temperature operation (versus heating at 70 °C for 25 min in the NBD-Cl method), and the inherently low power consumption of spectrofluorometers. When specific AGREE criteria were examined, particular strengths were demonstrated by the proposed method in the categories of reagent toxicity, energy consumption, and waste generation.










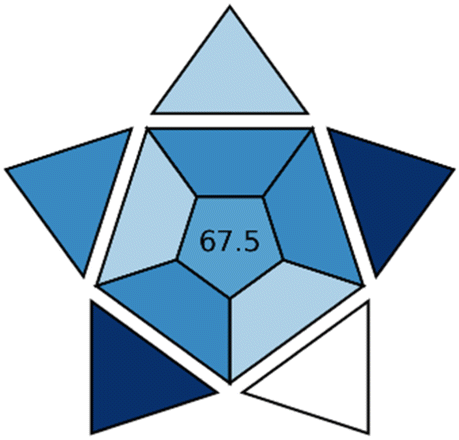
The MOGAPI assessment further confirmed these environmental benefits with a score of 83 compared to 68 for HPLC-UV, 66 for LC-MS/MS, and 68 for the NBD-Cl derivatization method (Table 5). In the pentagram analysis, predominantly green and yellow sections were revealed for the proposed approach, especially regarding reagent consumption, energy usage, and waste generation. The NBD-Cl derivatization method showed particular limitations in energy efficiency due to its heating requirements and higher reagent consumption associated with the derivatization step.
The practical applicability was assessed using BAGI, wherein a score of 77.5 was obtained for the proposed method, which was positioned between HPLC-UV (80), LC-MS/MS (75.0), and NBD-Cl fluorescence method (67.5) (Table 5). Although slightly lower than HPLC-UV, both the more sophisticated UPLC-MS/MS approach and the derivatization-based fluorescence method were outperformed by the developed direct quenching method in terms of overall practical utility. The NBD-Cl method offered better sensitivity but required longer analysis time (25 min versus 5 min) and additional derivatization steps.
Several practical advantages were demonstrated by the proposed method, including significantly reduced instrumentation costs compared to chromatographic systems, minimal maintenance requirements, rapid analysis time (approximately 5 min per sample versus 25 min for NBD-Cl method), and low reagent consumption. These practical benefits can be particularly valuable for laboratories with limited resources where sophisticated chromatographic instrumentation may not be accessible. Certain limitations were reflected in the comparative BAGI scores, primarily related to single-analyte determination capability (compared to Elmasry's method that allows simultaneous determination of linagliptin and empagliflozin) and manual sample preparation requirements. However, these limitations were counterbalanced by high analytical performance characteristics and simpler methodology.
3.7. Comparison with reported literature
The developed fluorescence-based method for linagliptin determination using eosin Y quenching offers specific advantages compared to previously reported methods. As shown in Table 6, the method provides sensitivity with LOD and LOQ values of 0.0315 and 0.0946 μg mL−1, respectively, which meets the requirements for pharmaceutical quality control and most clinical applications.| Method | Method parameters | Linearity | LOD | LOQ | Advantages | Limitations | Ref. |
|---|---|---|---|---|---|---|---|
| Proposed method (eosin Y fluorescence quenching) | λex/λem: 305/544 nm, pH 5.25, room temperature reaction | 0.1–3.0 μg mL−1 | 0.03 μg mL−1 | 0.09 μg mL−1 | No derivatization required, rapid analysis (5 min), environmentally friendly, high sensitivity | Lower sensitivity compared to derivatization methods | |
| HPLC-UV | C18 column, methanol (40:60, v/v) with 0.3% TEA, pH 4.5, flow rate 1 mL min−1, detection at 225 nm |
1.0–50 μg mL−1 | 0.3 μg mL−1 | 1.0 μg mL−1 | Stability-indicating, suitable for quality control | Longer analysis time (17 min), higher solvent consumption | [Mourad et al., 2016]16 |
| LC-MS/MS | Cyano column, 2 mM ammonium acetate buffer and acetonitrile (gradient), flow rate 0.4 mL min−1, MRM transitions 473.2 → 420.2 | 0.015–15.0 ng mL−1 | 0.015 ng mL−1 | 0.045 ng mL−1 | Highest sensitivity, rapid analysis (5 min) | Expensive instrumentation and complex extraction procedure | [Shah et al., 2019]19 |
| Spectrophotometric (PDAB) | λmax: 407 nm, heating at 70–75 °C for 35 min | 5–45 μg mL−1 | 1.58 μg mL−1 | 4.79 μg mL−1 | Simple equipment, economical | Requires heating, longer analysis time (45 min), lower sensitivity, limited to bulk powder analysis | [Sahloul & Salami, 2023]22 |
| Spectrofluorimetric (isoindole formation) | λex/λem: 337.8/434.3 nm, borate buffer pH 8.5, o-phthalaldehyde | 0.2–2.0 μg mL−1 | 0.079 μg mL−1 | 0.241 μg mL−1 | Simple procedure, no heating required | Limited to tablet analysis and content uniformity | [Abu-hassan et al., 2023]24 |
| Spectrofluorimetric (ninhydrin) | λex/λem: 385/475 nm, heating at 70–75 °C for 35 min | 20–460 ng mL−1 | 1.50 ng mL−1 | 4.50 ng mL−1 | Good sensitivity, applicable for tablets and plasma | Requires heating, longer analysis time (35 min), derivatization step | [El-Gizawy et al., 2024]25 |
| Spectrofluorimetric (NBD-Cl) | λex/λem: 469/538 nm, heating at 70 °C for 25 min, borate buffer pH 8 | 3–700 ng mL−1 | 0.884 ng mL−1 | 2.69 ng mL−1 | Allows simultaneous analysis of two drugs (linagliptin and empagliflozin) | Requires heating, longer analysis time (25 min), derivatization step | [Elmasry et al., 2021]26 |
| Spectrofluorimetric (NBD-Cl) | λex/λem: 459/529 nm, heating at 70–75 °C for 25 min, borate buffer pH 8.5 | 1.0–100 ng mL−1 | 0.60 ng mL−1 | 1.82 ng mL−1 | Good sensitivity, applicable for human plasma | Requires heating, longer analysis time (30 min), derivatization step | [Aref et al., 2020]27 |
The sensitivity of the proposed method is lower than the work reported by Elmasry et al. using the NBD-Cl derivatization approach (LOD = 0.884 ng mL−1, linear range 3–700 ng mL−1 for linagliptin) and other derivatization-based methods, as presented in Table 6. This difference in sensitivity is attributed to the inherent amplification effect achieved through derivatization, where each linagliptin molecule is converted to a highly fluorescent derivative. In contrast, the proposed method relies on a direct quenching approach that depends on the intrinsic interaction between linagliptin and eosin Y without signal amplification. Despite the lower sensitivity, the proposed method offers advantages in terms of simplicity (derivatization steps are avoided), speed (5 minutes versus 25–45 minutes including derivatization), and environmental impact (room temperature operation versus heating at 70 °C) as highlighted in Table 6. For pharmaceutical quality control and most clinical applications, the sensitivity of the method is sufficient. However, for specialized applications requiring ultrahigh sensitivity, such as pharmacokinetic studies examining terminal elimination phases, derivatization approaches might be preferred despite their procedural complexities.
The spectrophotometric method using p-dimethylaminobenzaldehyde (PDAB) demonstrates acceptable precision and accuracy, but its sensitivity (LOD: 1.58 μg mL−1) is 50 times lower compared to the spectrofluorimetric methods. Complex mobile phase compositions and specialized instrumentation are required by chromatographic methods like HPLC-UV and LC-MS/MS, making them less accessible for routine analysis in some laboratories.
The optimization of reaction conditions using Box–Behnken experimental design in the proposed method represents a systematic approach to method development, ensuring optimal sensitivity and robustness within the limitations of a non-derivatized approach. In conclusion, when comparing the analytical performance parameters, the proposed fluorescence-based method using eosin Y quenching offers a rapid (5 min analysis time), room-temperature, and environmentally friendly alternative to existing methods for linagliptin determination in pharmaceutical and biological samples.
4. Conclusion
In this study, a highly sensitive and selective “turn-off” fluorescence quenching method was developed and validated for the determination of the antidiabetic drug linagliptin using eosin Y as the fluorescent probe. The sensing mechanism was elucidated through Stern–Volmer analysis, Job's method, and thermodynamic studies, revealing a static quenching process driven by the formation of a 1:1 non-fluorescent linagliptin–eosin Y complex. The influencing factors, including pH, buffer volume, eosin Y concentration, and incubation time, were optimized using a Box–Behnken experimental design, leading to enhanced analytical performance. The method demonstrated excellent linearity in the range of 0.1–3.0 μg mL−1, with a low limit of detection of 0.03 μg mL−1, and satisfactory accuracy and precision, in accordance with ICH guidelines. The developed fluorescence quenching method was successfully applied for the determination of linagliptin in pharmaceutical dosage forms and spiked human plasma samples, with reliable recovery and reproducibility. Furthermore, a comprehensive evaluation of the environmental impact and analytical practicality of the proposed method was conducted using the AGREE, MOGAPI, and BAGI assessment tools. The results confirmed the “green” and “blue” analytical profile of the developed fluorescence-based approach, which exhibited superior environmental sustainability and practical utility compared to conventional HPLC-UV and LC-MS/MS techniques. These findings establish the eosin Y-based fluorescence quenching method as a viable and environmentally friendly alternative for the routine analysis of linagliptin in various pharmaceutical and bioanalytical applications.
While several advantages are offered by the developed ‘turn-off’ fluorescence quenching method, its inherent limitations must be acknowledged. Unlike ‘turn-on’ systems, fluorescence reduction can potentially be caused by factors other than analyte–probe interaction, including environmental changes (particularly pH variations) or non-specific quenching mechanisms. These concerns were addressed through multiple control measures: measurements were performed against reagent blanks, pH was strictly controlled (±0.05 units), and selectivity was assessed with structurally diverse compounds at concentrations exceeding linagliptin by 10-fold. Additional limitations include the need for manual sample preparation and the single-analyte determination capability, which may be addressed through future investigations. Background interference in plasma samples due to incomplete quenching and matrix autofluorescence necessitated significant sample dilution, which may limit sensitivity compared to some ‘turn-on’ methodologies. Furthermore, the applicability of the method to other antidiabetic drugs or complex biological samples warrants further exploration. However, the overall results demonstrate the analytical and environmental merits of the developed fluorescence-based approach, offering a promising solution for the sensitive and sustainable determination of linagliptin particularly in resource-limited settings.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI file.†Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors extend their appreciation to the Deanship of Research and Graduate Studies at King Khalid University for funding this work through large groups program under grant number RGP2/384/46.References
- J. Yao, M. Yang and Y. Duan, Chem. Rev., 2014, 114, 6130–6178 CrossRef CAS PubMed.
- X. Zhang, J. Yin and J. Yoon, Chem. Rev., 2014, 114, 4918–4959 CrossRef CAS PubMed.
- M. Dutta and D. Das, TrAC, Trends Anal. Chem., 2012, 32, 113–132 CrossRef CAS.
- S. M. Derayea and D. M. Nagy, Rev. Anal. Chem., 2018, 37, 20170020 CAS.
- M. A. Abdel-Lateef, I. A. Darwish, H. Gomaa and N. S. Katamesh, J. Fluoresc., 2024, 1–12 Search PubMed.
- A. Alqahtani, T. Alqahtani, A. Alshehri and A. A. Almrasy, Luminescence, 2025, 40, e70076 CrossRef CAS.
- M. A. Omar, A. B. Ahmed, N. S. Abdelwahab, M. M. Abdelrahman and S. M. Derayea, Spectrochim. Acta, Part A, 2020, 236, 118337 CrossRef CAS.
- K. Kaur and A. K. Malik, J. Fluoresc., 2013, 23, 533–542 CrossRef CAS PubMed.
- H. Ahmed, Y. El-Shabrawy, A. Barseem and F. Belal, Microchem. J., 2019, 149, 104054 CrossRef CAS.
- A. Roshdy, H. Elmansi, S. Shalan and A. Elbrashy, Luminescence, 2020, 35, 788–796 CrossRef CAS.
- A. Gałuszka, Z. Migaszewski and J. Namieśnik, TrAC, Trends Anal. Chem., 2013, 50, 78–84 CrossRef.
- D. M. Nathan, Jama, 2015, 314, 1052–1062 CrossRef CAS PubMed.
- H. Sun, P. Saeedi, S. Karuranga, M. Pinkepank, K. Ogurtsova, B. B. Duncan, C. Stein, A. Basit, J. C. N. Chan, J. C. Mbanya, M. E. Pavkov, A. Ramachandaran, S. H. Wild, S. James, W. H. Herman, P. Zhang, C. Bommer, S. Kuo, E. J. Boyko and D. J. Magliano, Diabetes Res. Clin. Pract., 2022, 183, 109119 CrossRef PubMed.
- E. D. Deeks, Drugs, 2012, 72, 1793–1824 CrossRef CAS.
- P. P. Toth, Postgrad. Med., 2011, 123, 46–53 CrossRef PubMed.
- S. S. Mourad, E. I. El-Kimary, D. A. Hamdy and M. A. Barary, J. Chromatogr. Sci., 2016, 54, 1560–1566 CAS.
- J. C. Rajbangshi, M. M. Alam, M. S. Hossain, M. S. Islam and A. S. S. Rouf, Dhaka Univ. J. Pharm. Sci., 2018, 17, 175–182 CrossRef CAS.
- B. A. Moussa, M. A. Mahrouse and M. G. Fawzy, J. Pharm. Biomed. Anal., 2019, 163, 153–161 CrossRef.
- P. A. Shah, P. S. Shrivastav and A. George, Microchem. J., 2019, 145, 523–531 CrossRef CAS.
- A. K. G. Thallapalli and R. M. Manda, Anal. Sci., 2024, 40, 185–198 CrossRef CAS.
- R. I. El-Bagary, E. F. Elkady and B. M. Ayoub, Int. J. Biomed. Sci., 2013, 9, 41 CrossRef CAS.
- L. Sahloul and M. Salami, Sci. Rep., 2023, 13, 4083 CrossRef CAS.
- A. H. Kamal, S. F. Hammad, M. M. Salim, M. M. Elkhodary and A. A. Marie, Spectrochim. Acta, Part A, 2024, 305, 123556 CrossRef CAS.
- A. A. Abu-hassan, M. A. El Hamd, M. H. El-Maghrabey, W. A. Mahdi, S. Alshehri and B. Shaaban Mohammed, Spectrochim. Acta, Part A, 2023, 291, 122390 CrossRef CAS PubMed.
- S. M. El-Gizawy, N. N. Atia, D. H. Rushdy and M. F. B. Ali, Luminescence, 2024, 39, e4609 CrossRef CAS.
- M. S. Elmasry, M. A. Hasan, W. S. Hassan, H. A. Merey and I. M. Nour, Spectrochim. Acta, Part A, 2021, 248, 119258 CrossRef CAS.
- H. A. Aref, S. F. Hammad, M. S. Elgawish and K. M. Darwish, Luminescence, 2020, 35, 626–635 CrossRef CAS.
- F. Pena-Pereira, W. Wojnowski and M. Tobiszewski, Anal. Chem., 2020, 92, 10076–10082 CrossRef CAS.
- F. R. Mansour, J. Płotka-Wasylka and M. Locatelli, Analytica, 2024, 5, 451–457 CrossRef.
- N. Manousi, W. Wojnowski, J. Płotka-Wasylka and V. Samanidou, Green Chem., 2023, 25, 7598–7604 RSC.
- E. A. Slyusareva and M. A. Gerasimova, Russ. Phys. J., 2014, 56, 1370–1377 CrossRef CAS.
- M. H. Mahnashi, A. M. Mahmoud, M. M. El-Wekil and R. Y. Shahin, Microchem. J., 2023, 193, 109062 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01945a |
| This journal is © The Royal Society of Chemistry 2025 |