
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and structure–activity relationship studies of teixobactin analogues†
Chunlei Wu‡
,
Zhengyin Pan‡,
Guiyang Yao,
Wei Wang,
Lijing Fang* and
Wu Su *
*
Guangdong Key Laboratory of Nanomedicine, Institute of Biomedicine and Biotechnology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, 1068 Xueyuan Boulevard, Shenzhen 518055, China. E-mail: lj.fang@siat.ac.cn; wu.su@siat.ac.cn
First published on 12th January 2017
Abstract
A new series of teixobactin analogues were synthesized via an oxidative cyclative cleavage approach using aryl hydrazide resin as the solid support. Structure–activity relationship studies revealed that the guanidine or amine group at position 10, the hydroxyl group of Ser7 residue and the NH proton of the N-terminal Phe1 residue are critical to the antibacterial activities, while side chain size and functional group changes are tolerated at position 4. These findings will facilitate the development of new teixobactin analogues with enhanced pharmacological properties.
The antibiotics crisis derived from increasing bacterial resistance to existing antibiotic drugs has highlighted the need for the development of new classes of antibacterial agents.1–3 One molecule that has received great attention is teixobactin (1, Fig. 1), a peptidyl antibiotic discovered by Ling and co-workers in a screen of uncultured bacteria utilizing iChip technology in 2015.4 Teixobactin exhibits good activity against a wide range of Gram-positive pathogens as well as Mycobacterium tuberculosis (Mtb) including drug-resistant strains. Notably, resistance is less likely to develop against teixobactin which inhibits bacterial cell wall biosynthesis by binding to a highly conserved motif of lipid II and lipid III, two key precursors of bacterial cell-wall polymers (peptidoglycan and teichoic acid, respectively). As a consequence of its potent antibacterial activity and distinct mechanism of action, teixobactin is regarded as a very promising antibiotic candidate for the treatment of Gram-positive and Mtb infections.5
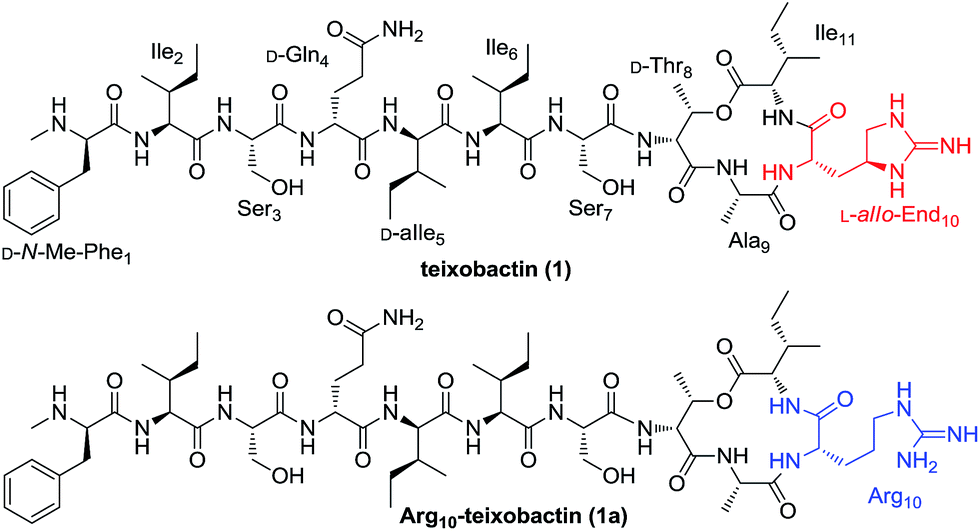 | ||
| Fig. 1 Structures of teixobactin (1) and Arg10-teixobactin (1a). | ||
Teixobactin is a side-to-tail cyclized depsipeptide possessing a 13-membered macrocycle. Apart from D-amino acids and N-Me-Phe, the guanidine-based uncommon amino acid L-allo-enduracididin (L-allo-End) is present in this unique undeca-peptide. Recently, the total synthesis of teixobactin was achieved by two independent groups employing a straightforward solid-phase synthetic strategy and a ligation-mediated convergent 6 + 5 strategy, respectively.6,7 With the aim to identify new teixobactin analogues with improved pharmacological properties, great efforts have also been devoted to the synthesis and bioactive evaluation of teixobactin analogues.8–12 These studies revealed that Arg10-teixobactin (1a, Fig. 1) where the difficultly obtained L-allo-End residue bearing the chiral cyclic guanidine was replaced by commercially available L-Arg analogue bearing a linear guanidine could maintain a modest antibacterial activity.8–10 Substitution of the D-allo-Ile residue by a D-Ile residue caused a significant reduction in activity.7 Other modifications such as substitution of D-Thr of the macrolactone ring by L-Thr and the substitution of the D-configured amino acids outside the macrocycle by L-configured residues resulted in the total loss of the activity.9–11 These findings suggest that teixobactin has a well defined conformational structure and is highly sensitive to both bone and side chain stereotopical modifications. In addition to the stereochemistry, it is valuable to elucidate the influence of other changes to certain structural aspects of the amino acid residues towards the antibacterial activity. For this purpose, we started a structure–activity relationship (SAR) study and report here the synthesis and evaluation of a new series of teixobactin analogues.
First our efforts were focused on developing a convenient strategy for the synthesis of Arg10-teixobactin (1a), which served as an alternative structural motif for the medicinal chemistry studies of teixobactin. As demonstrated in our previous research, aryl hydrazide resin has potential advantages over TCP resin and Wang resin currently used for the synthesis of structurally complex cyclodepsipeptides.13 The aryl hydrazide linker is stable in both strongly acidic and basic conditions and yet it can be cleaved under very mild oxidative conditions.14 The reactivity of the transient acyl diazene species towards nucleophiles allows cyclative cleavage, which can release the cyclized peptide directly into solution without the need for an additional cyclization reaction.15 Moreover, the possibility of partial racemization of the C-terminal residue during the cyclization step can be suppressed.16 Therefore, we anticipated that the application of aryl hydrazide resin would allow all of the coupling steps and the macrolactamization to occur on the solid support. Since the cyclative cleavage on the aryl hydrazide resin is highly sterically demanding, the macrocyclization site was chosen at Ala9-Arg10 junction for minimum steric hindrance. We envisioned that precursor peptide 3 (Scheme 1) could be obtained by assembly of the main peptidyl chain followed by introduction of Ile residue via ester-bond formation and elongation of the side chain on solid phase successively.
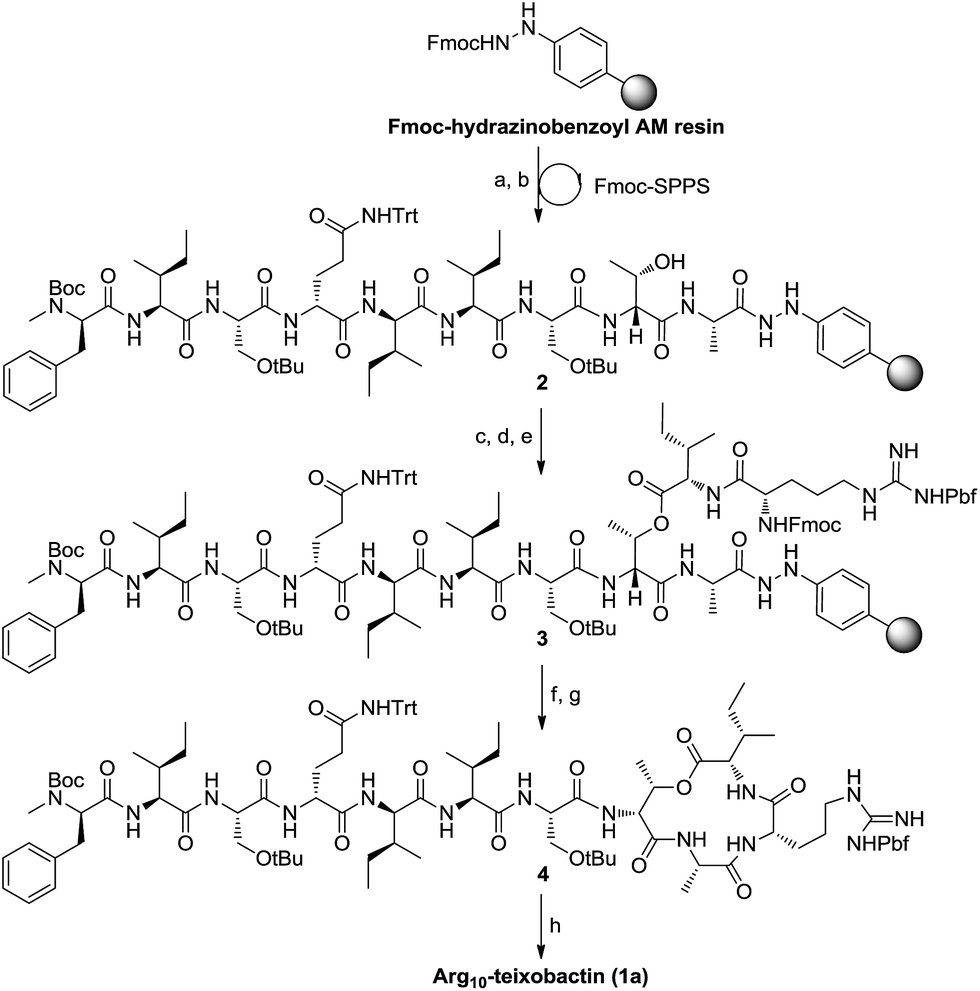 | ||
Scheme 1 Synthesis of Arg10-teixobactin (1a). Reagents and conditions: (a) 20% piperidine/DMF; (b) Fmoc-Xaa-OH (or Boc-D-N-Me-Phe-OH); HATU, DIEA, DMF; (c) Fmoc-Ile-OH; DCC, DMAP, CH2Cl2/DMF; (d) 20% piperidine/DMF; (e) Fmoc-Arg(Pbf)-OH, HATU, DIEA, DMF; (f) 20% piperidine/DMF; (g) Cu(OAc)2, Py, DMF; (h) TFA/TIS/H2O (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5:2.5). :2.5:2.5). | ||
Following this strategy, the synthesis was started by attaching Fmoc-Ala-OH to Fmoc-hydrazinobenzoyl AM resin after it was deprotected with 20% piperidine in DMF. A substitution level of 0.3 mmol g−1 was preferred to facilitate the ester bond formation and prevent unwanted side reactions during the on-resin macrocyclization. To avoid truncated products, pivalic anhydride was then added to cap any of the remaining hydrazine groups on the resin. As expected, the standard Fmoc-based solid-phase synthesis of the nonapeptide 2 proceeded smoothly employing HATU/DIEA as the coupling reagents. D-Thr8 was introduced without a protecting group at the hydroxy position. Boc-D-N-Me-Phe-OH was used to access the main peptidyl chain with N-terminal protected by Boc. O-Acylation of D-Thr8 was achieved using Fmoc-Ile-OH symmetric anhydride (formed by the reaction of Fmoc-Ile-OH and DCC in a ratio of 2:1). The following residue, Arg, was then incorporated into the side peptidyl chain to give precursor 3.
After removal of the Fmoc group from 3, cyclative cleavage of the cyclodepsipeptide from the resin was investigated. To our delight, after the transient acyl diazene species was formed by oxidation of the hydrazide with Cu(OAc)2, the following intramolecular nucleophilic attack by arginine amine took place simultaneously to release the cyclized product. Direct aminolysis of the resin-bound peptide in the presence of atmospheric O2 at elevated temperatures (55 °C) can also provide the cyclized depsipeptide. After global deprotection with TFA/TIS/H2O, Arg10-teixobactin (1a) was obtained in 18% yield with respect to the first loading of the resin. The spectroscopic data of 1a is identical to those reported by Yang et al.10
The general applicability of this approach was further demonstrated by the synthesis of analogues 1b–1i (Fig. 2). By replacing Arg10 with Lys10 and His10, Lys10-teixobactin (1b) and His10-teixobactin (1c) were synthesized, respectively. By replacing Ser7 with Ala7, replacing D-Gln4 with D-Asn4 and D-Arg4, and replacing D-N-Me-Phe1 with D-Phe1 and D-Tyr1, Ala7-Arg10-teixobactin (1d), D-Asn4-Arg10-teixobactin (1e), D-Arg4-Arg10-teixobactin (1f), D-Phe1-Arg10-teixobactin (1g) and D-Tyr1-Arg10-teixobactin (1i) were obtained accordingly. To prepare D-Me2Phe1-Arg10-teixobactin (1h), modification was made by using Fmoc protection for the D-Phe residue instead of Boc which allowed access of D-Me2Phe as the N-terminal via reductive amination of D-Phe in the presence of aqueous formaldehyde and NaBH3CN.17 All these compounds were characterized by HRMS and 1H NMR.
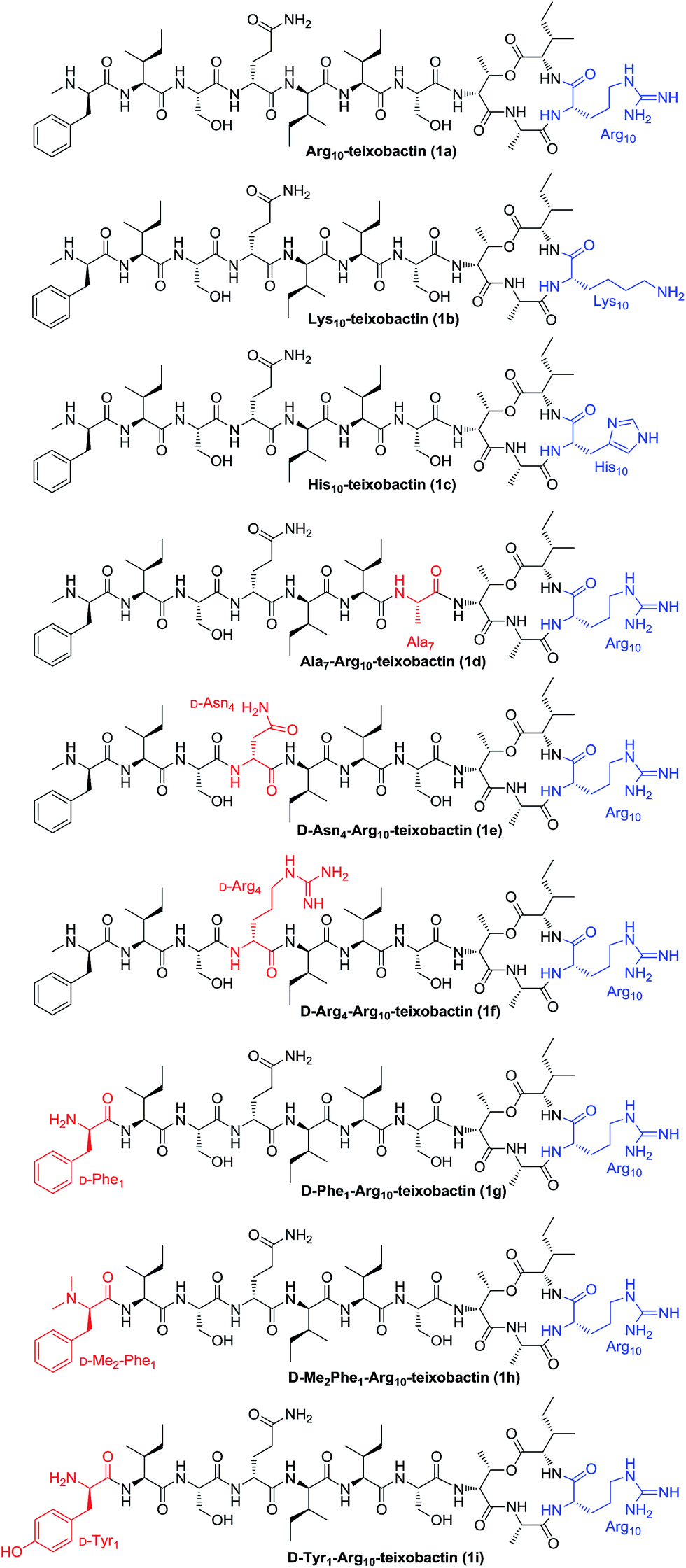 | ||
| Fig. 2 Structures of teixobactin analogues 1a–1i. | ||
The antibacterial properties of teixobactin analogues 1a–1i were evaluated against two Gram-positive bacteria (Staphylococcus aureus ATCC 29213 and Bacillus subtilis ATCC 6633) and one Gram-negative bacterium (Escherichia coli ATCC 25922). β-Lactam-type antibiotic meropenem was used as a positive control. The minimum inhibitory concentration assay (MIC) results are listed in Table 1 in comparison to meropenem and the reported activity of natural teixobactin.4
| Compound | Staphylococcus aureus | Bacillus subtilis | Escherichia coli |
|---|---|---|---|
| a The original published values of teixobactin in related bacteria from ref. 4. | |||
| 1a | 2 | 0.25 | >128 |
| 1b | 2 | 0.5 | >128 |
| 1c | 8 | 2 | >128 |
| 1d | 32 | 8 | >128 |
| 1e | 2 | 0.5 | >128 |
| 1f | 2 | 1 | 32 |
| 1g | 2 | 0.5 | >128 |
| 1h | 32 | 8 | >128 |
| 1i | 128 | 32 | >128 |
| Teixobactina | 0.16–0.31 | 0.02–0.06 | 25 |
| Meropenem | 0.125 | 0.25 | 0.0625 |
As found in other lipid II-binding depsipeptides, the guanidine side chain secures a positive charge and thus possibly plays an important role, while interacting with the phosphate moiety of lipid II.18 To explore the role of the guanidinium group at position 10, we compared the MIC of Lys10-teixobactin (1b) and His10-teixobactin (1c) to Arg10-teixobactin (1a). 1b containing Lys at position 10 was slightly inferior to 1a against Bacillus subtilis while the activity of 1b against Staphylococcus aureus was comparable to 1a and the Orn-containing analogue reported by Jin and coworkers.7 In contrast, analogue 1c containing His residue at this position exhibited decreased potency towards different types of Gram-positive bacteria. Since Arg and Lys are more basic than His, these observations indicate that the antibacterial activity may have positive correlation with the basicity of the amino acid side chain. Thus, guanidine or amine groups are preferred to maintain the cationic character of position 10.
Besides N-terminal D-N-Me-Phe, the linear peptide fragment is composed of six amino acids, including three Ile (two L and one D-allo), two Ser, and one D-Gln. Among these residues, Ser7 and D-Gln4 were selected to explore the influence of side chain size and functional group changes towards the antibacterial activity. Ala7-Arg10-teixobactin (1d), D-Asn4-Arg10-teixobactin (1e) and D-Arg4-Arg10-teixobactin (1f) were therefore designed and synthesized to compare with Arg10-teixobactin (1a). It was revealed that the hydroxyl group of the Ser residue at position 7 was required for the antibacterial activity, aliphatic substitution (Ala in 1d) significantly decreased the activity. The substitution of D-Gln residue at position 4 with D-Asn residue or D-Arg residue has limited influence to the antibacterial activity against Gram-positive bacteria. However, the D-Arg analogue showed remarkably improved potency toward Gram-negative bacterium, which can be attributed to the enhanced transmembrane capability of the Arg group.
In the previous work, Albericio's group demonstrated that substitution of the terminal N-Me group by N-Ac group resulted in the total loss of the antibacterial activity of teixobactin, since both the cationic character and the hydrogen bond pattern of the peptide were altered by introduction of the acetyl group on the N-terminal.11 To further explore the impact of the N-terminal Phe residue, we compared the MIC of D-Phe1-Arg10-teixobactin (1g), D-Me2Phe1-Arg10-teixobactin (1h) and D-Tyr1-Arg10-teixobactin (1i) with Arg10-teixobactin (1a). As a result, analogue with no terminal N-methyl group (1g) maintained the activity, while N,N-dimethyl substitution (1h) at this position caused a significant reduction in activity, which indicates that the terminal NH proton plays an important role towards its binding with lipid II or lipid III. Introduction of an extra hydroxyl group to the aromatic ring (1i) dramatically reduced the antibacterial activity. Although the cationic character did not change in the latter case, the presence of the free hydroxyl group could change the hydrogen bond pattern of the molecule as well as the hydrophobic interaction, preventing it from adopting the preferred conformation for interacting with the precursors of the cell wall polymers.
In conclusion, a convenient strategy for the synthesis of teixobactin analogues was developed employing an efficient on-resin macrocyclization as the key step. A new series of teixobactin analogues were prepared and the antibacterial activities were evaluated by using a minimum inhibitory concentration assay. Our preliminary structure–activity relationship (SAR) studies demonstrated the importance of maintaining the cationic character, the hydrogen bond pattern as well as the hydrophobic interaction to favor the bioactive conformation. The relevant trends of the structure–activity relationship disclosed in this study will facilitate the design of new teixobactin analogues with enhanced pharmacological properties.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21402232 and 21432003), Shenzhen Sciences & Technology Innovation Council (KQCX2015033117354154 and JCYJ20150316143416083) and the “Hundred Talents Program” of Chinese Academy of Sciences. The authors are grateful for the assistance of Mass facility from Peking University Shenzhen Graduate School.Notes and references
- G. Taubes, Science, 2008, 321, 356–361 CrossRef CAS PubMed; M. A. Cooper and D. Shlaes, Nature, 2011, 472, 32 CrossRef PubMed.
- J. M. A. Blair, M. A. Webber, A. J. Baylay, D. O. Ogbolu and L. J. V. Piddock, Nat. Rev. Microbiol., 2014, 13, 42 CrossRef PubMed.
- K. S. Long and B. Vester, Antimicrob. Agents Chemother., 2012, 56, 603–612 CrossRef CAS PubMed.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaeberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- G. Wright, Nature, 2015, 517, 442–443 CrossRef CAS PubMed; V. Ng and W. C. Chan, Chem.–Eur. J., 2016, 22, 12606–12616 CrossRef PubMed; G. Kumar, World J. Pharm. Res., 2015, 4, 1489–1490 Search PubMed.
- M. Giltrap, L. J. Dowman, G. Nagalingam, J. L. Ochoa, R. G. Linington, W. J. Brotton and R. J. Payne, Org. Lett., 2016, 18, 2788–2791 CrossRef PubMed.
- K. Jin, I. H. Sam, K. H. L. Po, D. Lin, E. H. G. Zadeh, S. Chen, Y. Yuan and X. Li, Nat. Commun., 2016, 7, 12394 CrossRef CAS PubMed.
- Y. E. Jad, G. A. Acosta, T. Naicker, M. Ramtahal, A. El-Faham, T. Govender, H. G. Kruger, B. G. de la Torre and F. Albericio, Org. Lett., 2015, 17, 6182–6185 CrossRef CAS PubMed.
- A. Parmar, A. Iyer, C. S. Vincent, D. V. Lysebetten, S. H. Prior, A. Madder, E. J. Taylor and I. Singh, Chem. Commun., 2016, 52, 6060–6063 RSC.
- H. Yang, K. H. Chen and J. S. Nowick, ACS Chem. Biol., 2016, 11, 1823–1826 CrossRef CAS PubMed.
- S. A. H. A. Monaim, Y. E. Jad, G. A. Acosta, T. Naicker, E. J. Ramchuran, A. El-Faham, T. Govender, H. G. Kruger, B. G. Torre and F. Albericio, RSC Adv., 2016, 6, 73827–73829 RSC.
- S. Dhara, V. B. Gunjal, K. L. Handore and D. S. Reddy, Eur. J. Org. Chem., 2016, 25, 4289–4293 CrossRef.
- G. Yao, Z. Pan, C. Wu, W. Wang, L. Fang and W. Su, J. Am. Chem. Soc., 2015, 137, 13488–13491 CrossRef CAS PubMed.
- Y. Woo, A. R. Mitchell and J. A. Camarero, Int. J. Pept. Res. Ther., 2007, 13, 181–190 CrossRef CAS.
- A. Shigenaga, J. A. Moss, F. T. Ashley, G. F. Kaufmann and K. D. Janda, Synlett, 2006, 4, 551–554 Search PubMed.
- C. Rosenbaum and H. Waldmann, Tetrahedron Lett., 2001, 42, 5677–5680 CrossRef CAS.
- G. A. Sable, J. Park, H. Kim, S. Lim, S. Jang and D. Lim, Eur. J. Org. Chem., 2015, 32, 7043–7052 CrossRef.
- F. Nussbaum and R. D. Süssmuth, Angew. Chem., Int. Ed., 2015, 54, 6684–6686 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthetic method, 1H NMR, HRMS. See DOI: 10.1039/c6ra26567g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |