
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of new photoswitchable azobenzene-containing poly(ε-caprolactones)†
Clement
Appiah
a,
Katrin R.
Siefermann
b,
Marcel
Jorewitz
b,
Haitham
Barqawi
a and
Wolfgang H.
Binder
*a
aInstitute of Chemistry, Chair of Macromolecular Chemistry, Faculty of Natural Science II (Chemistry, Physics and Mathematics), Martin-Luther University Halle-Wittenberg, D-06120, Halle (Saale), Germany. E-mail: wolfgang.binder@chemie.uni-halle.de
bLeibniz-Institute of Surface Modification, Permoserstrasse 15, Leipzig, Germany
First published on 5th January 2016
Abstract
A novel and efficient strategy in obtaining series of mono- and bi-armed azobenzene-containing poly(ε-caprolactone)s is described, starting from a commercially available azobenzene dye via azide/alkyne-“click”-reactions. The attachment of alkyne-telechelic poly(ε-caprolactone)s (1 kDa and 3 kDa), followed by chromatographic separation, allowed the attachment of either one or two PCl-chains to either side of the azobenzene-dye. The resulting mono- and bi-armed photo-switchable polymers are fully characterized by 2D-NMR techniques and show a high thermal stability. Additionally liquid chromatography at critical conditions (LCCC) coupled to ESI-TOF allowed us to prove the presence of either one or two polymer chains affixed onto the central azobenzene dye.
Introduction
Polymeric based molecular switches have recently attracted much interest due to their wide range of applications ranging from nanomedicine such as affinity separations, enzyme recovery and drug delivery1,2 to industrial applications including storage3 and communication elements in optical devices.4 The output of such photoresponsive polymeric materials mainly depends on a complex interplay between the chromophore5 (e.g. an azobenezene dye), the morphology of the polymeric network (amorphous, semicrystalline and liquid crystalline),3a the thermomechanical properties of the material (glassy or rubbery state modulus), and the molecular geometry of the sample.6 Thus, e.g. reversible solubility of azobenzene polymers7 or reversible contractions of thin films8 (see also e.g. the work of Eisenbach9 and Blair10) have been reported, stressing the importance of photochemically switchable polymers.The photoresponsive properties of azobenzene-containing polymers are based on the photo-induced fast and reversible isomerization between trans- and cis-isomers of the azobenzene moiety upon exposure to UV or visible light, which can trigger significant changes in the physicochemical properties of the corresponding polymeric materials.11–13 Similar to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds, the azobenzenes display two geometric isomers (cis and trans) around the NN double bond,14 where the trans isomer is the more stable isomer. Isomerization to the cis-isomer is accomplished upon irradiation at a wavelength of ∼320–350 nm. Due to the reversibility of this reaction, the trans-isomer can be easily recovered by irradiating the cis-isomer at a wavelength of 400–450 nm or via purely thermal relaxation.14 These two photochemical reactions usually occur on picoseconds scales, while the thermal relaxation from the cis- to trans-isomer is much slower (milliseconds to days).15–17 It is important to mention that after the photo-isomerization process, characteristic changes in the physical properties of the two isomers affect the molecular geometry and the dipole moment of the dye.15,16 Thus the attachment of polymeric chains onto the azobenzene in consequence leads to additional effects of the optoelectronic and relaxation properties of the dye,18,19 often influenced by the value of the chemical linkage between the azo-dye and the polymer chain.19 It has been demonstrated that the linkage between the azo chromophore and the polymer backbone can restrict both, the molecular mechanical properties and the motion of the azo-groups:20 polymers linked via a rigid attachment to the azo chromophore can confine both, the chromophore's motion and its subsequent re-orientation, but also can induce an increased stability of the formed cis-isomer after the light has been turned off.20 A large number of publications has been dealing with the influence of the polymeric chains on the physicochemical properties of the azo moiety,21–23 such as an elongation of the cis-lifetime.24,25 Lamarre and Sung25 have studied the physical ageing and molecular motion of azochromophoric labels attached to main chain poly(urethane)s, showing a lifetime of 4 days after switching to the cis-form. Furthermore, the conformational strain of macrocyclic azo compounds has also been used to lock the cis form for days.24,26 Intermolecular interactions, such as hydrogen bonds to generate cyclic structures can also lead to largely increased lifetimes of the cis-form of up to ∼40 days.27
C double bonds, the azobenzenes display two geometric isomers (cis and trans) around the NN double bond,14 where the trans isomer is the more stable isomer. Isomerization to the cis-isomer is accomplished upon irradiation at a wavelength of ∼320–350 nm. Due to the reversibility of this reaction, the trans-isomer can be easily recovered by irradiating the cis-isomer at a wavelength of 400–450 nm or via purely thermal relaxation.14 These two photochemical reactions usually occur on picoseconds scales, while the thermal relaxation from the cis- to trans-isomer is much slower (milliseconds to days).15–17 It is important to mention that after the photo-isomerization process, characteristic changes in the physical properties of the two isomers affect the molecular geometry and the dipole moment of the dye.15,16 Thus the attachment of polymeric chains onto the azobenzene in consequence leads to additional effects of the optoelectronic and relaxation properties of the dye,18,19 often influenced by the value of the chemical linkage between the azo-dye and the polymer chain.19 It has been demonstrated that the linkage between the azo chromophore and the polymer backbone can restrict both, the molecular mechanical properties and the motion of the azo-groups:20 polymers linked via a rigid attachment to the azo chromophore can confine both, the chromophore's motion and its subsequent re-orientation, but also can induce an increased stability of the formed cis-isomer after the light has been turned off.20 A large number of publications has been dealing with the influence of the polymeric chains on the physicochemical properties of the azo moiety,21–23 such as an elongation of the cis-lifetime.24,25 Lamarre and Sung25 have studied the physical ageing and molecular motion of azochromophoric labels attached to main chain poly(urethane)s, showing a lifetime of 4 days after switching to the cis-form. Furthermore, the conformational strain of macrocyclic azo compounds has also been used to lock the cis form for days.24,26 Intermolecular interactions, such as hydrogen bonds to generate cyclic structures can also lead to largely increased lifetimes of the cis-form of up to ∼40 days.27
Several synthetic strategies have so far been reported in obtaining azobenzene polymers either via the direct polymerization of a monovinyl azo monomer,28,29 or via a postfunctionalization-reaction of a polymer onto an azo-dye.3b,30 Based on this methodology both, side-chain31 and main-chain32 azobenzene polymers have been prepared, resulting in different architectures like block-,32,33 brush-,34,35 star-36 and dendritic-37–39 polymers. Besides a direct polymerization of suitable monomers, the subsequent attachment of azobenzenes onto polymer matrices40,41 or the covalent attachment of polymers onto azobenzenes42,43 has been reported. Despite the many reported methodologies, not always the expected polymers were obtained,44–46 often due to setbacks in preparation and purification.
We hereby report an efficient strategy in preparing a series of azobenzene containing poly(ε-caprolactone)s by chemical modification of a commercially available azobenzene dye into a poly(ε-caprolactone) backbone with different molecular weights (see Fig. 1). The chemical separations and purification of the resulting azobenzene polymers are described in detail.
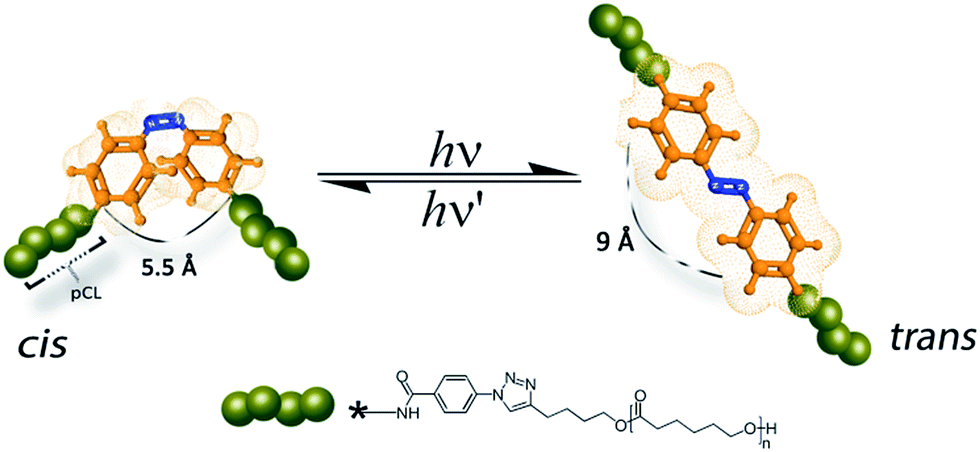 | ||
| Fig. 1 Isomerization of azobenzene polymers. | ||
Experimental
Materials
All chemicals used for the synthesis were purchased from Sigma-Aldrich and CHEMOS chemicals. ε-Caprolactone was stirred and distilled over calcium hydride (100 °C, 10 mbar). Tin(II)-2-ethylhexanoate and 5-hexyn-1-ol were also purified via the distillation procedures in the literature.47,48 Cu(I)Br was stirred for 2 days in acetic acid/anhydride, washed with dry diethyl ether several times, and dried in vacuo under an argon atmosphere. N,N-Diisopropylethylamine (DIPEA), N,N-dicyclohexylcarbodiimide (DCC), N,N-dimethylamino-4-pyridine (DMAP) and 2,2′-bipyridine were used without further purification. Tetrahydrofuran (THF) was predried over sodium hydroxide (NaOH) for several days followed by heating over sodium/benzophenone and final distillation under an argon atmosphere before use.Measurements
NMR spectroscopy was recorded on a Varian Gemini 2000 FT-NMR spectrometer (400 MHz). Chloroform (99.8 atom% D) and dimethyl sulfoxide-d6 (99.9 atom% D) were used as solvents, using MestrecC software version 4.7.0.0 for the FIDs analysis. Chemical shifts (δ) were recorded in parts per million (ppm) and referenced to residual protonated solvent CDCl3: 7.26 ppm (1H), 77.0 ppm (13C), (CD3)2SO: 2.54 ppm (1H), 39.52 ppm (13C); coupling constants (J) are given in hertz (Hz) using standard abbreviations (s = singlet, d = doublet, t = triplet, m = multiplet).GPC measurements were conducted on a Viscotek GPCmax VE 2001 with a Styragel linear column GMHHR. THF was used as a carrier solvent at 1 mL min−1 at room temperature. The sample concentration was approximately 3 mg mL−1. Poly(styrene) standards (in the range of 1050–1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 870000 g mol−1) were used for conventional external calibration, using a Waters RI 3580 refractive index detector at 35 °C.
870000 g mol−1) were used for conventional external calibration, using a Waters RI 3580 refractive index detector at 35 °C.
FTIR spectra were recorded with a Brucker Vertex70MIR spectrometer using an ATR Golden Gate unit with a diamond crystal. The scan number was 32 scans per spectrum with a resolution of 2 cm−1.
MALDI-TOF-MS was done on a Bruker Autoflex III Smartbeam using a nitrogen laser source (λ = 337 nm) in reflection and linear modes. The polymer samples were dissolved in THF at a concentration of 20 mg mL−1; 1,8,9-anthracenetriol in THF (20 mg mL−1) was used as matrix material; sodium trifluoroacetate (NaTFA) in THF (20 mg mL−1) was used as salt. The solutions of the polymer, the matrix, and the salt were mixed in a volume ratio of 100:20:2 and 2 μL of this mixture were spotted on the MALDI target plate. The instrument was calibrated with a poly(ethylene glycol) standard (Mp = 2000 g mol−1) using a quadratic calibration method.
ESI-TOF MS measurements were carried out on a Brucker Daltonics microgel time-of-flight LC-MS system. Spectra were recorded in a positive mode with an accelerator voltage of 4.5 kV, a transfer line with 190 °C and a scan range of 50–7000 m/z at the spectral rate of 1 Hz. The spectra were processed on Brucker Daltonics ESI compass 1.3 for microTOF (Data Analysis 4.0). Samples were prepared by dissolving 1–5 mg of the respective compound in 1 mL mixture of THF/methanol (92/8).
HPLC measurements were performed on Elite LaChrom HPLC, Hitachi VWR, equipped with an autosampler, a quaternary gradient pump, a degasser, a diode array detector (UV-DAD) operating at 190–900 nm and a column oven with temperature control (temperature = 20 °C). Temperatures were maintained constant (±0.2 °C) throughout all experiments, and the injected sample volume was 30 μL. Liquid chromatography at critical conditions (LCCC) was carried out on a non polar reversed-phase Atlantis-RP C18 column, 100 Å, 5 μm, dimension 4.6 × 250 mm; methanol/DCM = 8/92 was used as the mobile phase with a flow rate of 0.30 mL min−1. The DAD signals were recorded on EZchrom Elite software version 3.3.2 SP2 with an operating wavelength from 190 to 900 nm at a sampling width of 200 ms to obtain sufficient data points across peaks.
Synthesis of alkyne-functionalized PCL (PCL-alkyne)
Alkyne-functionalized PCL was prepared via coordination insertion ring opening polymerization (ROP) using e-caprolactone as a monomer, 5-hexyn-1-ol as an initiator, and tin(II) 2-ethylhexanoate (Sn(Oct)2) as a catalyst according to the literature.47 Briefly, a Schlenk flask was carefully dried by heating (500 °C), purged with argon, cooled down, charged with the initiator 5-hexyn-1-ol (55 μL, 5.0 × 10−4 mol), adding the catalyst tin(II) 2-ethylhexanoate (6.0 × 10−6 mol, 2 μL), and a solvent mixture of 3 mL toluene and 1 mL THF. This mixture was stirred at room temperature for 30 min to promote the complex formation between catalyst and initiator. Afterwards, the solution was treated with ε-caprolactone (3 mL, 0.026 mol) and stirred at 110 °C for 3 h. Finally, the product was dissolved in dry DCM (10 mL) and precipitated into cold methanol (150 mL), filtered off and dried under vacuum overnight.1H-NMR (400 MHz, CDCl3, δ, ppm, PCL-alkyne3, DP = 29): 4.04 (t, 59H, J = 6.7 Hz), 3.63 (t, 2H, J = 6.5 Hz), 2.28 (t, 59H, J = 6.7 Hz), 1.94 (t, 1H, J = 2.6 Hz), 1.65 (m, 118H), 1.38 (m, 59H). 13C-NMR (100 MHz, CDCl3, δ, ppm, PCL-alkyne3): 173.4, 64.1, 34.1, 28.3, 25.5, 24.4.
Synthesis of 4-(azidobenzamido)-4′-(amino) azobenzene (2) and 4,4′-bis(benzamido) azobenzene (3)
To a solution of 4,4′-bis(amino)azobenzene (1) (1 mol eq.) and DIPEA (2.2 mol eq.) in THF at 0 °C was added 4-azidobenzoyl chloride (3 mol eq.) dissolved in THF. The mixture was stirred for 30 min, and finally warmed to room temperature with constant stirring for 24 h. The obtained crude product was filtered and purified via column chromatography (DCM/ETOAc 3:1, Rf values: 4,4′-bis(benzamido) azobenzene = 0.82, 4-(azidobenzamido)-4′-(amino)azobenzene = 0.61).
Alternatively,49 into a dried and nitrogen purged flask was added a solution of 4-azidobenzoic acid (2 mol eq.) and DCC (1 mol equiv.) in THF (5 mL). The mixture was stirred for 6 h at 0 °C to form bis-azidobenzoic anhydride. In a second dried round bottom flask a solution of 4,4′-bis(amino)azobenzene (1 mol equiv.) and DMAP (0.5 mol equiv.) in THF (5 mL) were added. The two solutions were then mixed and stirred at room temperature for 48 h. The obtained crude product was purified in a similar faction as the previous method.
1H-NMR (400 MHz, DMSO, δ, ppm, 2): 10.42, (s, 1H), 8.04 (d, 2H, J = 8.03 Hz), 7.93 (d, 2H, J = 8.23 Hz), 7.76 (d, 2H, J = 8.30 Hz), 7.63 (d, 2H, J = 8.81 Hz), 7.28 (d, 2H, J = 8.02 Hz), 6.66 (d, 2H, J = 8.12 Hz), 6.00 (s, 2H). 13C-NMR (100 MHz, DMSO, δ, ppm, 2): 165.0, 152.9, 148.8, 143.3, 140.8, 131.6, 130.2, 125.3, 122.8, 120.9, 119.5, 113.9.
1H-NMR (400 MHz, DMSO, δ, ppm, 3): 12.95, (s, 2H), 7.95 (d, 8H, J = 8.35 Hz), 7.20 (d, 4H, J = 8.48 Hz), 5.58 (d, 4H, J = 8.03 Hz). 13C-NMR (100 MHz, DMSO, δ, ppm, 3): 207.0, 157.1, 131.6, 119.5.
IR (cm−1, 3): 3385.8, 3074.8, 2989.9, 2811.2, 2703.9, 2407.8, 2119.8, 1649.9, 1601.9, 1420.5, 1345.1, 1313.2, 1180.0, 1129.4, 1098.1, 1067.9, 1037.7, 930.6, 851.2, 822.5, 773.8, 689.5, 570.5.
Synthesis of 4-(poly(ε-caprolactone)-benzamido)-4′-(amino)azobenzene (polymer-1a and polymer-1b) and 4,4′-bis (poly (ε-caprolactone)-benzamido)azobenzene (polymer-2a and polymer-2b)
The functionalization of PCL with 4-(azidobenzamido)-4′-(amino)azobenzene and 4,4′-bis(benzamido) azobenzene was conducted via an azide/alkyne click-reaction, using 2,2′-bipyridine as base and Cu(I)Br as a catalyst. The reaction was accomplished in a one-necked flask carefully dried and purged with argon. Alkyne-functionalized PCL (PCL-alkyne) (1 mol equiv.) and azide-functionalized azobenzene dyes (4 mol equiv.) were dissolved in 6 mL of dry THF, then a solution of 2,2′-bipyridyl (20 mol equiv.) in dry THF (2 mL) was added via a syringe and the mixture was degassed with argon for 30 min to remove oxygen. Subsequently, a catalytic amount of Cu(I)Br (1 mol equiv.) was weighed into the flask. The flask was closed with a septum, and the mixture was degassed again with argon for another 30 min. The reaction proceeded for 24 h by stirring at 80 °C. Afterwards, the reaction mixture was cooled down to room temperature and the product was purified by chromatography via a silica gel packed column (ethyl acetate:DCM 1:3). The crude product was dissolved in 10 mL of DCM, slowly added into methanol (200 mL) and left overnight for complete precipitation. The precipitation step was repeated two more times to completely remove the excess of the azide-functionalized azobenzene dyes and the 2,2′-bipyridine, which are all well soluble in methanol. The pure products were filtered off and dried under vacuum for 3 days (yields: 1a = 71%, 1b = 75%, 2a = 50% and 2b = 58%).
1H-NMR (400 MHz, DMSO, δ, ppm, polymer-1b): 10.55, (s, 1H), 8.70, (s, 1H), 8.17 (d, 2H, J = 8.03 Hz), 8.07 (d, 2H, J = 8.23 Hz), 7.96 (d, 2H, J = 8.30 Hz), 7.77 (d, 2H, J = 8.81 Hz), 7.64 (d, 2H, J = 8.02 Hz), 6.66 (d, 2H, J = 8.12 Hz), 6.00 (s, 2H), 4.30 (t, 2H, J = 6.5 Hz), 3.98 (t, 65H, J = 6.7 Hz), 3.28 (t, 2H, J = 6.4 Hz), 2.74 (t, 2H, J = 6.1 Hz), 2.24 (t, 65H, J = 6.7 Hz), 1.54 (m, 130H), 1.28 (m, 65H).
1H-NMR (400 MHz, DMSO, δ, ppm, polymer-2b): 8.58, (s, 4H), 8.08 (m, 8H), 7.85 (m, 8H), 4.32 (t, 4H, J = 6.0 Hz), 3.94 (t, 238H, J = 5.9 Hz), 3.31 (m, 4H), 2.72 (m, 4H), 2.24 (t, 238H, J = 6.7 Hz), 1.54 (m, 476H), 1.28 (m, 238H).
IR (cm−1, 2b): 3475.6, 2945.9, 2866.1, 2439.6, 1802.6, 1727.9, 1646.4, 1451.1, 1344.6, 1315.4, 1185.5, 1108.6, 1044.1, 963.1, 812.9, 741.7.
Results and discussion
Synthesis of photoswitchable PCL-azobenzene polymers
Conceptually, we planned to address possible synthetic strategies for obtaining switchable mono-functional- (polymer-1) and bi-functional polymers (polymer-2) via a grafting-to approach by “click-chemistry” (see Scheme 1). For this purpose, polymerization of ε-caprolactone using a coordination insertion ring opening polymerization47 method was carried out as described in the Experimental section. Subsequently, the polymers were attached to azido functionalized azobenzene dyes via the azide/alkyne “click” reaction.50,51 The developed synthetic route is presented in Scheme 1. The first step for the synthesis of the photoswitchable polymeric compounds was an amidation reaction between a commercially available azobenzene dye (1) and 4-azidobenzoylchloride in the presence of a base (DIPEA) to yield a mono-substituted 4-(azidobenzamido)-4′-(amino)azobenzene (2) and a di-substitued 4,4′-bis(benzamido) azobenzene (3). Click-chemistry between the dyes (2 and 3) and an already synthesized PCL-alkyne (Table 1) with varying molecular weights (1000 g mol−1 and 3000 g mol−1) were performed in THF to yield the products (polymer-1a, polymer-1b, polymer-2a and polymer-2b) respectively. After several purification techniques, including column chromatography, precipitation, extraction, the final purified products were characterized using HPLC, ESI-TOF-MS, MALDI-TOF-MS, IR, 1H-NMR 1H-1H-COSY NMR and GPC techniques.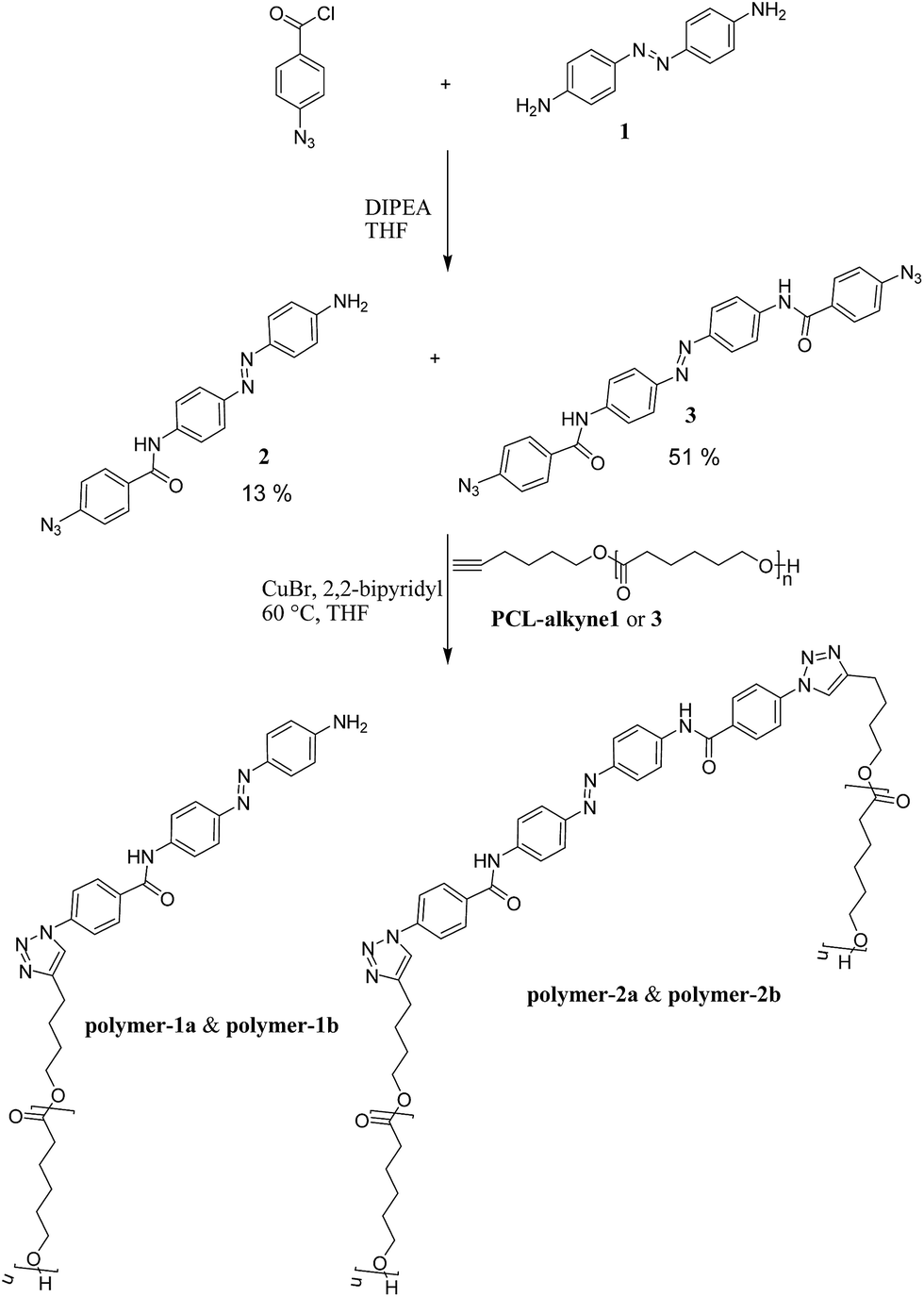 | ||
| Scheme 1 Synthetic approach for the modified azobenzenes (2 and 3) and the polymer incorporated azobenzenes (polymer-1 and polymer-2). | ||
| Samples | M n (NMR) (g mol−1) | DPa (NMR) | M n (GPC) b (g mol−1) | M w/Mn (GPC) | Yieldc (%) |
|---|---|---|---|---|---|
| a Degree of polymerization obtained via NMR spectroscopy. b Corrected molecular weight from GPC (correction factor = 0.59). c Isolated yields after purification. | |||||
| PCL-alkyne1 | 1111 | 9 | 985 | 1.2 | 95 |
| PCL-alkyne3 | 3306 | 29 | 2900 | 1.1 | 98 |
| Polymer-1a | 1920 | 9 | 1700 | 1.4 | 71 |
| Polymer-1b | 3800 | 29 | 3220 | 1.2 | 75 |
| Polymer-2a | 2850 | 18 | 2500 | 1.4 | 50 |
| Polymer-2b | 7200 | 58 | 7100 | 1.1 | 58 |
Characterization of the photoswitchable PCL-azobenzenes
The structures of all the photoswitchable PCL-azobenzenes were initially confirmed by GPC and 1H-NMR. The GPC curves were monomodal with molecular weight distributions of 1.2–1.4. Fig. 2 shows the 1H-NMR spectra of the synthesized compounds including the photoswitchable PCL-azobenzenes. All compounds showed the expected resonances in the NMR spectroscopy. Importantly the molecular weights from GPC and NMR of the photoswitchable PCL-azobenzenes matched well, proving a complete endgroup transformation of the compounds (Table 1). The degree of the end functionalization was obtained by comparing the intensity of the signal ‘h’ and signal ‘j’ for the case of the mono-armed photoswitchable polymer-1 and the peaks ‘d’ and ‘c’ for the bi-armed photoswitchable polymer-2. The result showed the completeness of the ‘click’ chemistry. The structures were further confirmed by FTIR spectroscopy (ESI†), in which the vibration peaks of –OH and –NH– (the broad peak at around 3475 cm−1) both shifted to higher wavenumbers compared to their starting precursors (<3300 cm−1).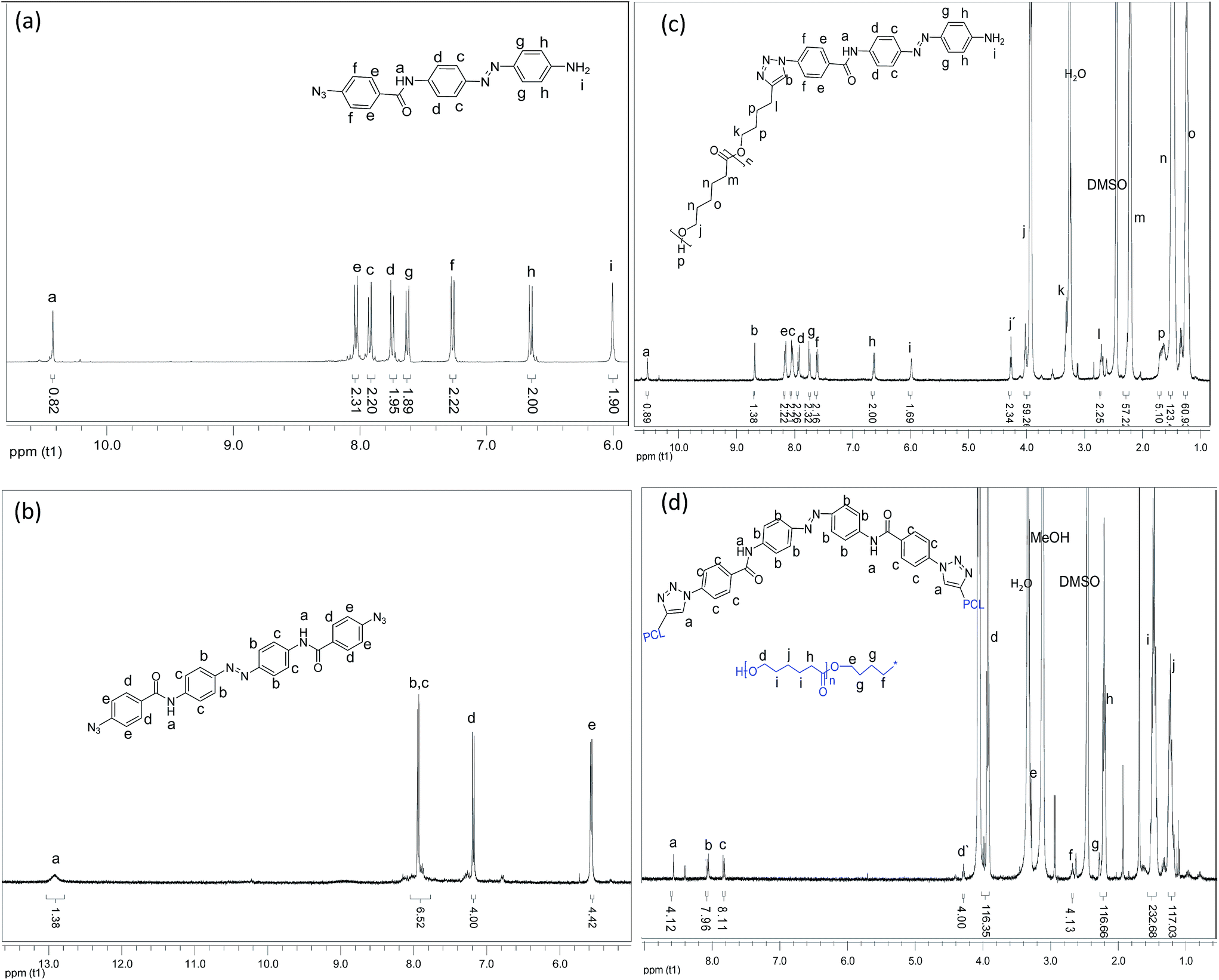 | ||
| Fig. 2 1H-NMR spectra of all the synthesized compounds; (a) 4-(azidobenzamido)-4′-(amino) azobenzene (2), (b) 4,4′-bis(benzamido) azobenzene (3), (c) 4-(poly(ε-caprolactone)-benzamido)-4′-(amino)azobenzene (polymer-1b), (d) 4,4′-bis (poly (ε-caprolactone)-benzamido)azobenzene (polymer-2b). | ||
Two-dimensional H/H COSY NMR analysis of the photoswitchable azobenzene polymers
Based on the complexity of the photoswitchable PCL-azobenzene polymers, we conducted 2D-NMR techniques, including 1H-1H COSY NMR. Fig. 3 shows the proton homonuclear correlated 2-dimensional 1H-1H COSY NMR of polymer-1b. The backbone azobenzene moiety of this photoswitchable polymer could be traced through its J-coupling.52 In the H,H-COSY of polymer-1b, a long-range coupling by arrangement of protons is observed between the doublet at 7.77 ppm (2H, J = 8.81 Hz) and the doublet at 6.66 ppm (2H, J = 8.12 Hz). The upfield shifted benzylic doublet is assigned to the protons ortho- to the amide functionality (i.e., 2H, signal-e), whereas the low field doublet is assigned to the protons ortho- to the amine group (i.e., 2H, signal-h). The aromatic benzylic protons are associated to their neighboring protons via a 1,3 bond with a strong J-coupling distance of about ∼8.03 Hz. Protons a, b and i are separated by their neighboring protons via a weak 1,4 bonding interaction and therefore show a very weak coupling effect, thus, appearing as singlet in the spectrum. While the coupling behavior of the mono-armed polymer-1 is well observable in the COSY spectrum, the bi-armed polymers (polymer-2) are significantly more difficult to resolve. The signals appear as multiplets instead of individual doublet structures (see Fig. 2d and ESI†). The COSY correlations helped to precisely assign the protons of the compounds observed in the 1-dimensional 1H-NMR especially in the mono-armed polymers and to further confirm the obtained structures (Fig. 4).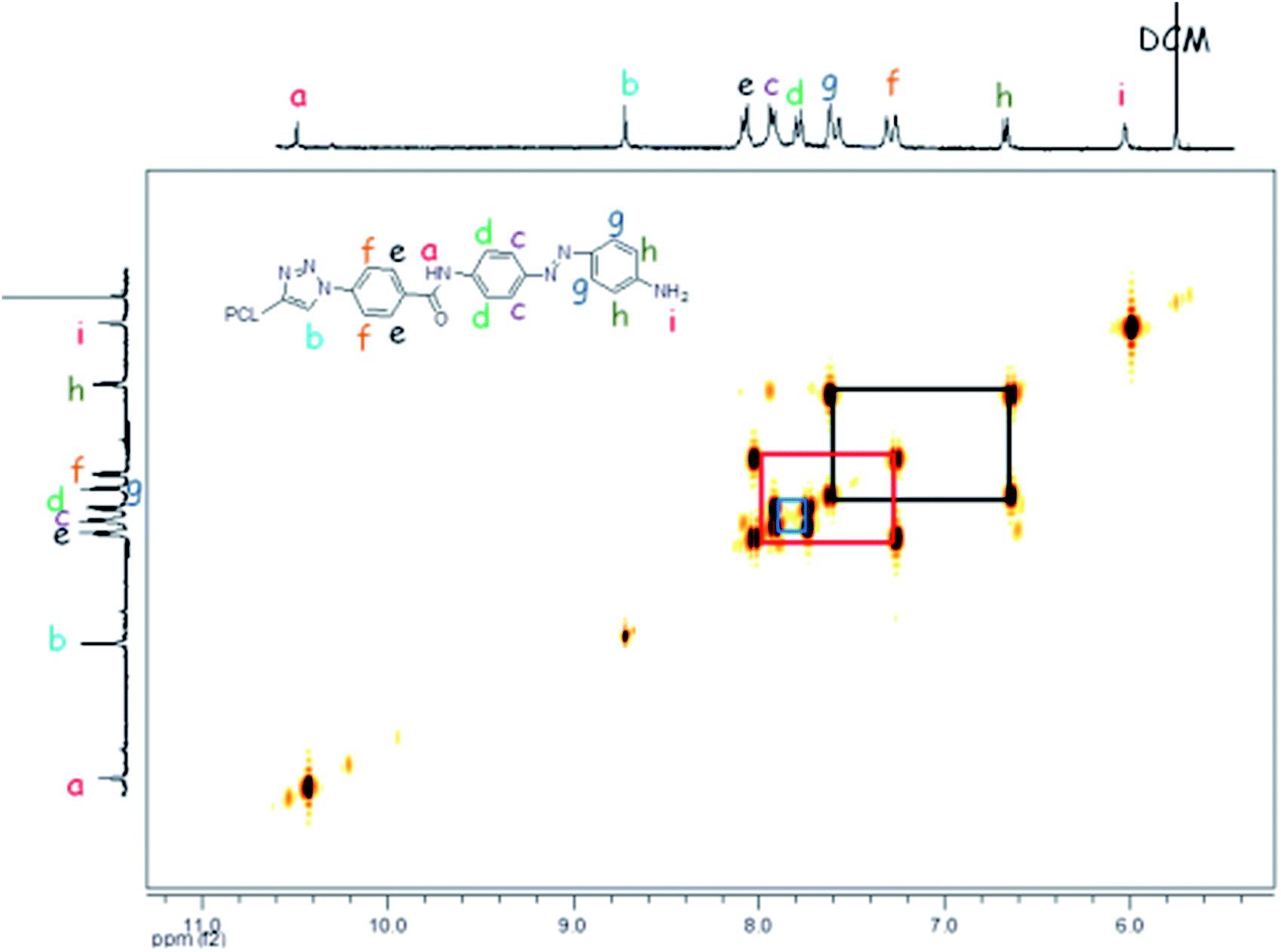 | ||
| Fig. 3 Expanded proton homonuclear correlated 2-dimensional 1H-1H COSY NMR of polymer-1b. | ||
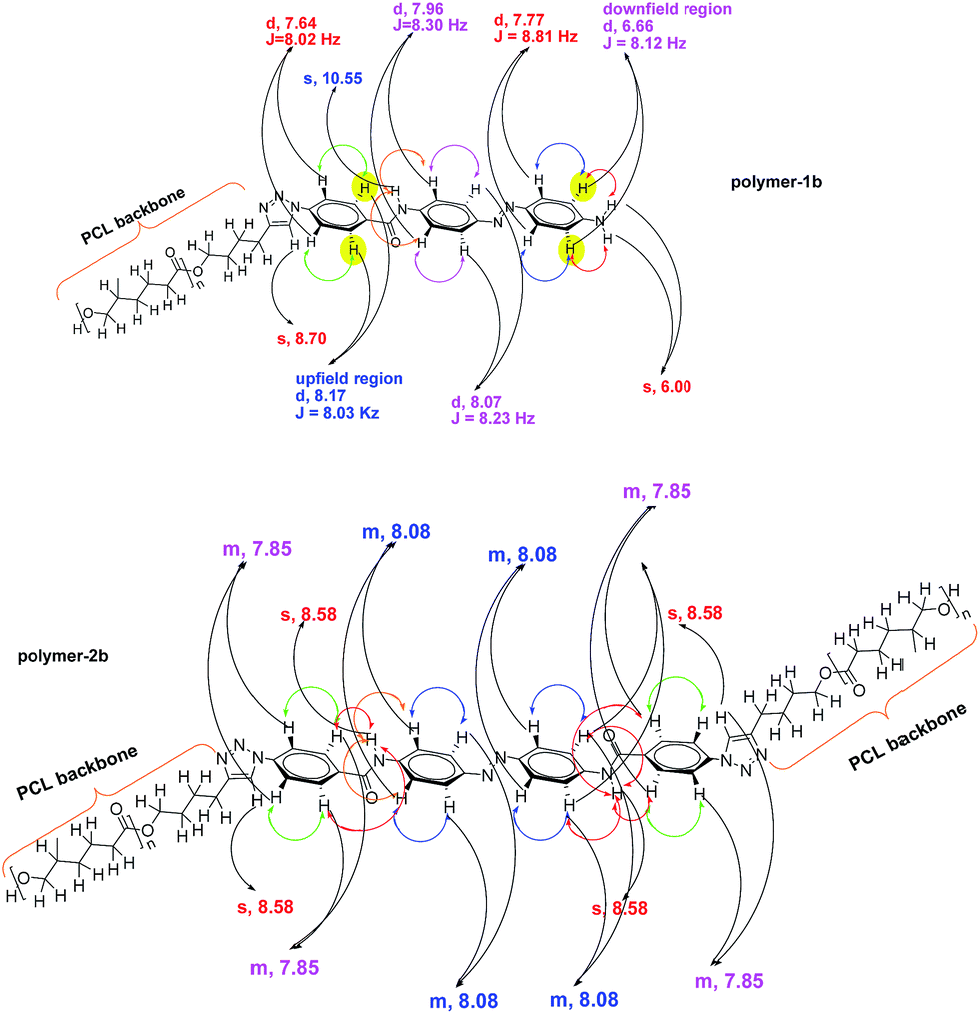 | ||
| Fig. 4 Main 2D-H/H-COSY correlations of polymer-1b and polymer-2b. The colored double headed arrows represent the long and short range couplings of protons from the H,H-COSY analysis, with the red ones showing the weak 1,4-coupling interactions. | ||
MALDI-TOF MS and ESI-TOF MS analysis of the photoswitchable azobenzene polymers
In order to prove the expected chemical composition of the photoswitchable polymers, a straight forward MALDI-TOF-MS analysis was conducted. Fig. 5, depicts the MALDI-TOF spectra of the photoswitchable polymers. Three main series are observed for polymer-1a, while polymer-2a shows four series. Each series shows a difference of 114 units, corresponding to the PCL repeating unit and is observed to be PCL containing the photoswitchable azobenzene dye moiety with different ions attached. According to Fig. 5 the signal appearing at 2161.9 g mol−1 can be simply assigned to a PCL containing the photoswitchable dye moiety with the chemical formula [C109H165N7O30Ag]+ (with n = 14), while the signal at 2131.0 g mol−1 can be represented by [M + 2K]2+. The signal at 2115.7 g mol−1 is also identified by the chemical formula [M + Na + K]2+.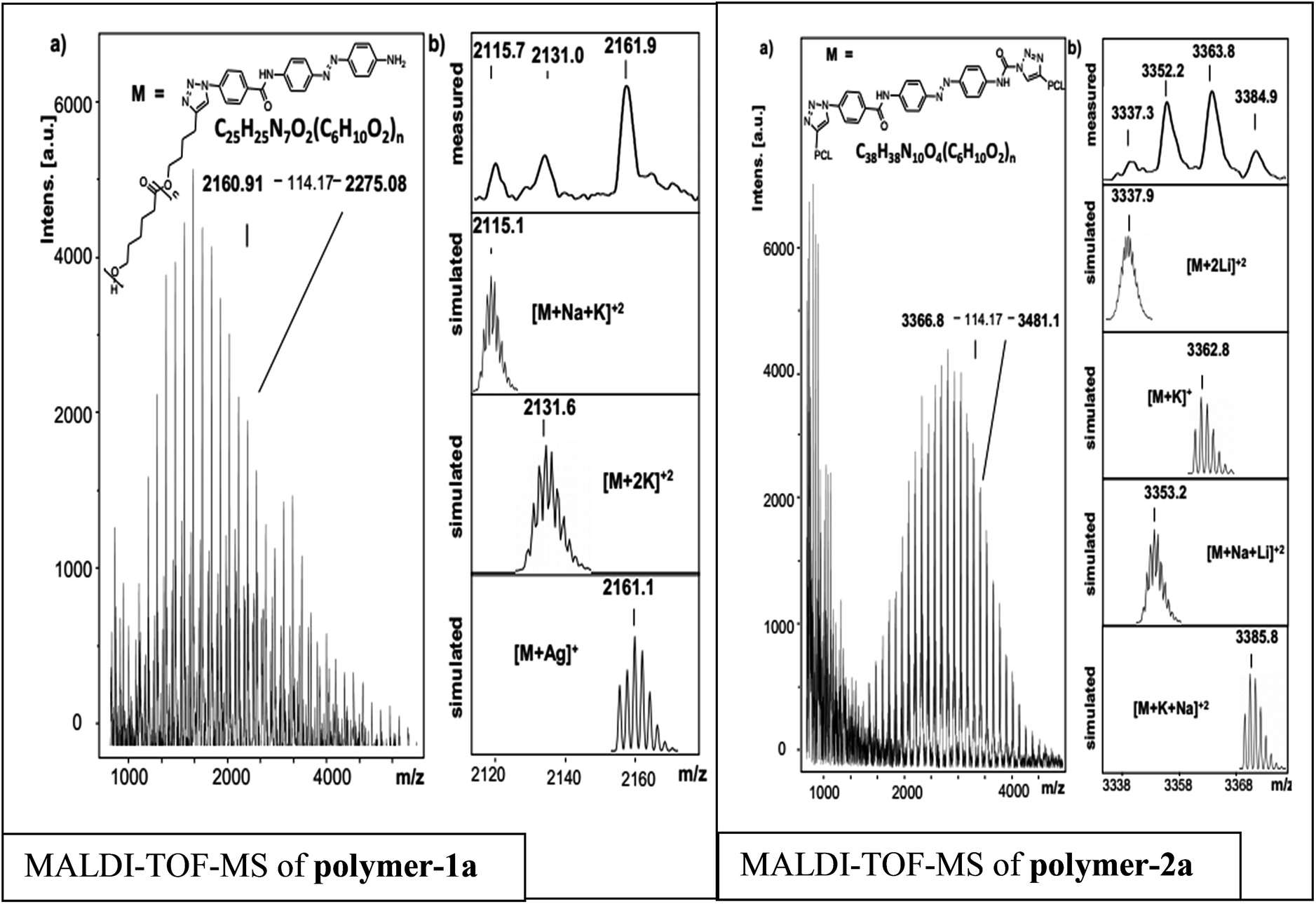 | ||
| Fig. 5 (a) Full MALDI-TOF-MS spectrum of polymer-1a and polymer-2a and (b) measured and simulated patterns of different series observed. | ||
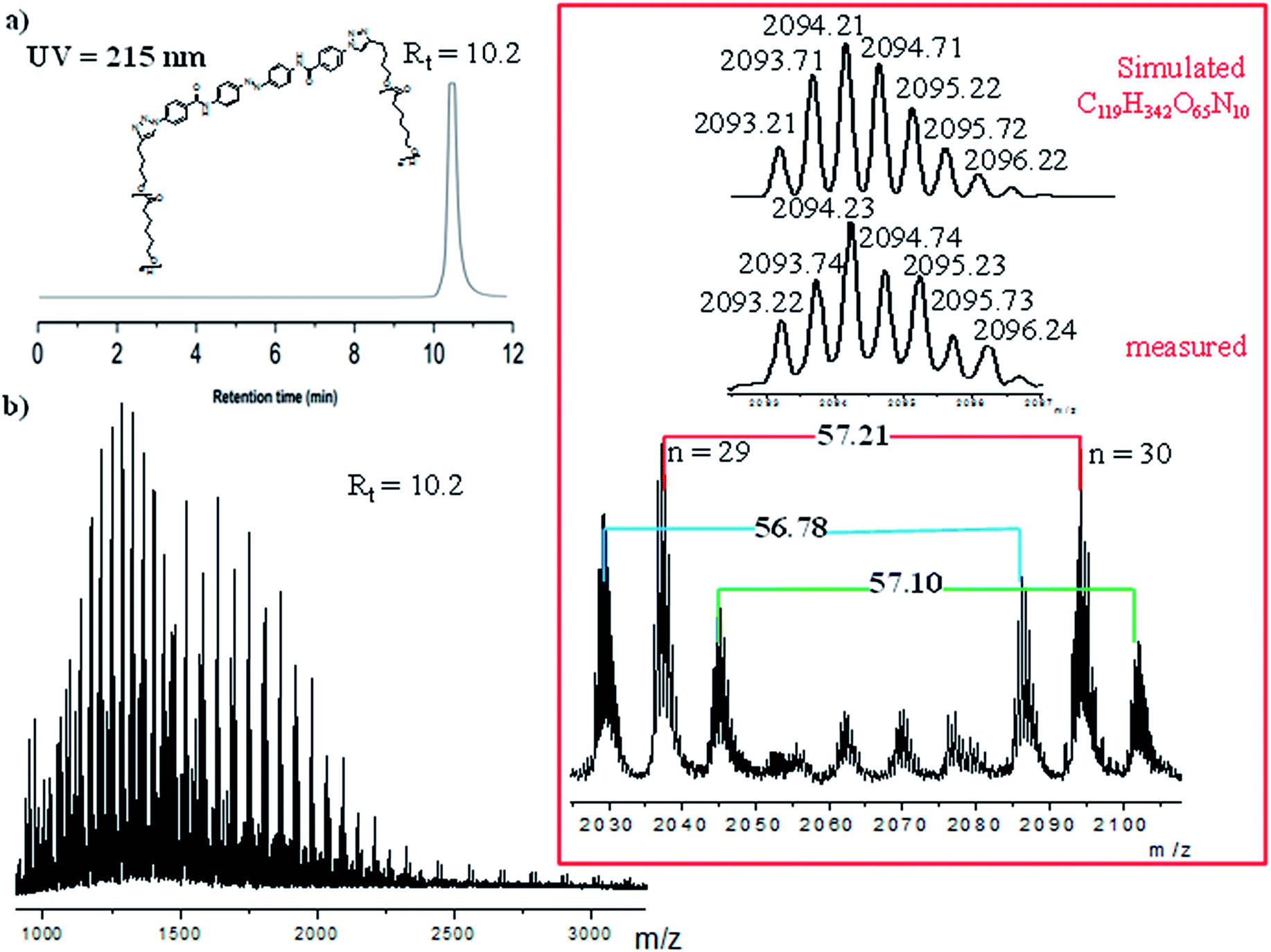
However, to fully proof the structures being presented in Scheme 1, we embarked on ESI-TOF MS investigations to further support the chemical composition of the two photoswitchable polymers. Fig. 6 and S12 (ESI†) show detailed spectra of all the different series of polymer-1a and polymer-2a. Whereas the monofunctional polymer-1a shows a single charged state, the bifunctional polymer-2a shows multiple charged states (1+, 2+ and 3+ ions). All the observed peaks were successfully simulated to match to the corresponding species. Thus e.g. the isotopic peaks of polymer-2a at m/z value of 723.37 g mol−1 correspond to fragments generated during the analysis (see ESI for the fragmented structures†). The isotopic peak at m/z = 2036.20 g mol−1 with a charge state of 2+ is observed to correspond to the species [M + H + CH3OH]2+, while the molecular ions peak at m/z = 1283.75 g mol−1 is identified as the high charge state of +3, corresponding to the species [M + 2Na + H]3+. The proof of an existence of a purely mono-armed polymer (polymer-1) and a bi-armed polymer (polymer-2) was evident from the presence of one detected 1,2,3-triazole group and two detected 1,2,3-triazole groups for polymer-1 and polymer-2 respectively. From the measured and simulated pattern results, it was obvious that the ESI-TOF analysis truly confirmed the structures of the compounds being presented.
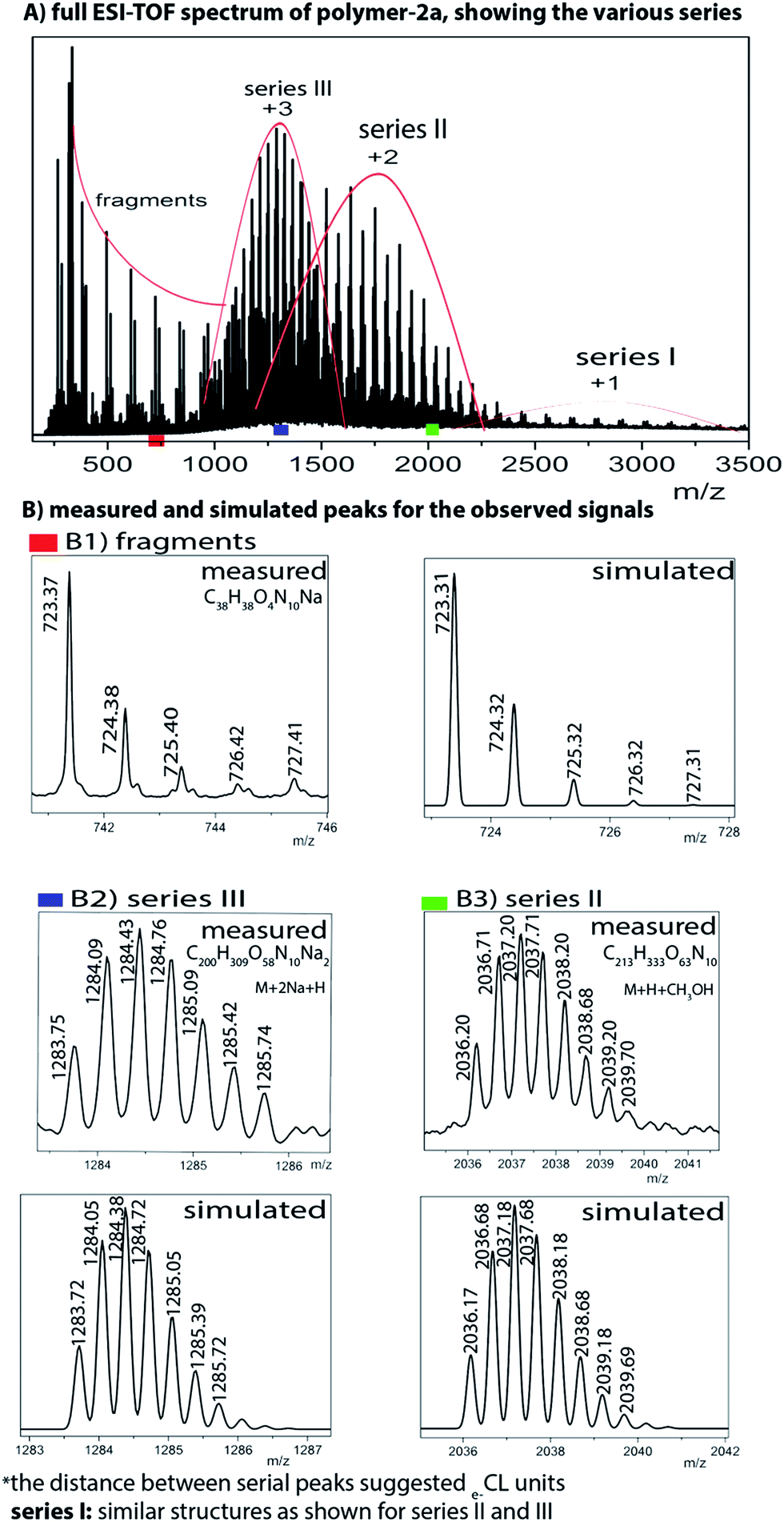 | ||
| Fig. 6 ESI-TOF MS spectra of the bifunctional photoswitchable polymer-2a, showing the observed and simulated signals. | ||
One-dimensional liquid chromatography (SEC and HPLC) and UV-Vis investigations
:DCM = 8:92). Fig. 7 shows the GPC traces of the photoswitchable polymers-1b and -2b as well as the alkyne-functionalized PCL precursors (PCL-alkyne3). The peak maximum of the polar sample polymer-1b shifted slightly towards higher retention time (Rt = 8.12 min) compared to the retention time of polymer-2b (Rt = 7.74 min). The symmetric monomodal distribution nature of the curves with different retention times, coupled to an exact match of the Mn values from NMR and GPC allowed the conclusion that the investigated samples were highly pure and were void of contaminants from their starting precursors.
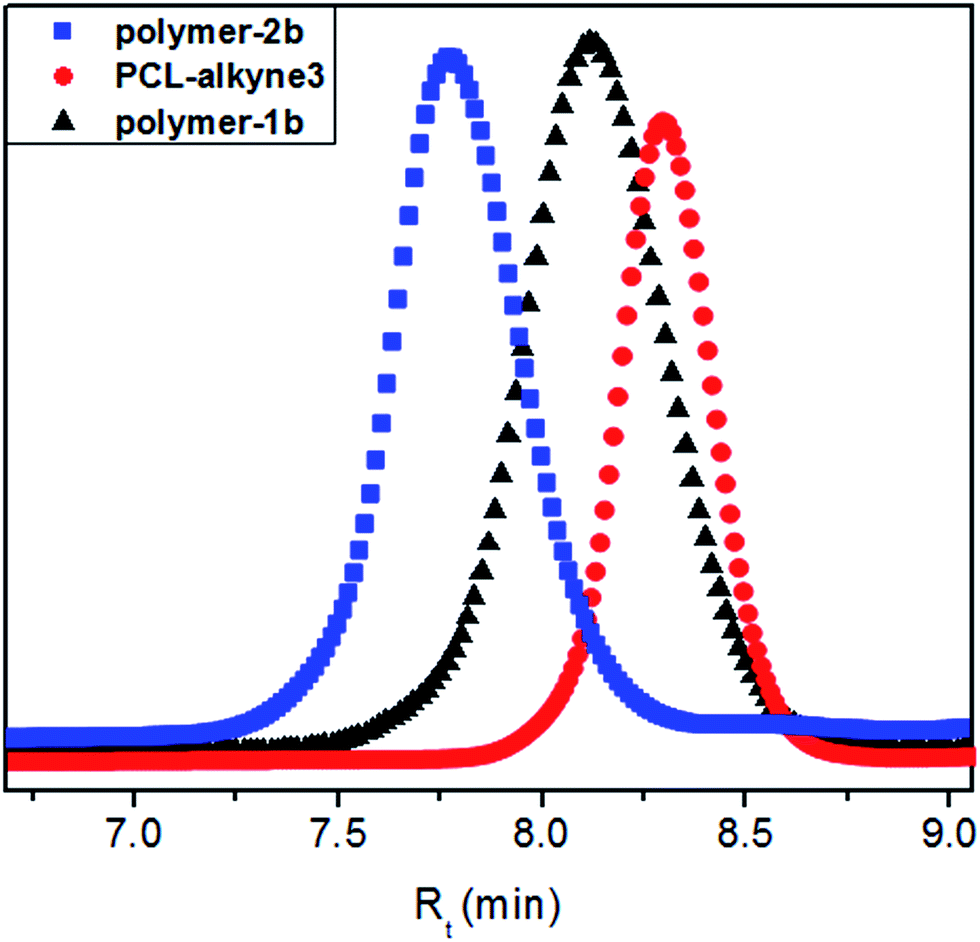 | ||
| Fig. 7 SEC traces of PCL-alkyne3, polymer-1b and polymer-2b. | ||
:DCM = 8:92, using non polar reverse phase Atlantis-RP C18 column) were furthermore conducted to confirm the purity of the obtained compounds, mainly to prove whether the structures presented in Scheme 1 were truly void of contaminants from their starting precursors and also not a mixture of the respective mono- and bi-substituted products. Fig. 8 shows the HPLC separation chromatogram of polymer-1b and -2b, as well as their starting precursors (2 and 3). The elution peaks of both polymer-1b and polymer-2b are well separated from each other. Polymer-2b elutes at a retention time of 10.2 min, 2.7 min higher than the retention time of polymer-1b. This difference is basically due to the PCL-unit being attached to the both ends of polymer-2b. Thus, incorporation of the PCL group onto both ends of polymer-2b reduces its polarity and hence leads to increased interaction within the Atlantis column, thereby resulting in higher retention times. The elution times of the polymers with similar functionalities (e.g.polymer-2a and -2b) were nearly the same, proving the LCCC conditions at these experiments.
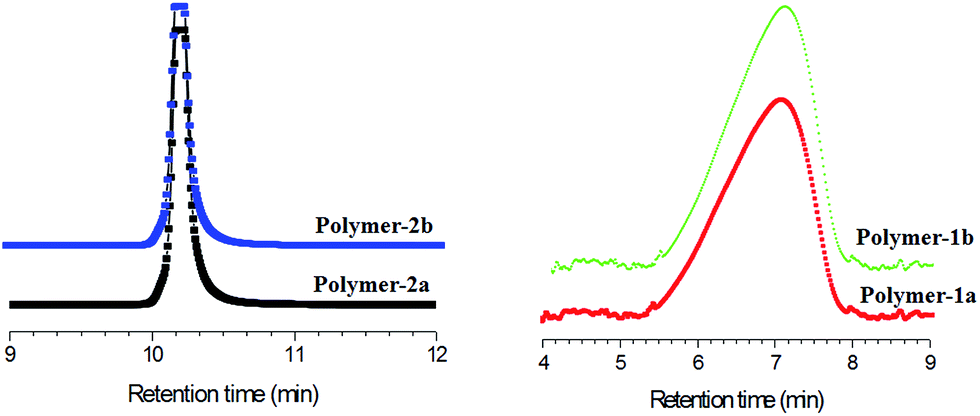 | ||
| Fig. 8 LC-UV traces analysis of the purified PCL-azobenzene polymers at LCCC conditions. | ||
 | ||
| Fig. 9 (a) LC-ESI-TOF-MS of polymer-2a indicating the purity of the sample. (b) Comparison between the measured and simulated isotopic pattern of a doubly charged species (Rt = 10.2 min) proving the final structure. | ||
Multidimensional chromatography (2D-LC/SEC) of the photoswitchable polymers
2D LC/SEC experiments were also conducted on the azobenzene polymers (see Fig. 10) with different molecular weights and end group functionalities. Fig. 10b shows the experiments on polymer-1a and -1b, having similar end group functionalities but different molecular weights. Thus on the LC-trace one observes nearly equal elution volumes for the two polymers at ∼0.97 mL retention volume. This proves that the experiments are separated under LCCC conditions and thus retention times of the polymers at this point are independent on the molecular masses. Thus changes in the functionalities of the compounds should be the only reason of the noticeable shift in the 2D contour diagram. Hence a comparison between polymer-1 and -2 having different endgroup functionalities should result in a significant change in the retention times. Fig. 10a shows a comparison of polymer-1a and -2a having different endgroup functionalities. The experiments unveiled a complete separation of the two photoswitchable polymers based on their functionality type distributions. Thus, as polymer-1a elutes at a retention volume of 0.97 mL, polymer-2a elutes at 1.43 mL retention volume, which clearly indicates that the two polymers were different in terms of their functionalities and also proves the structures presented in Scheme 1 to be pure and void of contaminants from each other.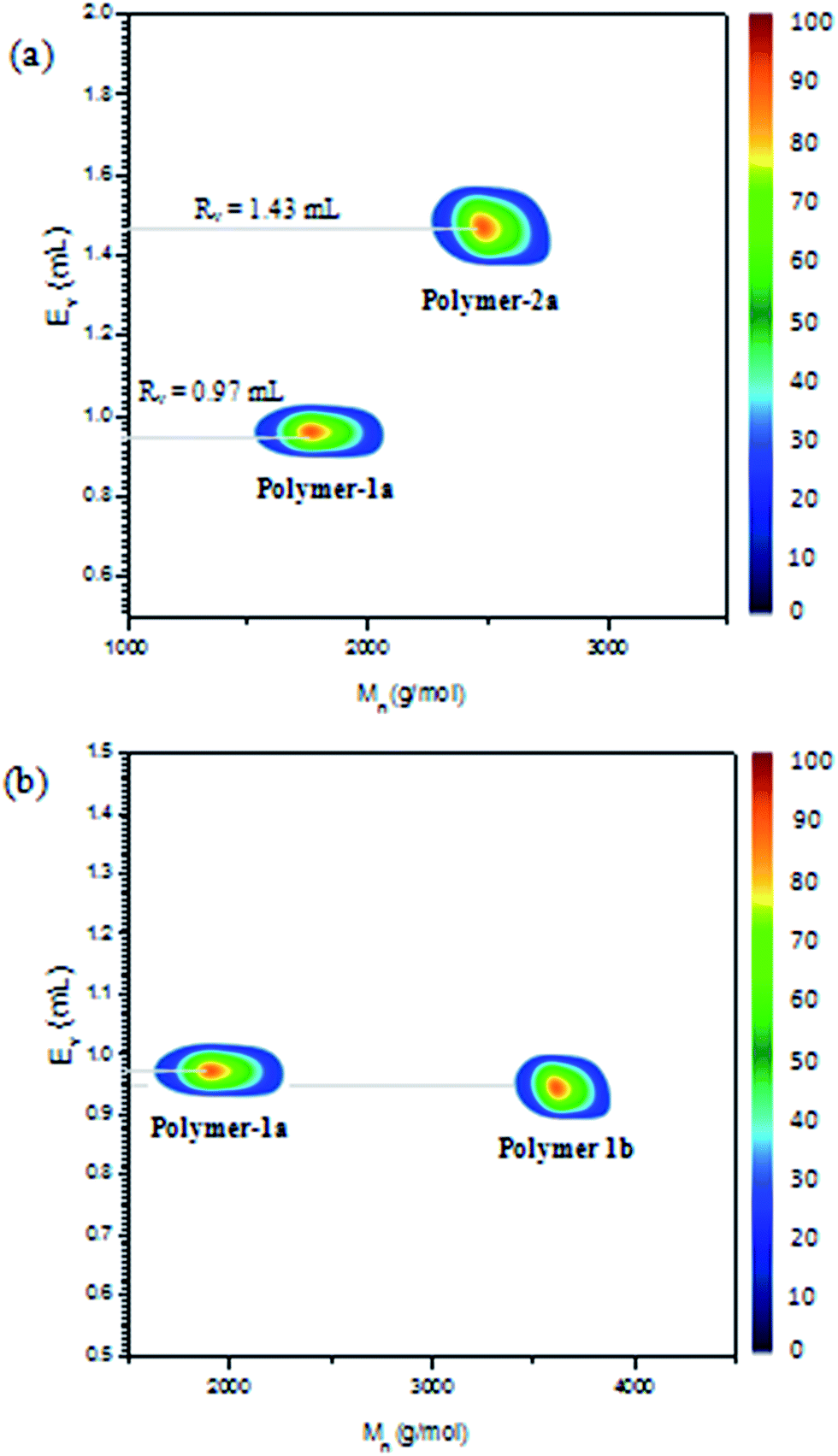 | ||
| Fig. 10 2D LC/SEC plots of (a) polymer-1a and polymer-2a showing separation based on their functionalites and (b) polymer-1a and polymer-2a with similar endgroup functionalities showing no noticeable changes in their LC-retention volumes, but a significant difference in their molecular weight in SEC. | ||
Conclusion
We here report a simple and efficient synthesis to obtain mono- and bi-armed azobenzene-containing polymers using “click” chemistry. The mono- and bi-armed PCL-azobenzene polymers were synthesized using the copper catalyzed azide-alkyne Huisgen cycloaddition reactions, attaching different molecular weights, ranging from 1000 g mol−1 to 7000 g mol−1, onto the azobenzene-backbone. The structure of the mono- and bi-armed polymers was proven via chromatographic techniques (HPLC under LCCC-conditions, LC/ESI-TOF MS) as well as 2D H/H COSY NMR experiments, also proving the purity of all of the samples and the excellent separation of the mono-armed polymers (polymer 1) from the bi-armed polymers (polymer 2). The investigated samples showed a significant thermal stability, when compared to their starting PCL-alkyne precursors.Acknowledgements
The authors express thanks for Grant SFB TRR 102 (CA, WHB) (Project A03) for financial support.References
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468 RSC.
- F. D. Jochum, L. zur Borg, P. J. Roth and P. Theato, Macromolecules, 2009, 42, 7854 CrossRef CAS.
- (a) J. García-Amorós and D. Velasco, Beilstein J. Org. Chem., 2012, 8, 1003 CrossRef PubMed; (b) L. Fang, H. Zhang, Z. Li, Y. Zhang, Y. Zhang and H. Zhang, Macromolecules, 2013, 46, 7650 CrossRef CAS.
- S. Moller, U. Pliquett and C. Hoffmann, RSC Adv., 2012, 2, 4792 RSC.
- H. Yu, J. Mater. Chem. C, 2014, 2, 3047 RSC.
- B. K. M. Lee, H. Koerner, D. H. Wang, L.-S. Tan, T. J. White and R. A. Vaia, Macromolecules, 2012, 45, 7527 CrossRef CAS.
- M. Irie and H. Tanaka, Macromolecules, 1983, 16, 210 CrossRef CAS.
- F. Agolini and F. P. Gay, Macromolecules, 1970, 3, 349 CrossRef CAS.
- C. D. Eisenbach, Polymer, 1980, 21, 1175 CrossRef CAS.
- H. S. Blair, H. I. Pague and J. E. Riordan, Polymer, 1980, 21, 1195 CrossRef CAS.
- X. Li, R. Wen, Y. Zhang, L. Zhu, B. Zhang and H. Zhang, J. Mater. Chem., 2009, 19, 236 RSC.
- C. Weber, T. Liebig, M. Gensler, L. Pithan, S. Bommel, D. Bléger, J. P. Rabe, S. Hecht and S. Kowarik, Macromolecules, 2015, 48, 1531 CrossRef CAS.
- K. G. Yager and C. J. Barrett, J. Photochem. Photobiol., A, 2006, 182, 250 CrossRef CAS.
- (a) C. M. Stuart, R. R. Frontiera and R. A. Mathies, J. Phys. Chem. A, 2007, 111, 12072 CrossRef CAS PubMed; (b) D. Fanghänel, G. Timpe and V. Orthman, in Organic Photochromes, ed. A. V. El'tsov, Consultants Bureau, New York, 1990, ch. 3 Search PubMed.
- K. G. Yager and C. Barrett, J. Mater. Chem., 2008, 424 CAS.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234 CrossRef CAS PubMed.
- C. R. Crecca and A. E. Roitberg, J. Phys. Chem. A, 2006, 110, 8188 CrossRef CAS PubMed.
- C. Barrett, A. Natansohn and P. Rochon, Chem. Mater., 1995, 7, 899 CrossRef CAS.
- G. S. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915 CrossRef CAS.
- X. Meng, A. Natansohn and P. Rochon, J. Polym. Sci., Part B: Polym. Phys., 1996, 34, 1461 CrossRef CAS.
- H.-K. Kim, X.-S. Wang, Y. Fujita, A. Sudo, H. Nishida, M. Fujii and T. Endo, Macromol. Rapid Commun., 2005, 26, 1032 CrossRef CAS.
- R. H. Lambeth and J. S. Moore, Macromolecules, 2007, 40, 1838 CrossRef CAS.
- N. Lomadze, A. Kopyshev, J. Rühe and S. Santer, Macromolecules, 2011, 44, 7372 CrossRef CAS.
- Y. Norikane, K. Kitamoto and N. Tamaoki, J. Org. Chem., 2003, 68, 8291 CrossRef CAS PubMed.
- L. Lamarre and C. S. P. Sung, Macromolecules, 1983, 16, 1729 CrossRef CAS.
- S. A. Nagamani, Y. Norikane and N. Tamaoki, J. Org. Chem., 2005, 70, 9304 CrossRef CAS PubMed.
- M. S. Vollmer, T. D. Clark, C. Steinem and M. R. Ghadiri, Angew. Chem., Int. Ed. Engl., 1999, 38, 1598 CrossRef CAS.
- M. S. Ho, C. Barrett, J. Paterson, M. Esteghamatian, A. Natansohn and P. Rochon, Macromolecules, 1996, 29, 4613 CrossRef CAS.
- A. Natansohn, P. Rochon, M. Pezolet, P. Audet, D. Brown and S. To, Macromolecules, 1994, 27, 2580 CrossRef CAS.
- X. Wang, S. Balasubramanian, J. Kumar, S. K. Tripathy and L. Li, Chem. Mater., 1998, 10, 1546 CrossRef CAS.
- B. Gallot, M. Fafiotte, A. Fissi and O. Pieroni, Macromol. Rapid Commun., 1996, 17, 493 CrossRef CAS.
- N. K. Viswanathan, D. Yu Kim, S. Bian, J. Williams, W. Liu, L. Li, L. Samuelson, J. Kumar and S. K. Tripathy, J. Mater. Chem., 1999, 9, 1941 RSC.
- X. Meng, A. Natansohn, C. Barrett and P. Rochon, Macromolecules, 1996, 29, 946 CrossRef CAS.
- M. Sisido, Y. Ishikawa, K. Itoh and S. Tazuke, Macromolecules, 1991, 24, 3993 CrossRef CAS.
- M. Sisido, Y. Ishikawa, M. Harada and K. Itoh, Macromolecules, 1991, 24, 3999 CrossRef CAS.
- V. E. Campbell, I. In, D. J. McGee, N. Woodward, A. Caruso and P. Gopalan, Macromolecules, 2006, 39, 957 CrossRef CAS.
- A. Momotake and T. Arai, Polymer, 2004, 45, 5369 CrossRef CAS.
- J. del Barrio, L. Oriol, R. Alcalá and C. Sánchez, Macromolecules, 2009, 42, 5752 CrossRef CAS.
- H. Ma, S. Liu, J. Luo, S. Suresh, L. Liu, S. H. Kang, M. Haller, T. Sassa, L. R. Dalton and A. K. Y. Jen, Adv. Funct. Mater., 2002, 12, 565 CrossRef CAS.
- R. Birabassov, N. Landraud, T. V. Galstyan, A. Ritcey, C. G. Bazuin and T. Rahem, Appl. Opt., 1998, 37, 8264 CrossRef CAS PubMed.
- F. Lagugné Labarthet, T. Buffeteau and C. Sourisseau, J. Phys. Chem. B, 1998, 102, 2654 CrossRef.
- S. Shinkai, T. Minami, Y. Kusano and O. Manabe, J. Am. Chem. Soc., 1983, 105, 1851 CrossRef CAS.
- I. Willner and S. Rubin, Angew. Chem., Int. Ed. Engl., 1996, 35, 367 CrossRef CAS.
- X. Yu, Y. Luo, Y. Deng, Q. Yan, G. Zou and Q. Zhang, Eur. Polym. J., 2008, 44, 881 CrossRef CAS.
- C. Xu, B. Wu, M. W. Becker, L. R. Dalton, P. M. Ranon, Y. Shi and W. H. Steier, Chem. Mater., 1993, 5, 1439 CrossRef CAS.
- M. Irie and W. Schnabel, Macromolecules, 1981, 14, 1246 CrossRef CAS.
- E. Ostas, K. Schröter, M. Beiner, T. Yan, T. Thurn-Albrecht and W. H. Binder, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 3404 CrossRef CAS.
- C. Appiah, J. Akbarzadeh, H. Peterlik and W. H. Binder, Eur. Polym. J., 2015, 64, 138 CrossRef CAS.
- L. Mespouille, M. Vachaudez, F. Suriano, P. Gerbaux, O. Coulembier, P. Degée, R. Flammang and P. Dubois, Macromol. Rapid Commun., 2007, 28, 2151 CrossRef CAS.
- H. Barqawi and W. H. Binder, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 4855 CrossRef CAS.
- A. Stojanovic, C. Appiah, D. Dohler, J. Akbarzadeh, P. Zare, H. Peterlik and W. H. Binder, J. Mater. Chem. A, 2013, 1, 12159 CAS.
- B. t. Baptiste, C. l. Douat-Casassus, K. Laxmi-Reddy, F. d. r. Godde and I. Huc, J. Org. Chem., 2010, 75, 7175 CrossRef CAS PubMed.
- H. Barqawi, E. Ostas, B. Liu, J.-F. Carpentier and W. H. Binder, Macromolecules, 2012, 45, 9779 CrossRef CAS.
- H. Barqawi, M. Schulz, A. Olubummo, V. Saurland and W. H. Binder, Macromolecules, 2013, 46, 7638 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed NMR analysis of 4-azidobenzoic acid, 4-azidobezoylchloride, PCL-alkyne3, cpd. 1, cpd. 2 and cpd. 3; H/H-COSY of polymer-1b; TGA curves; FTIR experiments and ESI-TOF MS of polymer 1. See DOI: 10.1039/c5ra25216d |
| This journal is © The Royal Society of Chemistry 2016 |