
A thermally remendable and reprocessable crosslinked methyl methacrylate polymer based on oxygen insensitive dynamic reversible C–ON bonds†
Ze Ping Zhanga,
Yan Lua,
Min Zhi Rong*b and
Ming Qiu Zhang*b
aKey Laboratory for Polymeric Composite and Functional Materials of Ministry of Education, GD HPPC Lab School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China
bMaterials Science Institute, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: cesrmz@mail.sysu.edu.cn; ceszmq@mail.sysu.edu.cn
First published on 11th January 2016
Abstract
To improve the room temperature application stability of a dynamically reversible self-healing system based on alkoxyamine moieties, and to provide the polymer with oxygen insensitive self-healability, a crosslinked methyl methacrylate polymer with embedded alkoxyamine moieties that contain amido groups is prepared. Owing to synchronous fission/radical recombination of C–ON bonds, cracks in the polymer can be repeatedly healed without an additional catalyst, and the polymer itself can be remolded in the solid state or even decomposed by nitroxides in a controlled manner. The homolysis temperature of the dynamic reversible alkoxyamine derivative, which is closely related to crack healing and the reprocessing temperature of the polymer, is adjusted to be higher than room temperature by means of the appropriate electron-absorbing feature of amido groups. Meanwhile, the healing chemistry is coupled with air resistance. Besides, thiol–ene click chemistry is used to synthesize the target polymer. The possible side reaction that reduces the concentration of reversible bonds during polymerization is minimized accordingly.
Introduction
Intrinsic self-healing covalent polymers take effect through disconnection and reconnection of reversible bonds under certain stimuli (e.g., heat, light, pH, etc.).1,2 Although room temperature intrinsic self-healing has been enabled by dynamic reversible bonds,3–14 which is analogous to extrinsic self-healing polymers containing microencapsulated healing agents,15 the actual scope of application has to be narrowed accordingly. This is because the ceaseless scrambling of the reversible bonds at ambient temperature would lead to structural instability when bearing load, and the materials are not suitable for working under long-term stress conditions. In this context, imparting intrinsic self-healing polymers with a healing temperature higher than their operation temperature is desired.16Our previous investigation revealed that polymers carrying alkoxyamine moieties as intra- or intermacromolecular linkages are allowed for self-healing.8,9,17–19 The damage healing is completed at a single temperature owing to synchronous fission and radical recombination of the C–ON bonds, which is different from the healing through Diels–Alder reaction that has to proceed at two temperatures.20 In addition, self-healing at either low temperature8 (15 and 25 °C) or elevated temperature17 (130 °C) offers high healing efficiencies. On the basis of these pilot studies, we continue to explore the following two issues in this work: (i) facile strategy of tuning homolysis temperature of alkoxyamines (which is directly related to healing temperature of ultimate polymers) and increasing their oxidation resistance, and (ii) promising approach of preparing polymers containing alkoxyamine moieties. The research is of particular importance for material design, as a toolbox may be generated for coupling oxygen insensitive self-healability with different polymers (i.e. polymers with different working temperatures).
The authors believe that synthesis of alkoxyamines by using azo initiators can easily adjust homolysis temperature of the former because different groups attached to the carbon atom of C–ON bonds exert different electronic effects and conjugation effects. In general, the factors (i.e. electronic effects, conjugative effect and steric effect) that are able to reduce the bond energy of C–ON bonds would increase the stability of carbon-centered radicals and decrease homolysis temperature of C–ON bonds. Dozens of commercially available azo initiators imply that the tunable temperature range could be rather wide. Hereinafter amide-containing azo initiator, 2,2′-azobis(2-methyl-N-(2-hydroxyethyl) propionamide) (AMHEPA), is applied to produce the dynamic reversible monomer, 2-(4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yloxy)-N-(2-hydroxyethyl)-2-methylpropanamide (Diol, Scheme 1a). Since amide possesses poorer electron-withdrawing conjugation effect property than nitrile, the homolysis temperature of Diol should be higher than that of the alkoxyamine derivative made from nitrile azo initiator,8 while anti-oxidization property of such alkoxyamine might remain.
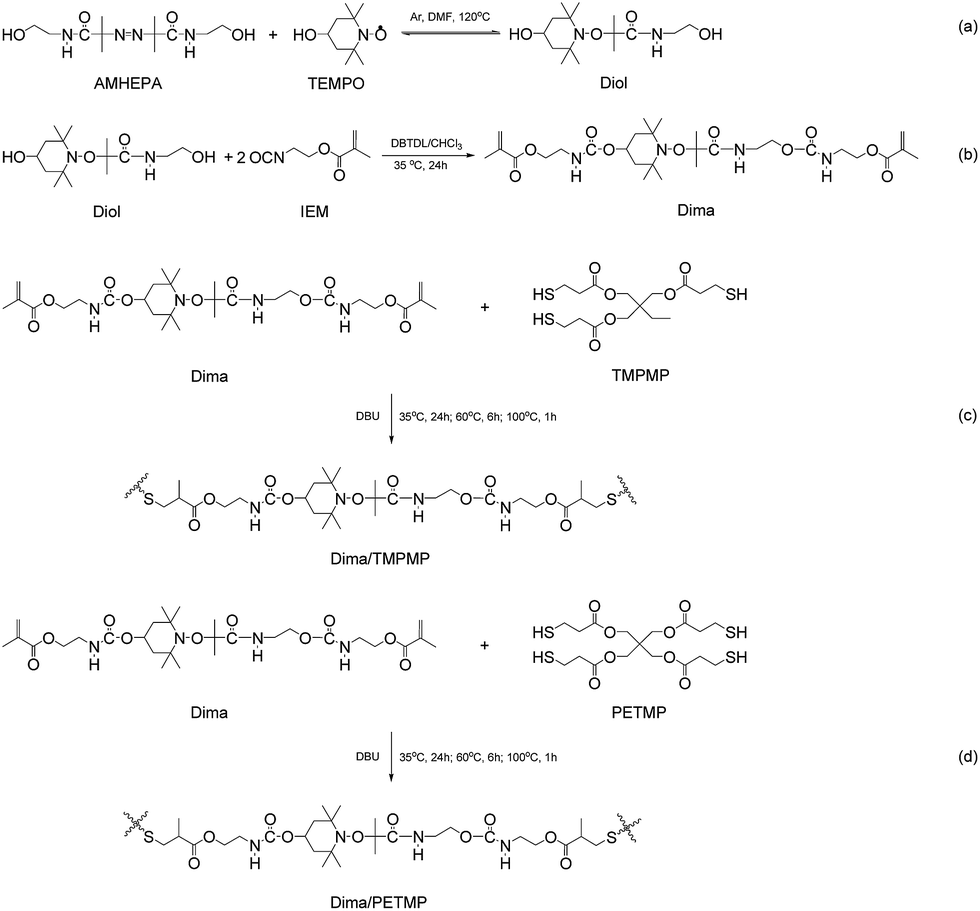 | ||
| Scheme 1 Synthesis of (a) Diol, (b) Dima, (c) Dima/TMPMP and (d) Dima/PETMP. | ||
On the other hand, synthesis of alkoxyamine-containing macromolecules toward introduction of dynamic reversible bonds should avoid irreversible combination of carbon-centered radicals. Otherwise, molecular weight of target polymer would be hard to be increased and the concentration of dynamic reversible bonds has to be low.21 Considering that homolysis temperature of C–ON bonds can be raised by using amide-containing azo initiator as the raw material, we construct the self-healing polymer via thiol–ene click chemistry that proceeds rapidly in air at room temperature. Naturally, the above-mentioned unwanted side reaction would be avoided during polymerization. In reality, the Diol is converted to urethane methacrylate (2-(4-(2-(carbamate)ethylmethacrylate)-2,2,6,6-tetramethylpiperidin-1-yloxy)-N-(2-hydroxyethyl)-2-methylpropanamide, Dima) by the reaction with 2-isocyanatoethyl methacrylate (IEM, Scheme 1b). Then, Dima reacts with trimethylol-propane tri(3-mercaptopropionate) (TMPMP) under catalysis of nucleophile 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU), forming crosslinked methyl methacrylate elastomer (Dima/TMPMP, Scheme 1c). There are also quite a few thiol curing agents with different functionalities and structures, which favors property control.
The following discussion is mainly dedicated to verification of the proposed ideas. Structure and properties relationship of the crosslinked version of the above-mentioned methyl methacrylate polymer is studied in detail.
Experimental
Materials and reagents
4-Hydroxy-2,2,6,6-tetramethylpiperidinyloxy (TEMPO, 98%) was purchased from Aldrich. 2,2′-Azobis(2-methyl-N-(2-hydroxyethyl) propionamide) (AMHEPA, 98%) was supplied by Wuhan Fude Chemical Co., Ltd, China. Trimethylol-propane tri(3-mercaptopropionate) (TMPMP, 95%), pentaerythritol tetra(3-mercaptopropionate) (PETMP, 95%) and poly(ethyleneglycol)dimethacrylate (PEGDMMA, Mn = 750 g mol−1) were obtained from Sigma Aldrich. Dibutyltindilaurate (DBTDL, 95%), 1,8-diazabicyclo(5.4.0)undec-7-ene (DBU, 98%), and 2-isocyanatoethyl methacrylate (IEM, 98%) were supplied by TCI. Chloroform was dried over activated molecular sieves (4 Å) prior to use. All the other chemicals and solvents were used as received.Synthesis of hydroxyl-terminated alkoxyamine (Diol)
A typical process of synthesis of 2-(4-hydroxy-2,2,6,6-tetramethylpiperidin-1-yloxy)-N-(2-hydroxyethyl)-2-methylpropanamide (Diol) is described as follows. 2,2′-Azobis(2-methyl-N-(2-hydroxyethyl) propionamide) (AMHEPA, 2.883 g, 0.01 mol) and 4-hydroxy-2,2,6,6-tetramethylpiperidinyloxy (TEMPO, 1.72 g, 0.01 mol) were completely dissolved in 100 ml dimethyl formamide (DMF). Next, the mixture was heated up to 120 °C and stirred in argon for 5 h. Afterwards, the colorless solution was concentrated under vacuum and purified by column chromatography (silica gel) gradient eluted with 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 hexane–ethyl acetate (v/v) and pure ethyl acetate in turn. Proton nuclear magnetic resonance spectrum (1H NMR, 400 MHz, dimethyl sulfoxide (DMSO), 25 °C, Fig. S1, ESI†) with the following characteristic peaks demonstrates chemical structure of the product: δ/ppm 0.998, 1.154, 1.337, 1.701, 1.853, 1.969, 3.151, 3.370, 3.760, 4.444, 4.712, 7.394 and 7.874. 0.998–1.969 (complex, 23H, CH3, CH2, and CH of the TEMPO group, CH3 linked to the quaternary carbon atom), 3.151 (1H, HO–CH–(CH2)2), 3.370 (2H, –CO–NH–CH2), 3.760 (2H, CH2–CH2–OH), 4.444 (1H, reactive hydrogen of –OH connect to TEMPO group) and 4.712 (1H, reactive hydrogen of another –OH), 7.394 (1H, reactive hydrogen of amido, hydrogen bond), and 7.874 (1H, reactive hydrogen of amido, non-hydrogen bond). Carbon-13 nuclear magnetic resonance spectrum (13C NMR, 400 MHz, DMSO, 25 °C, Fig. S2, ESI†): δ/ppm 19.732, 21.544, 25.087, 33.526, 39.762, 41.703, 42.260, 49.510, 59.922, 61.275, 82.241, and 175.824. Fourier transform infrared spectrum (FTIR, KBr/cm−1, Fig. S3, ESI†): 3363, 2987, 2940, 2877, 1658, 1538, 1467, 1377, 1365, 1221, 1180, 1147, 1052, 956, and 738. Mass spectrum exact mass calculated for [M + 1]+ C15H30N2O4 302.41; found 303.40 (Fig. S4, ESI†). Elea. anal. calcd for C15H30N2O4: C, 59.6%; H, 9.9%; N, 9.3%. Found: C, 59.1%; H, 9.7%; N, 9.5%.
:3 hexane–ethyl acetate (v/v) and pure ethyl acetate in turn. Proton nuclear magnetic resonance spectrum (1H NMR, 400 MHz, dimethyl sulfoxide (DMSO), 25 °C, Fig. S1, ESI†) with the following characteristic peaks demonstrates chemical structure of the product: δ/ppm 0.998, 1.154, 1.337, 1.701, 1.853, 1.969, 3.151, 3.370, 3.760, 4.444, 4.712, 7.394 and 7.874. 0.998–1.969 (complex, 23H, CH3, CH2, and CH of the TEMPO group, CH3 linked to the quaternary carbon atom), 3.151 (1H, HO–CH–(CH2)2), 3.370 (2H, –CO–NH–CH2), 3.760 (2H, CH2–CH2–OH), 4.444 (1H, reactive hydrogen of –OH connect to TEMPO group) and 4.712 (1H, reactive hydrogen of another –OH), 7.394 (1H, reactive hydrogen of amido, hydrogen bond), and 7.874 (1H, reactive hydrogen of amido, non-hydrogen bond). Carbon-13 nuclear magnetic resonance spectrum (13C NMR, 400 MHz, DMSO, 25 °C, Fig. S2, ESI†): δ/ppm 19.732, 21.544, 25.087, 33.526, 39.762, 41.703, 42.260, 49.510, 59.922, 61.275, 82.241, and 175.824. Fourier transform infrared spectrum (FTIR, KBr/cm−1, Fig. S3, ESI†): 3363, 2987, 2940, 2877, 1658, 1538, 1467, 1377, 1365, 1221, 1180, 1147, 1052, 956, and 738. Mass spectrum exact mass calculated for [M + 1]+ C15H30N2O4 302.41; found 303.40 (Fig. S4, ESI†). Elea. anal. calcd for C15H30N2O4: C, 59.6%; H, 9.9%; N, 9.3%. Found: C, 59.1%; H, 9.7%; N, 9.5%.
Synthesis of vinyl-difunctionalized alkoxyamine (Dima)
2-(4-(2-(Carbamate)ethylmethacrylate)-2,2,6,6-tetramethylpiperidin-1-yloxy)-N-(2-hydroxyethyl)-2-methylpropanamide (Dima) is synthesized through the following procedure. 1.51 g (0.005 mol) Diol with hydroxyl groups and 0.15 g 2-isocyanatoethyl methacrylate (IEM, 0.01 mol) were dissolved in 20 ml CHCl3 (over freshly activated molecular sieves for 1 week at room temperature) and placed in a 50 ml two-necked flask. 20 mg DBTDL was then dropped into the flask, and the mixture was heated up to 35 °C and stirred in argon for 24 h until the reaction is completed. Afterwards, the solution was concentrated under vacuum to obtain a yellow liquid with yield of 97.4%. 1H NMR (400 MHz, DMSO, 25 °C, Fig. S5, ESI†): δ/ppm 0.986, 1.190, 1.336, 1.717, 1.886, 1.966, 3.281, 3.997, 4.011, 4.082, 5.681, 6.067, 7.208–7.530, 7.969. 0.986–1.966 (complex, 27H, CH3 and CH2 of the TEMPO group, CH3 linked to quaternary carbon atom, CH3 linked to ethylene linkage), 3.281 (4H, –OOC–NH–CH2), 3.997 (1H, –CH of the TEMPO group), 4.011 (2H, –CH2–COO–NH), 4.082 (2H, –CH2–OOC–), 5.681 and 6.067 (4H,![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2), 7.208–7.530 (3H, reactive hydrogen of amido, hydrogen bond), and 7.969 (3H, reactive hydrogen of amido, non-hydrogen bond). 13C NMR (400 MHz, DMSO, 25 °C, Fig. S6, ESI†): δ/ppm 18.309, 19.147, 21.847, 25.090, 38.753, 39.011, 45.770, 49.581, 59.920, 62.573, 63.455, 64.515, 82.199, 126.589, 136.375, 156.253, 158.427, 166.866, 176.101. FTIR (KBr/cm−1, Fig. S7, ESI†): 3357, 3085, 2975, 2955, 2928, 2865, 1717, 1661, 1637, 1533, 1457, 1405, 1376, 1321, 1298, 1260, 1220, 1166, 1048, 950, and 738. Mass spectrum exact mass calculated for [M + 1]+ C29H48N4O10 612.67; found 613.10. Elea. anal. calcd for C29H48N4O10: C, 56.8%; H, 7.9%; N, 9.1%. Found: C, 59.1%; H, 9.7%; N, 9.5%.
CH2), 7.208–7.530 (3H, reactive hydrogen of amido, hydrogen bond), and 7.969 (3H, reactive hydrogen of amido, non-hydrogen bond). 13C NMR (400 MHz, DMSO, 25 °C, Fig. S6, ESI†): δ/ppm 18.309, 19.147, 21.847, 25.090, 38.753, 39.011, 45.770, 49.581, 59.920, 62.573, 63.455, 64.515, 82.199, 126.589, 136.375, 156.253, 158.427, 166.866, 176.101. FTIR (KBr/cm−1, Fig. S7, ESI†): 3357, 3085, 2975, 2955, 2928, 2865, 1717, 1661, 1637, 1533, 1457, 1405, 1376, 1321, 1298, 1260, 1220, 1166, 1048, 950, and 738. Mass spectrum exact mass calculated for [M + 1]+ C29H48N4O10 612.67; found 613.10. Elea. anal. calcd for C29H48N4O10: C, 56.8%; H, 7.9%; N, 9.1%. Found: C, 59.1%; H, 9.7%; N, 9.5%.
Preparation of crosslinked methacrylate ester with alkoxyamine and the references
Cross-linked methacrylate esters with alkoxyamine were synthesized by amine-catalyzed thiol–ene click reaction.22 A stoichiometric mixture of the thermo-reversible monomer Dima and TMPMP was mixed under stirring. Then, 0.5 wt% DBU was added to the system. Finally, the polymer sheet (Dima/TMPMP) was obtained by curing at 35 °C for 24 h, 60 °C for 6 h and 100 °C for 1 h in a silicone mold. FTIR (ATR/cm−1, Fig. S8, ESI†): 3366, 2977, 2932, 1729, 1665, 1527, 1460, 1376, 1353, 1155, 1046, and 771.Dima/PETMP polymer with higher crosslinking density was also prepared following similar procedures (Scheme 1d) in order to examine its influence on healing behavior. FTIR (ATR/cm−1, Fig. S9, ESI†): 3373, 2980, 2933, 1724, 1666, 1516, 1458, 1377, 1350, 1157, 1047, and 771.
The reference material (PEGDMMA/TMPMP) that excludes alkoxyamine moiety was prepared by replacing Dima with poly(ethyleneglycol)dimethacrylate (PEGDMMA, Scheme 2), because the latter possesses similar molecular length as Dima but more flexibility. FTIR (ATR/cm−1, Fig. S10, ESI†): 2876, 1736, 1469, 1458, 1348, 1250, 1099, and 851.
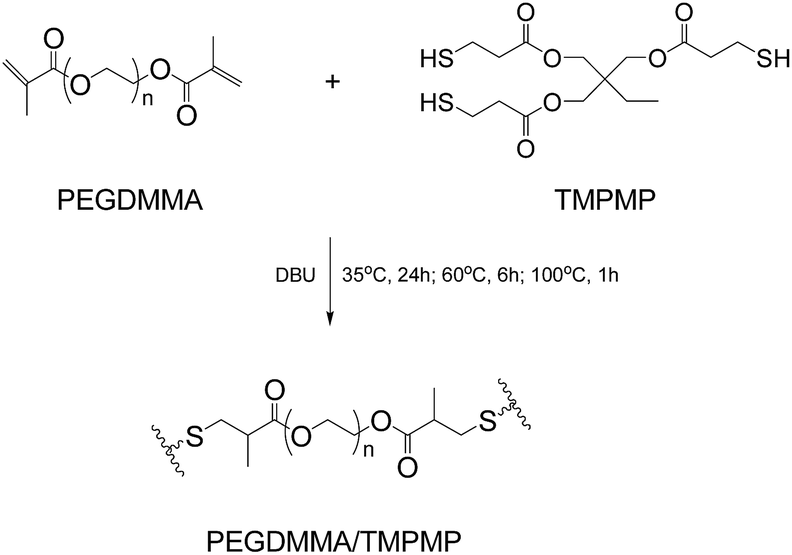 | ||
| Scheme 2 Synthesis of PEGDMMA/TMPMP. | ||
Characterization
1H NMR and 13C NMR spectra were measured by an AVANCE III 400 MHz (400 MHz) with DMSO as solvent. FTIR spectra were recorded by a Bruker EQUINOX55 Fourier transformation infrared spectrometer coupled with an infrared microscope spectrometer. Mass spectra were obtained from a Thermo LCQ DECA XP spectroscopy. FT-Raman spectroscopy was measured by a Nicolet NXR 9650, and the spectra were collected by placing the reaction mixture or polymer in a quartz NMR tube. 128 spectra with a resolution of 8 cm−1 over a spectral range of 50–3500 cm−1 were signal-averaged. Electron spin resonance (ESR) spectroscopy study was conducted on a Bruker A300-10-12 spectrometer equipped with nitrogen heating setup operating at 8.85 Hz. Modulation frequency and amplitude were 100 kHz and 0.1 mT, respectively. Dynamic mechanical analysis (DMA) was carried out on a METRA-VIBDMA25 using tension mode under 1 Hz at a heating rate of 3 °C min−1 in nitrogen. Molecular weight between crosslinks of the crosslinked methacrylate ester, Mc, was calculated from:23| Mc = 3(1 − 2/ϕ)ρRT/E′ | (1) |
Stress relaxation behavior was recorded by METRA-VIBDMA25 (DMA) using tension mode. It performed at 10% strain applied at t = 0 min. Creep was measured by the same machine using tension mode at a stress that is 30% of the static ultimate tensile strength of each specimen.
To evaluate self-healing ability of the materials, tensile test was performed on dumbbell-shaped specimens according to ISO527-2 (20 × 4 × 2 mm3, grip to grip separation: 20 mm) at 50 mm min−1 on a SANSCMT6103 universal tester. Efficiency of healing is defined as the ratio of tensile strengths of healed and virgin materials. Healing was conducted at 80 °C by bringing cut samples together. Gentle pressure of about 0.2 MPa was applied to ensure that the faces of the two halves were kept in alignment and intimate contact.24 Finally, the healed specimens were tested to failure following the above procedure to check the effect of healing. Each batch included five specimens to yield an average value.
Results and discussion
Fig. 1 gives FT-Raman spectra of the stoichiometric mixture of Dima/TMPMP before and after curing. The strong absorptions at 2571 and 1637 cm−1, which are assigned to the stretching vibration of thiol group and carbon–carbon double bond, respectively, are absent on the spectrum of the cured version. It means that the thiol hardener has been completely consumed and the Dima is fully cured. The deduction is supported by the results of multiple DMA scans of the cured Dima/TMPMP (Fig. 2), which show that there is only a single tanδ peak on each curve and the peak temperature (i.e. Tg) of each curve is almost identical (∼27 °C). Meanwhile, Dima/PETMP with higher crosslinking density (Fig. S11, ESI† and 2) and the control PEGDMMA/TMPMP (Fig. S12 and S13, ESI†) are also completely cured, achieving Tg of 34 °C and −41 °C, respectively.
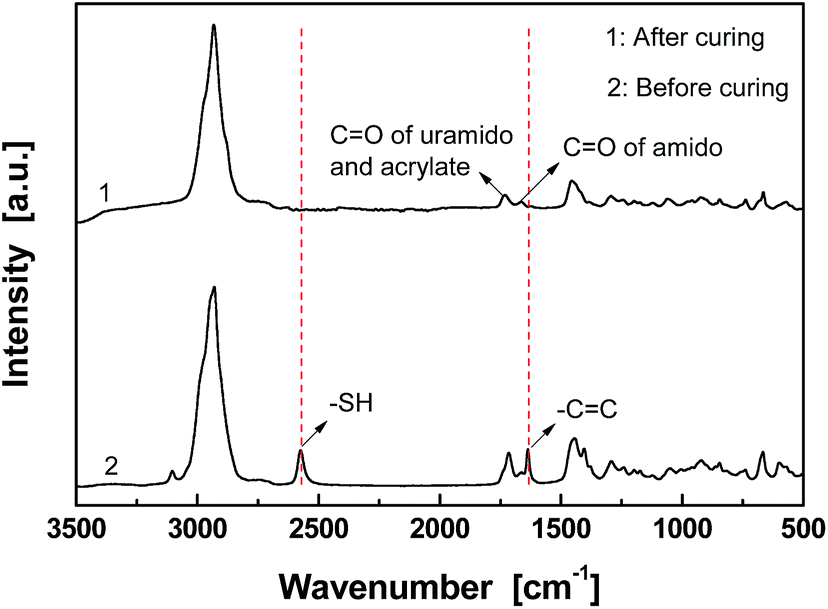 | ||
| Fig. 1 FT-Raman spectra of Dima/TMPMP before and after curing. Before curing, catalyst DBU was absent in the mixture of Dima/TMPMP so that no crosslinking reaction took place. | ||
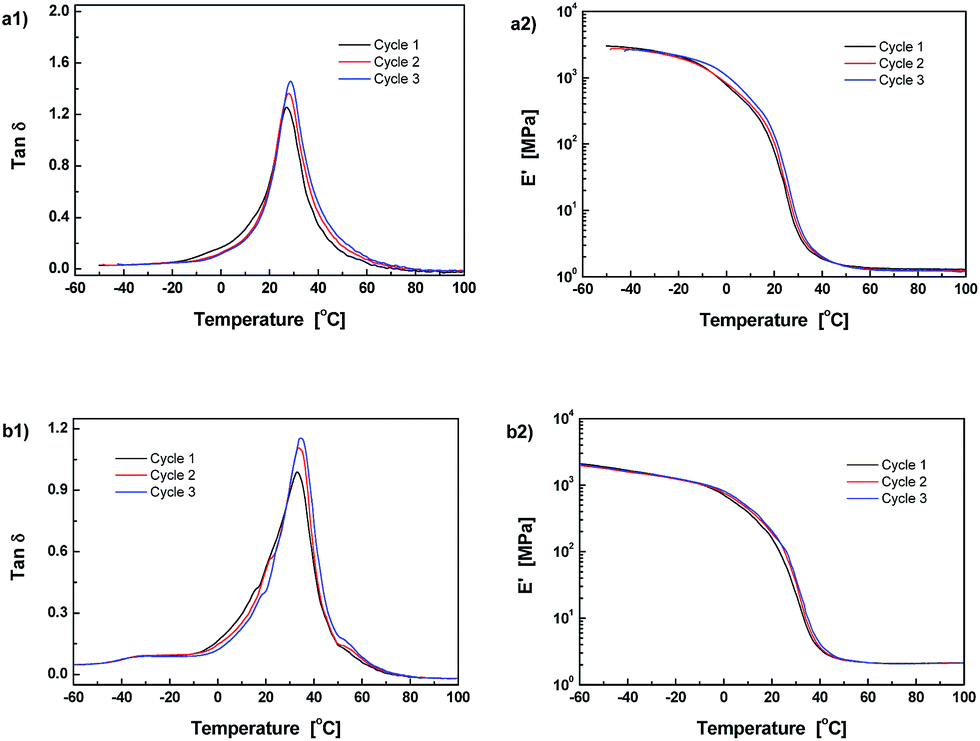 | ||
| Fig. 2 Temperature dependences of (a1) and (b1) tanδ and (a2) and (b2) storage modulus measure by repeated DMA scans. Materials: (a1) and (b1) Dima/TMPMP, (a2) and (b2) Dima/PETMP. | ||
As the dynamic reversibility of the alkoxyamines would affect microstructure and macroscopic properties of the crosslinked methacrylate esters, their storage moduli, E′, were repeatedly measured as a function of temperature between −40 and 100 °C (Fig. 2). By using eqn (1) and the data of Fig. 2, molecular weight among crosslinks, Mc, of the crosslinked polymers were calculated (Table 1). In comparison with the reference material PEGDMMA/TMPMP, both Dima/TMPMP and Dima/PETMP have much higher Mc. Moreover, their Mc values gradually increase with the test cycles, which form sharp contrast to the case of the reference material. The Mc values of the latter keep almost constant during the repeated scanning. It reveals that Dima/TMPMP and Dima/PETMP possess lower crosslinking density than the reference. With a rise in temperature, a certain amount of C–ON bonds in the reversibly bonded polymers are disconnected so that the measured Mc becomes higher. Additionally, a few permanently dissociated alkoxyamines appear after cyclic DMA measurements resulting in slightly increased Mc and reduced crosslinking density, due to the difficulty of recombination of carbon-centered radicals and nitroxide radicals in solid state. As for the reference materials, their crosslinking densities do not change with the heating and cooling cycles because of lack of reversible bonds like C–ON.
| Polymers | Mc (g mol−1) | ||
|---|---|---|---|
| Cycle 1 | Cycle 2 | Cycle 3 | |
| Dima/TMPMP | 2270 | 2381 | 2446 |
| Dima/PETMP | 1973 | 2268 | 2280 |
| PEGDMMA/TMPMP | 650 | 648 | 647 |
In the case of the same reversible bonds, nevertheless, trifunctional thiol leads to lower crosslinking density than tetrafunctional thiol. It follows the universal law of crosslinked polymers. That is, crosslinking density of crosslinked polymers increases with a rise of average functionality of monomers.25,26
The dynamic reversible characteristics of C–ON bonds can also be reflected by the stress relaxation and creep behaviors of the materials. It is seen from Fig. 3a that there is almost complete stress relaxation within 800 s for Dima/TMPMP. Higher temperature corresponds to faster relaxation. In contrast, the control PEGDMMA/TMPMP does not show any stress relaxation within the same time span. Evidently, the crosslinked networks of Dima/TMPMP are allowed to be rearranged to a less stretched state owing to bond fission/recombination of the alkoxyamines. Similar results were found in polymers with dynamic reversible disulfide bonds.5,6
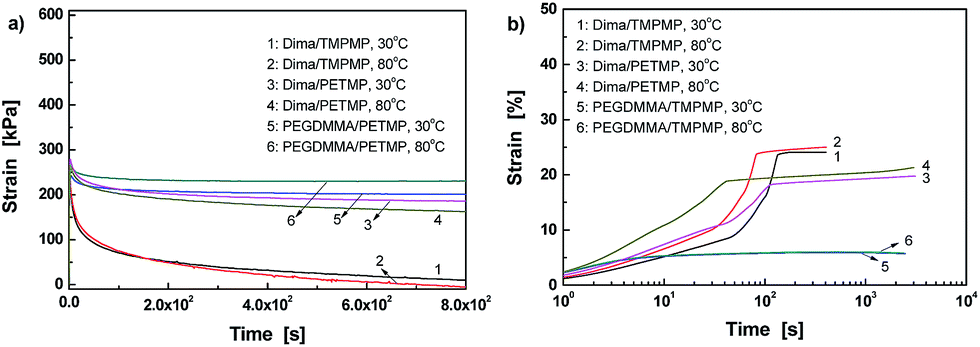 | ||
| Fig. 3 (a) Stress relaxation and (b) creep profiles of crosslinked methacrylate esters and the control measured at 30 and 80 °C. | ||
Fig. 3a further indicates that the higher crosslinking density of Dima/PETMP hinders the reversible reaction of C–ON bonds. The relaxation greatly slows down as compared to Dima/TMPMP.
Creep tests were also carried out to gain a better understanding (Fig. 3b). Generally, a creep strain versus time curve of plastic material consists of four stages:27 (i) initial rapid elongation, which is independent of time and arises from elastic and plastic deformations of the polymer; (ii) primary creep, during which the creep rate starts at a relatively high value and then decreases with time; (iii) secondary creep; and (iv) tertiary creep, in which the creep rate increases rapidly and eventually results in creep fracture. The Dima/TMPMP exhibits an insignificant initial rapid elongation stage. Then, the primary creep is observed, while the transition to the secondary creep is inconspicuous. Finally, the tertiary creep stage lasts for short time. When temperature increases, the appearance time of the creep failure is moved up. In contrast, the reference PEGDMMA/TMPMP only shows the first three stages of creep within the time range of interests, and no tertiary creep appears. Moreover, its strain due to the secondary creep is nearly time independent and lower than those of Dima/TMPMP and Dima/PETMP. Comparatively, the creep rate of Dima/PETMP at the primary stage is the highest for unknown reason. It is significantly reduced during the secondary creep, and the tertiary creep is also undetected probably because of the increased crosslinking density.
To monitor dynamic reversible bonding habits of the materials at the molecular level, ESR was employed. As the signal at each temperature is kept on certain level within a fixed period of time, the ESR intensity can be used to describe radical concentration in material.28 Fig. 4a shows typical ESR spectra of Diol measured at various temperatures. In addition, Fig. 4b gives the normalized absorption lines corresponding to the ESR curves in Fig. 4a, from which integrated areas of the relative signal intensities can be calculated (Fig. 4c). Evidently, the radical intensity gradually increases after 60 °C due to cleavage of C–ON bonds. For purposes of highlighting the influence of temperature, heating time dependence of relative ESR signal intensities calculated from integral areas of the normalized ESR absorption curves collected at different temperatures (Fig. S14, ESI†) are plotted in Fig. 4d. The radical intensity at 60 °C keeps on increasing within the testing time frame, and more obvious changes are detected at the temperatures higher than 80 °C. It can thus be concluded that homolysis of Diol starts at 60 °C. Compared with the dynamic reversible monomer derived from nitrile azo initiator,9 the initial fission temperature of the present Diol is higher because of the weaker electron-absorbing capability of amido group attached to the C atom in C–ON bond. It means that manipulation of the electronic effect can effectively tune homolysis temperature of C–ON bond.
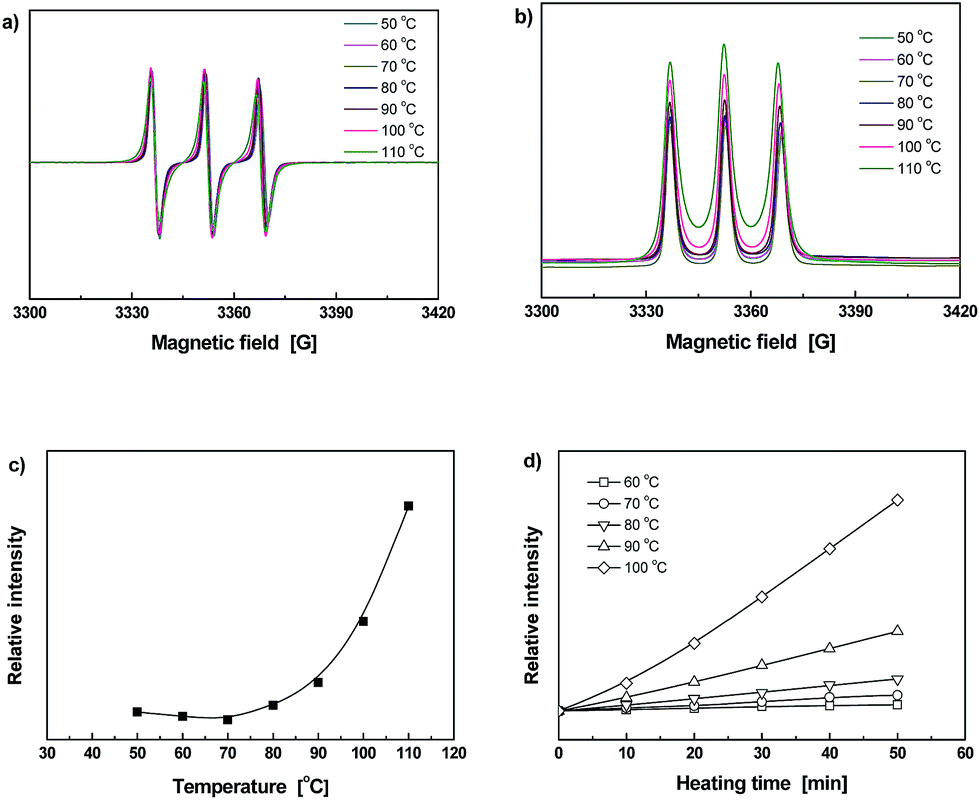 | ||
| Fig. 4 (a) Typical ESR spectra of Diol measured at various temperatures. (b) Normalized absorption lines obtained from the data of (a). (c) Temperature dependence of relative ESR signal intensities calculated from integral areas of the normalized absorption curves in (b). (d) Heating time dependences of relative ESR signal intensities calculated from integral areas of the normalized ESR absorption curves measured at various temperatures. | ||
The ESR spectra of Dima/TMPMP are shown in Fig. 5. Similarly, the signal increases with a rise in temperature (Fig. 5a–c), meaning crosslinking and de-crosslinking of the crosslinked polymer would be more effective at elevated temperature. The ESR spectra recorded during repeated heating–cooling cycles further demonstrate the thermal reversibility of the material (Fig. 5d). The radical concentration changes up and down over the thermal cycling between 20 and 80 °C as a result of reversible C–ON bond fission and radical recombination. Although the ESR signal intensity at 20 °C slightly increases due to permanent dissociation of a few alkoxyamines, the difference between the data at 80 and 20 °C remains nearly constant.
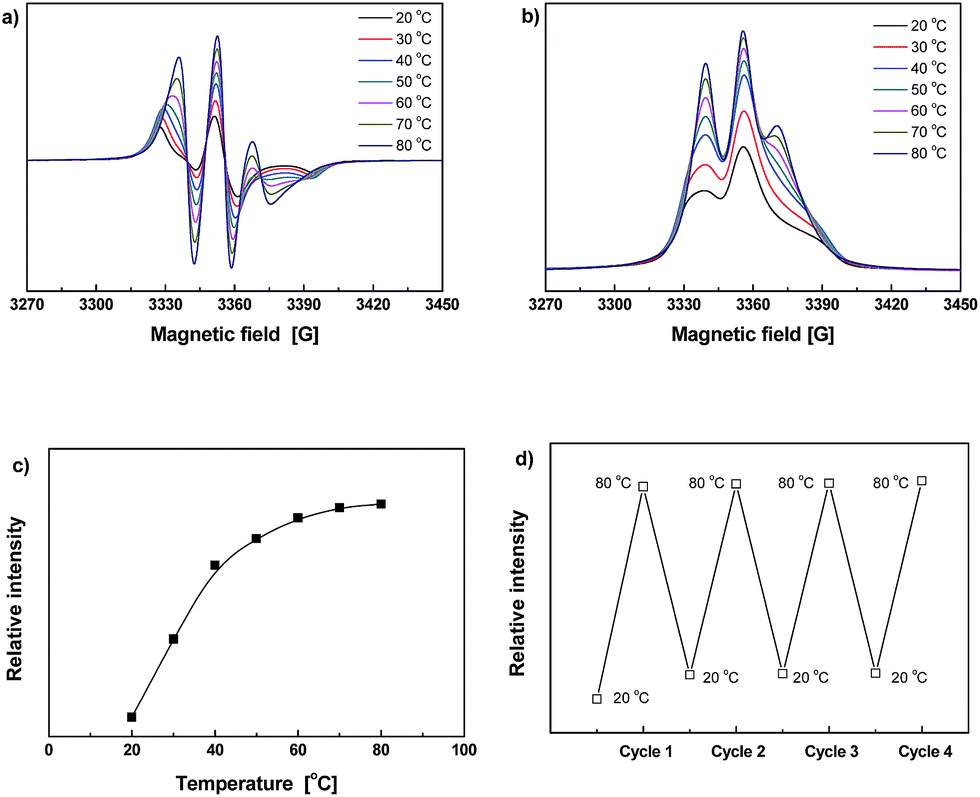 | ||
| Fig. 5 (a) Typical ESR spectra of Dima/TMPMP measured at various temperatures. (b) Normalized absorption lines obtained from the data of (a). (c) Temperature dependence of relative ESR signal intensities calculated from integral areas of the normalized absorption curves in (b). (d) Relative ESR signal intensities measured during heating–cooling cycles between 20 and 80 °C. | ||
By taking advantage of the reversible reaction, the crosslinked polymer can be transformed into solution (Fig. 6). The original Dima/TMPMP networks (Fig. 6a) are nearly not swellable in DMF (Fig. 6b), but behave differently after being exposed to an excess amount of nitroxides such as 4-OH-TEMPO (20 equiv./alkoxyamine unit). The polymer is completely dissolved within 6 h at 100 °C (Fig. 6c). The results not only prove that the crosslinked sites are thermally dissociable (Fig. 6d), but also suggest a feasible way of controllable de-crosslinking, reprocessing and recycling. That is, cradle-to-cradle design of polymers might be possible with the help of dynamic reversible bonds, which would eventually reduce fossil oil consumption.
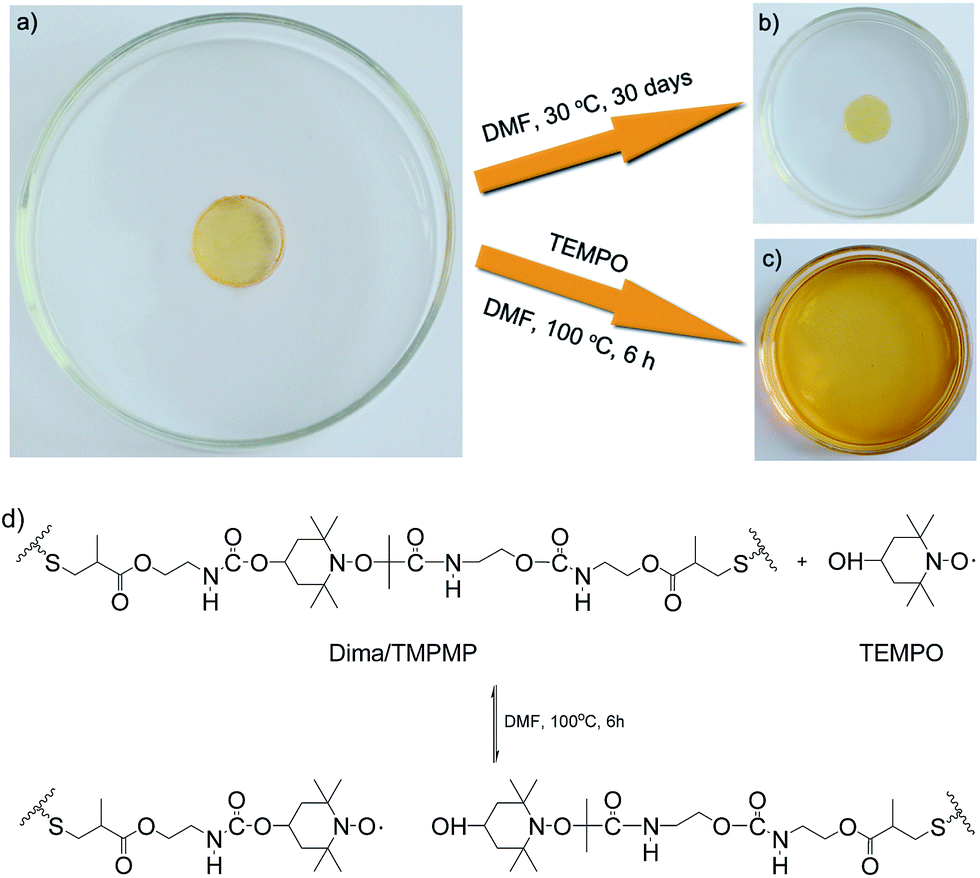 | ||
| Fig. 6 Optical images of Dima/TMPMP (a) immersed in DMF (b) and dissolved in DMF (c) with an excess amount of 4-OH-TEMPO (20 equiv./alkoxyamine unit) due to de-crosslinking (d). | ||
The above results and discussion prove that the crosslinked methacrylate ester elastomer has acquired reversibility of C–ON bonds. Therefore, it should be intrinsically self-healable. Fig. 7 illustrates that Dima/TMPMP can be repeatedly healed at 80 °C as characterized by three successive cut-reconnection processes. The thermal remendability has been coupled with the polymer as expected. Moreover, the quantitative examination in terms of tensile test evidences the self-healability. As shown in Fig. 8a, healing efficiency of Dima/TMPMP increases with healing time and reaches the maximum of 100% at 4 h. The healing is enabled for multiple times (Fig. 8b). The mild decline in healing efficiency with healing/re-fracture cycles (Fig. 8c) might originate from misalignment of the specimens and/or heterogeneity of dynamic recombination of alkoxyamine moieties at the fractured surfaces. It is interesting to see from Fig. 8d that the healability of Dima/TMPMP is nearly independent of the waiting time prior to reconnection of the broken specimens. It means that (i) the radicals' recombination can hardly take place at the same side of the cracked surface, and (ii) the crack healing is not governed by hydrogen bonds. If hydrogen bonds predominated, the broken materials would lose healability after long time separation due to self-combination of neighboring free hydrogen bonds.29
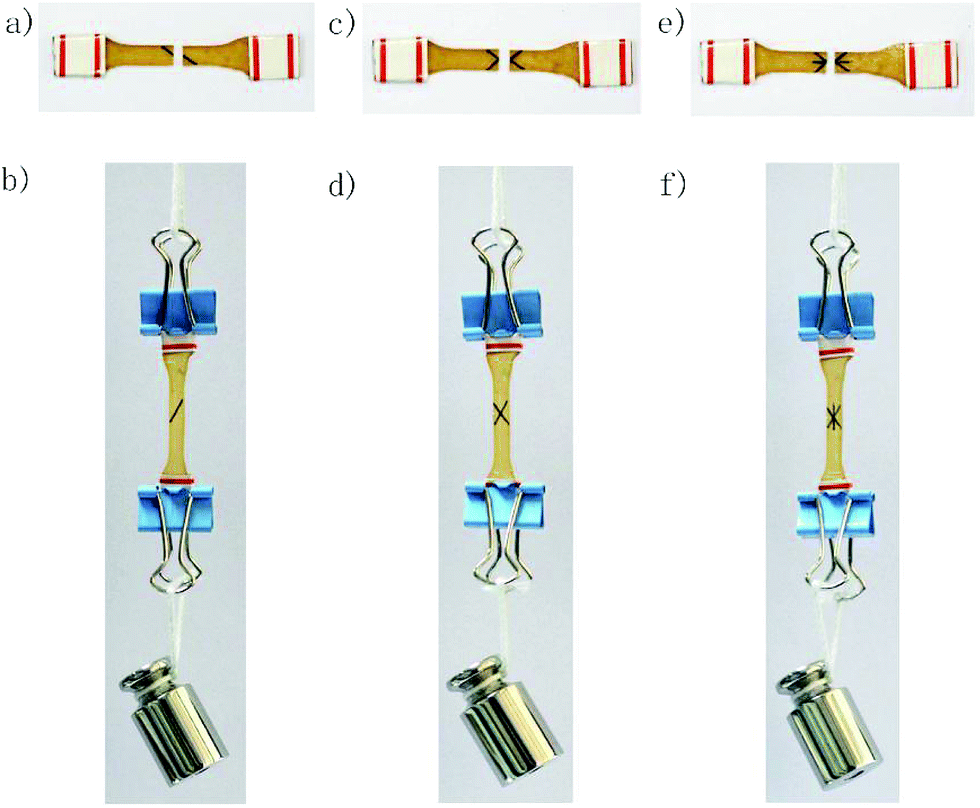 | ||
| Fig. 7 Repeated macro-repairing of a Dima/TMPMP specimen at 80 °C in air. (a) The virgin specimen was firstly cut, and then the broken halves were recombined for healing. (b) After 4 h, the specimen looks like the original one without any shape change, and can bear a weight of 50 g. (c) The first-healed specimen was subjected to a tensile stress higher than its original strength and ruptured again, and then repaired under the same conditions. (d) The second-healed specimen also possesses load bearing ability. (e) The second-healed specimen was broken under a tensile stress higher than its original strength, and healed as before. (f) The third-healed version behaves like the former two shown in (b) and (d). | ||
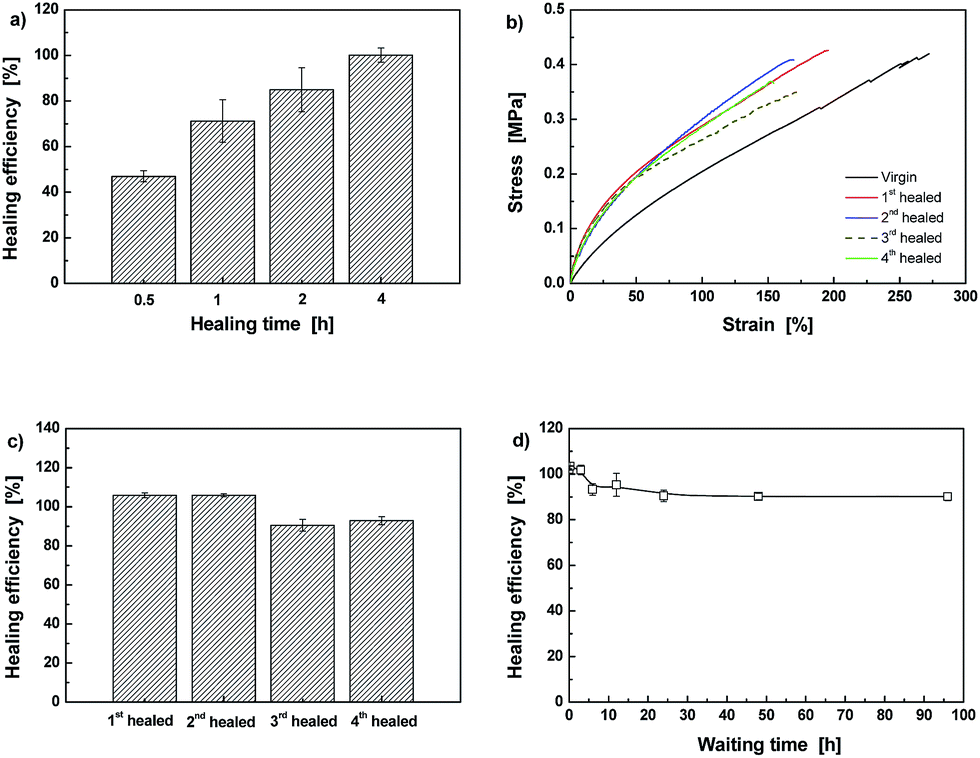 | ||
| Fig. 8 (a) Effect of healing time on healing efficiency of Dima/TMPMP measured by tensile test. Healing was conducted at 80 °C in air. (b) Stress–strain curves of a Dima/TMPMP specimen subjected to repeated fracture-healing at 80 °C in air. (c) Healing efficiencies calculated from the results of (b). (d) Healing efficiencies of Dima/TMPMP specimens as a function of waiting time, which is counted from specimen breakage to rejoining. | ||
The reference material PEGDMMA/TMPMP also experiences the same healing experiment. As it does not contain any alkoxyamines but carries CC end groups that are cured by the same hardener, no healing effect can be detected under the same conditions. The entanglement of dangling chains across the fractured surfaces is negligible, which does not make any contribution to detectable restoration of the material strength.
The influence of crosslinking density on healing behavior is demonstrated by comparing the results of Dima/TMPMP (Fig. 8) with those of Dima/PETMP (Fig. 9). It is evident that the higher crosslinking density restricts reversible reaction of C–ON bonds. Lower healing efficiency is perceived as a result of lower degree of recombination of C–ON across the interface (Fig. 9).
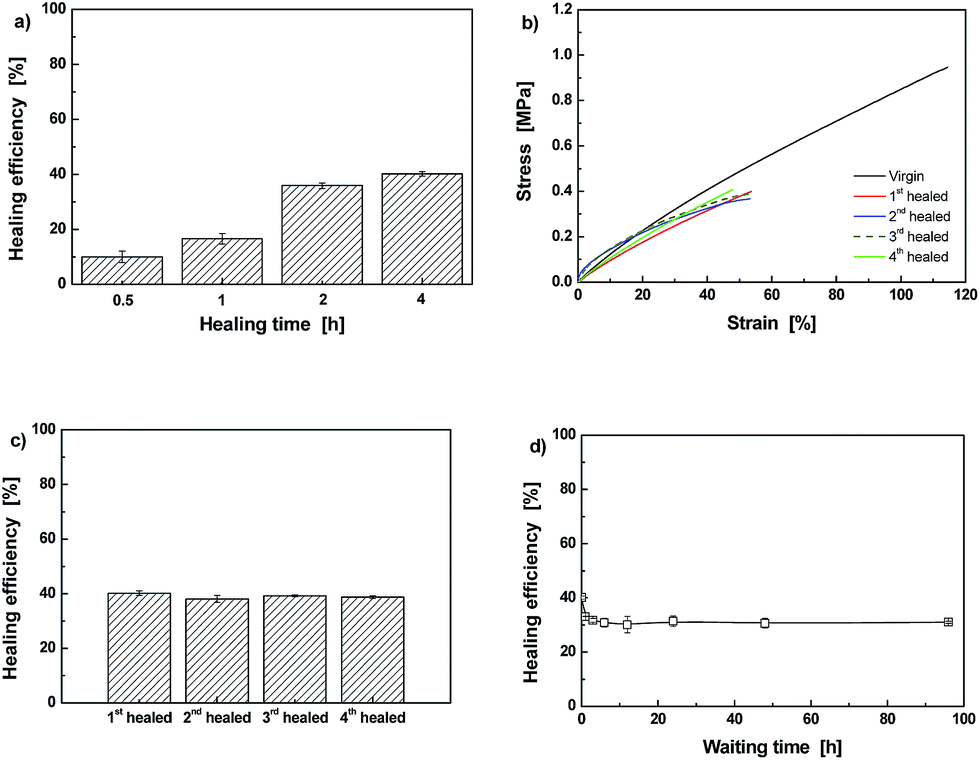 | ||
| Fig. 9 (a) Effect of healing time on haling efficiency of Dima/PETMP measured by tensile test. Healing was conducted at 80 °C in air. (b) Stress–strain curves of a Dima/PETMP specimen subjected to repeated fracture-healing at 80 °C in air. (c) Healing efficiencies calculated from the results of (b). (d) Healing efficiencies of Dima/PETMP specimens as a function of waiting time, which is counted from specimen breakage to rejoining. | ||
It is worth noting that the above healing experiments are conducted in air. Evidently, the system is insensitive to oxygen. Although the specimens are healed at a relatively high temperature (80 °C), the weaker electron-absorbing feature of amido group can still provide the healing reaction with air resistance. In this context, homolysis temperature of C–ON bond can be purposely changed with the fear of affecting its performance in air.
When reconsidering the thermal remendability of Dima/TMPMP represented by reconnecting the broken specimens (Fig. 7), we can reasonably suppose that the fragmented Dima/TMPMP should also be reformed by the same mechanism. In other words, the traditional insoluble and infusible crosslinked polymer can be simply reclaimed in solid state, even without the necessity of degradation as shown in Fig. 6.
Fig. 10 proves that it is true. Firstly, the disk-shaped specimen of Dima/TMPMP (Fig. 10a) was pulverized in liquid nitrogen (Fig. 10b). Then, the powders were remolded into disk-shaped specimen at 80 °C (Fig. 10c and d). In comparison, the reference PEGDMMA/TMPMP failed to be reprocessed even when the compression molding time was extended from 2.5 to 24 h. Mechanical test indicates that the recycled Dima/TMPMP has an average tensile strength of 0.36 MPa and failure strain of 196% (Fig. 11a), which are 86.0 and 73.6% of the values of the original version, respectively. Besides, Dima/PETMP can also be reprocessed in the same way. The average tensile strength and failure strain of the recycled Dima/PETMP are 0.72 MPa and 120%, respectively, which are 73.9% and 108% of the values of the original material (Fig. 11b). To date, there is no report about thermosets that exhibit solvent-free reprocessability based on reversible C–ON bonds.
 | ||
| Fig. 10 (a) Original filmy sample of Dima/TMPMP. (b) Powdered Dima/TMPMP obtained by frozen pulverization. (c) Mold for reprocessing (b). (d) Remolded Dima/TMPMP film. Conditions of reprocessing: 80 °C, 2.5 h, and 3 MPa. | ||
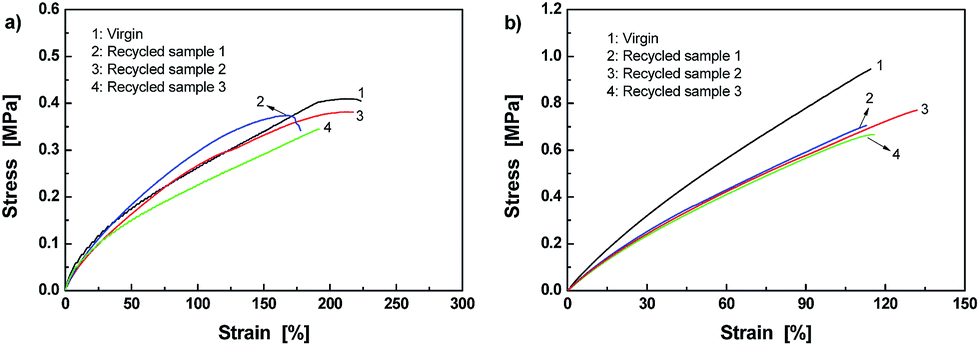 | ||
| Fig. 11 Stress–strain curves of original and reprocessed (a) Dima/TMPMP and (b) Dima/PETMP. | ||
Conclusions
The present work is focused on giving full play to thermally reversible fission/recombination of C–ON bonds in self-healing polymers. Strategies of tuning their homolysis temperature and synthesis of polymers with embedded reversible bonds were proposed and verified. A novel amide-containing alkoxyamine with moderate homolysis temperature and air-insensitivity was obtained by making use of proper electron withdrawing effect. Moreover, thiol–ene click chemistry was applied to prepare crosslinked methyl methacrylate polymer carrying alkoxyamine moieties below the homolysis temperature of the latter. Accordingly, the irreversible combination of carbon-centered radicals was minimized, and the polymer has acquired satisfactory thermal remendability as characterized by the capability of repeated strength restoration in air.The number of thiol group of the curing agent significantly affected crosslinking density of the resultant polymer networks. Tetrafunctional thiol led to higher crosslinking density but lower healing efficiency than trifunctional thiol.
Owing to the dynamic reversible nature of C–ON bonds, the crosslinked methyl methacrylate polymer can be easily decomposed into small molecules in a controlled manner, or remolded in solid state. Both approaches would be hopefully developed into facile reprocessing and reclaiming techniques.
Acknowledgements
The authors thank the support from the Natural Science Foundation of China (Grants: 51273214 and 51333008), the Natural Science Foundation of Guangdong (Grants: 2010B010800021 and S2013020013029), the Science and Technology Program of Guangzhou (Grant: 2014J4100121), and the Basic Scientific Research Foundation in Colleges and Universities of Ministry of Education of China (Grant: 12lgjc08).Notes and references
- M. Q. Zhang and M. Z. Rong, Self-healing Polymers and Polymer Composites, John Wiley & Sons, Inc., Hoboken, 2011 Search PubMed.
- M. Q. Zhang and M. Z. Rong, Polym. Chem., 2013, 4, 4878 RSC.
- Y. X. Lu and Z. B. Guan, J. Am. Chem. Soc., 2012, 134, 14226 CrossRef CAS PubMed.
- G. Deng, F. Li, H. Yu, F. Liu, C. Liu, W. Sun, H. Jiang and Y. Chen, ACS Macro Lett., 2012, 1, 275 CrossRef CAS.
- Z. Q. Lei, H. P. Xiang, Y. J. Yuan, M. Z. Rong and M. Q. Zhang, Chem. Mater., 2014, 26, 2038 CrossRef CAS.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955 RSC.
- K. Imato, M. Nishihara, T. Kanehara, Y. Amamoto, A. Takahara and H. Otsuka, Angew. Chem., Int. Ed., 2012, 51, 1138 CrossRef CAS PubMed.
- Z. P. Zhang, M. Z. Rong, M. Q. Zhang and C. E. Yuan, Polym. Chem., 2013, 4, 4648 RSC.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 3936 CrossRef CAS.
- J. J. Cash, T. Kubo, A. P. Bapat and B. S. Sumerlin, Macromolecules, 2015, 48, 2098 CrossRef CAS.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975 CrossRef CAS PubMed.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., Int. Ed., 2011, 123, 1698 CrossRef.
- J. Ling, M. Z. Rong and M. Q. Zhang, Chin. J. Polym. Sci., 2014, 32, 1286 CrossRef CAS.
- Z. Q. Lei, P. Xie, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2015, 3, 19662 CAS.
- D. Y. Zhu, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2015, 49–50, 175 CrossRef CAS.
- H. P. Xiang, H. J. Qian, Z. Y. Lu, M. Z. Rong and M. Q. Zhang, Green Chem., 2015, 17, 4315 RSC.
- C. E. Yuan, M. Z. Rong, M. Q. Zhang, Z. P. Zhang and Y. C. Yuan, Chem. Mater., 2011, 23, 5076 CrossRef CAS.
- C. E. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782 CrossRef.
- C. E. Yuan, M. Q. Zhang and M. Z. Rong, J. Mater. Chem. A, 2014, 2, 6558 CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698 CrossRef PubMed.
- F. Wang, M. Z. Rong and M. Q. Zhang, J. Mater. Chem., 2012, 22, 13076 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540 CrossRef PubMed.
- M. J. He, W. X. Chen and X. X. Dong, Polymer Physics, Fudan University Press, Shanghai, 2nd edn, 2000 Search PubMed.
- S. A. Hayes, W. Zhang and F. R. Jones, J. R. Soc., Interface, 2007, 4, 381 CrossRef PubMed.
- M. Morton, L. J. Fetters, J. Inomata, D. C. Rubio and R. N. Young, Rubber Chem. Technol., 1976, 49, 303 CrossRef.
- R. Y. Yee and A. Adicoff, J. Appl. Polym. Sci., 1976, 20, 1117 CrossRef.
- X. B. Shi, C. L. Wu, M. Z. Rong, T. Czigany, W. H. Ruan and M. Q. Zhang, Chin. J. Polym. Sci., 2013, 31, 377 CrossRef.
- S. Marque, C. Le Mercier, P. Tordo and H. Fischer, Macromolecules, 2000, 33, 4403 CrossRef.
- P. Cordier, F. Tournilhac, C. Soulie-Ziakovic and L. Leibler, Nature, 2008, 451, 977 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Proton nuclear magnetic resonance (1H NMR) spectra (Fig. S1, Fig. S5), carbon-13 nuclear magnetic resonance (13C NMR) spectra (Fig. S2, Fig. S6), Fourier transform infrared (FTIR) spectra (Fig. S3, Fig. S7, Fig. S8, Fig. S9, Fig. S10), electron-impact mass (EI-MS) spectrum (Fig. S4), Fourier transform Raman (FT-Raman) spectra (Fig. S11, Fig. S12), temperature dependences of tanδ and storage modulus (Fig. S13), and electron spin resonance (ESR) spectra (Fig. S14). See DOI: 10.1039/c5ra22275c |
| This journal is © The Royal Society of Chemistry 2016 |