
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Using a biomimetic membrane surface experiment to investigate the activity of the magnetite biomineralisation protein Mms6†
Scott M. Bird‡
a,
Andrea E. Rawlings‡a,
Johanna M. Gallowayb and
Sarah S. Staniland*a
aUniversity of Sheffield, Department of Chemistry, Dainton Building, Sheffield, S3 7HF, UK. E-mail: s.s.staniland@sheffield.ac.uk
bUniversity of Bristol, School of Chemistry, Cantock's Close, Bristol, BS8 1TS, UK
First published on 7th January 2016
Abstract
Magnetotactic bacteria are able to synthesise precise nanoparticles of the iron oxide magnetite within their cells. These particles are formed in dedicated organelles termed magnetosomes. These lipid membrane compartments use a range of biomineralisation proteins to nucleate and regulate the magnetite crystallisation process. A key component is the membrane protein Mms6, which binds to iron ions and helps to control the formation of the inorganic core. We have previously used Mms6 on gold surfaces patterned with a self-assembled monolayer to successfully produce arrays of magnetic nanoparticles. Here we use this surface system as a mimic of the interior face of the magnetosome membrane to study differences between intact Mms6 and the acid-rich C-terminal peptide subregion of the Mms6 protein. When immobilised on surfaces, the peptide is unable to reproduce the particle size or homogeneity control exhibited by the full Mms6 protein in our experimental setup. Moreover, the peptide is unable to support anchoring of a dense array of nanoparticles to the surface. This system also allows us to deconvolute particle binding from particle nucleation, and shows that Mms6 particle binding is less efficient when supplied with preformed magnetite nanoparticles when compared to particles precipitated from solution in the presence of the surface immobilised Mms6. This suggests that Mms6 binds to iron ions rather than to magnetite surfaces in our system, and is perhaps a nucleating agent rather than a controller of magnetite crystal growth. The comparison between the peptide and the protein under identical experimental conditions indicates that the full length sequence is required to support the full function of Mms6 on surfaces.
1. Introduction
Iron is an essential element in many organisms. It plays a vital role in critical biological processes.1–3 A host of proteins have evolved to coordinate, transport, and harness its useful chemical properties. Examples include: haemoglobin for oxygen transport in erythrocytes, iron storage proteins such as ferritin, and in enzymes which use the change in oxidation state of iron as the basis of electron transport and redox reactions.1,2 Although an essentially useful transition metal, the presence of iron within cells is strictly controlled. High levels of iron can result in the production of damaging oxygen radicals3 or biogenic iron oxide particles associated with neurodegenerative disorders such as Alzheimer's disease.4,5 However, some organisms have developed methods to exploit the inherent magnetic characteristics of certain iron oxides by forming magnetic nanoparticles. A convenient model system to study iron accumulation and subsequent biomineralisation is the controlled formation of magnetite nanoparticles in magnetotactic bacteria (MTB).6–8These specialised bacteria contain internal vesicle structures termed magnetosomes9,10 which act as nanoreactors for the synthesis of precise nanoparticles of the iron oxide magnetite (Fe3O4).8,11 Crystallisation of magnetite is closely regulated by dedicated biomineralisation proteins located within the lipid membrane of the magnetosome.12,13 These proteins control many aspects of the forming crystal, from its specific nucleation to the size and shape of the resulting particle. Within a single MTB strain a highly uniform population of nanoparticles is produced with homogeneous size, shape, and chemical composition. However, between strains these properties can differ significantly. There is intense ongoing research to identify and understand the role of biomineralisation Mms (magnetosome membrane specific) proteins and generate detailed mechanisms for iron oxide biomineralisation. Several key proteins have been discovered tightly bound to the magnetite particles of the MTB Magnetospirillum magneticum AMB-1.13 One of these, Mms6, is a 6 kDa protein comprising a hydrophobic N-terminal region and a hydrophilic C-terminal region (KSRDIESAQSDEEVELRDALA) rich in acidic residues.13–15 When the Mms6 gene is deleted from MTB, the resulting nanoparticles are smaller and the shape is less well controlled.16 Importantly, this protein has been used as an additive in synthetic chemical precipitations of magnetite nanoparticles, where it also appears to affect their size, formation and mineral type.13,17–19 This has led to the assignment of Mms6 as a key size and morphology controlling magnetite binding protein.
Mms6 has been found to self-assemble in solution to form micelle-like structures with a high number of protein subunits.14 This is likely to be due to the amphiphilic nature of the protein sequence. These protein micelles are able to bind and accumulate iron ions in solution. This is thought to trigger iron oxide nucleation, which in turn aids magnetite crystal growth.13,14,20,21 The acidic C-terminal region of Mms6 has also been investigated, with the peptide displaying some similar properties to the full length protein in terms of both iron binding and some ability to control iron oxide crystal growth.14,22
We have previously demonstrated a novel approach for the formation of magnetite nanoparticles (MNPs) on surfaces, through the patterning of the Mms6 protein and subsequent biomineralisation of magnetite.23,24 This generated consistent microscale MNP arrays when patterned onto functionalised gold surfaces using micro-contact printing (μCP).23,24 Recently we published a variation of this approach to pattern a version of Mms6 engineered to contain an N-terminal cysteine, binding directly to gold and biotemplating MNP arrays of magnetite and magnetically harder cobalt-doped magnetite.25 In this case, a protein resistant polyethylene glycol (PEG) self-assembled monolayer (SAM) was patterned onto gold surfaces via μCP, with the remaining space backfilled with cysteine-tagged Mms6.25 The regions of the surface with a locally high Mms6 concentration were surrounded by a dense monolayer of PEG molecules. In this context, our system can be considered as a mimic of the arrangement of Mms6 thought to exist on the interior surface of the magnetosome membrane. Clusters of Mms6 are anchored in the magnetosome membrane through their hydrophobic membrane interacting region, and the C-terminal acid-rich region is exposed within the magnetosome lumen. The N-terminal cysteine–gold attachment allows control over the orientation of the protein on the surface, ensuring that the active C-terminal region is displayed to the reaction solution. This surface based biomineralisation experiment therefore offers a unique in vitro method of studying Mms6 in an environment similar to the native state, anchored in the magnetosome membrane. This is in contrast to the previously performed solution based experiments,14,20,21 where the protein is thought to self-assemble into micelle structures, which have the opposite curvature to that found on the interior of the magnetosome membrane.
We used this biomimetic system to investigate the differences between the Mms6 C-terminal peptide and the intact Mms6 protein in MNP synthesis to determine if the peptide can be effectively substituted for the intact protein (Fig. 1). Being able to utilise a synthetic peptide offers advantages over the full protein, as peptides are cheaper and easier to produce, which would make the biotemplating properties of Mms6 more industrially amenable. This comparison also uncovers interesting differences between the activity of the protein and the peptide, which provides insight into the function of Mms6 in vivo. Previous studies have found Mms6 tightly associated with the isolated MNPs of magnetotactic bacteria,13 suggesting that the protein has a strong affinity for MNPs. To test this property in vitro we probed whether Mms6, or its C-terminal peptide region, was able to bind magnetite when supplied with pre-formed MNPs suspended in solution and successfully anchor them to the surface (Fig. 1). These experiments allowed the magnetite templating and magnetite binding activities of Mms6 to be analysed separately.
 | ||
| Fig. 1 Experimental scheme. (a) Stamping PEG-thiols onto a gold surface using micro-contact printing. (b) Formation of the protein resistant self-assembled monolayer. (c) Backfilling with the cysteine tagged Mms6 (cys-Mms6) or cysteine tagged C-terminal Mms6 peptide (cys-pep). The surface is then subjected to either a magnetite precipitation reaction (Scheme I) or supplied with preformed magnetite nanoparticles (Scheme II). | ||
2. Experimental
Synthesis of MNP arrays
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O2 30% v/v) for 10 minutes, rinsed with Milli-Q water and dried in nitrogen. 5 nm of chromium, followed by 50 nm of gold was evaporated onto the slides in an Edwards Auto 360 thermal evaporator. Following this, the slides were scribed and sectioned before patterning.:1 (w/w) mixture of Sylgard 184 poly-(dimethylsiloxane) (PDMS) (Dow Corning) prepolymer to curing agent. This mixture was stirred, vacuum degassed to remove trapped air bubbles, poured over micropatterned silicon masters and cured at 60 °C for >24 hours. The cured stamps were then cut from the masters and soaked in ethanol for >16 hours. Gold surfaces were cleaned in a piranha solution for 5 minutes, rinsed in Milli-Q water, dried in a nitrogen stream, rinsed in ethanol and dried in nitrogen. Stamps were then covered with a 5 mM solution of PEG thiol (11-mercaptoundecyl tetra(ethylene glycol), HS(CH2)11(OCH2CH2)4OH) (Sigma-Aldrich) in ethanol and incubated for 4 minutes. The excess PEG solution was then pipetted off the stamps, which were then dried in nitrogen, and placed in conformal contact with clean gold surfaces. After 4 minutes the stamps were removed, allowing time for an ordered SAM to form.
:H2O2 30% v/v) for 10 minutes, rinsed with Milli-Q water and dried in nitrogen. 5 nm of chromium, followed by 50 nm of gold was evaporated onto the slides in an Edwards Auto 360 thermal evaporator. Following this, the slides were scribed and sectioned before patterning.:1 (w/w) mixture of Sylgard 184 poly-(dimethylsiloxane) (PDMS) (Dow Corning) prepolymer to curing agent. This mixture was stirred, vacuum degassed to remove trapped air bubbles, poured over micropatterned silicon masters and cured at 60 °C for >24 hours. The cured stamps were then cut from the masters and soaked in ethanol for >16 hours. Gold surfaces were cleaned in a piranha solution for 5 minutes, rinsed in Milli-Q water, dried in a nitrogen stream, rinsed in ethanol and dried in nitrogen. Stamps were then covered with a 5 mM solution of PEG thiol (11-mercaptoundecyl tetra(ethylene glycol), HS(CH2)11(OCH2CH2)4OH) (Sigma-Aldrich) in ethanol and incubated for 4 minutes. The excess PEG solution was then pipetted off the stamps, which were then dried in nitrogen, and placed in conformal contact with clean gold surfaces. After 4 minutes the stamps were removed, allowing time for an ordered SAM to form.Cys-Mms6 or cys-pep patterned surfaces were rinsed in anaerobic Milli-Q water and added either directly to the POFHK reaction just after the water was added, or added to a 20 mL solution of the MNPs that were collected magnetically, dispersed into Milli-Q water and placed onto a tilt stirrer for >16 hours (concentration of MNP is 45 mg mL−1). Once the surfaces were removed from either the POFHK reaction or the MNPs dispersed in water, they were rinsed with anaerobic Milli-Q water and dried with nitrogen.
Characterisation
All experiments were performed with a flow rate of 50 μL min−1, and at a temperature of 22 °C. Changes in frequency (Δf) and dissipation (ΔD) were recorded for the 3rd, 5th, 7th, 9th, 11th and 13th overtones as Milli-Q water was flowed into the system until these values stabilised. Following this, a 10 μg mL−1 solution of cys-Mms6 or cys-pep in PBS was flowed into the system for 1 hour, and then the flow was returned to Milli-Q water again until the recorded values stabilised. Modelling was performed following the methods used by Krzemiński et al.26 using Qtools 2 Qsense software under the assumptions of the Kelvin Voigt model,27 a hydrodynamic protein layer density of 1200 kg m−3,28 a buffer viscosity of 0.001 kg m−3, and a buffer density of 1000 kg m−3.
3. Results and discussion
We used Quartz Crystal Microbalance with dissipation (QCM-D) to determine if the cysteine labelled Mms6 peptide (cys-pep) was able to interact with a gold surface as we have previously shown for the full length form of Mms6 (cys-Mms6).25 QCM-D systems are used extensively to study interactions of biomolecules with surfaces.31 In this experiment, a thin piezoelectric quartz crystal coated with a layer of gold oscillates at its resonant frequency. When material is applied and deposited on the surface, there is a corresponding shift in the resonant frequency. This shift, described by the Sauerbrey equation, is negative if the mass on the surface increases. The dissipated energy loss from the surface can also be measured, which gives an indication of the changes to the visco-elastic properties of the surface adsorbed material.The QCM-D analysis is presented in Fig. 2 for the cysteine labelled Mms6 peptide and, for comparison, the Mms6 protein. These both show the characteristic decrease in frequency associated with adsorption. In phase A, the system is washed with ultra-pure water. During phase B, the peptide or protein is allowed to flow into the system in PBS, before the surface is washed again with ultra-pure water to remove unbound material. The peptide appears to rapidly reach an equilibrium state, with the protein taking longer to plateau in phase B. This is probably because the molar concentration of the protein is less than for the peptide, as the Mms6 protein is larger than the peptide. As both solutions contained 10 μg mL−1 of the respective biomolecule, the molar concentration of cysteines for attachment is higher for the peptide than the protein, leading to more rapid adsorption of the peptide. Using the Sauerbrey equation32 and estimates of the dimensions of both of the peptide and the protein (obtained from models generated by the Quark protein prediction server33) the coverage of the gold surface by the biomolecule could be calculated (Table 1). This indicates that both molecules produce an almost monolayer coverage of the surface. The Voight model (ESI Table 1†) provides an estimated thickness of this layer of 8 nm. This suggests both the protein and the peptide are packed orthogonally to the surface. Furthermore, the return to baseline dissipation in the case of the peptide is consistent with available examples of rigid biopolymer deposition.31 To confirm that the peptide itself does not interact with the PEG-thiol SAM we conducted further QCM-D experiments (ESI Fig. 1 and ESI Table 2†). These clearly show that there is no detectable interaction between the PEG passivated surface and the cysteine tagged biomolecules occurring. Our previous study reveals that Mms6 also does not directly interact with the PEG layer.25
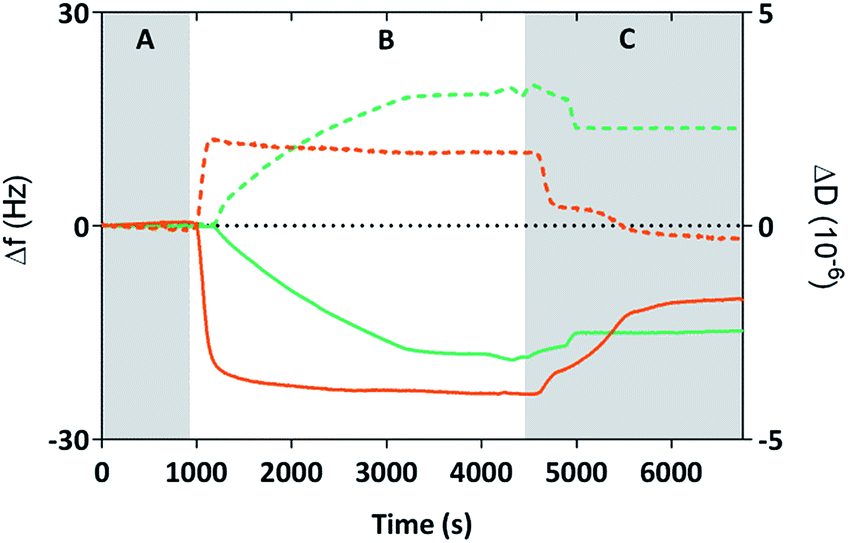 | ||
| Fig. 2 Frequency (Δf, solid lines) and dissipation (ΔD, dotted lines) changes of the 7th overtone recorded with QCM-D during adsorption of cys-Mms6 (green) or cys-pep (orange) onto clean gold coated quartz crystals. Grey regions show when Milli-Q water was applied (A and C), and the white region (B) shows when a PBS buffer containing cys-Mms6 or cys-pep at a concentration of 10 μg mL−1 was applied (flow rate 50 μL min−1). | ||
| Sauerbrey values | Cys-Mms6 | Cys-pep |
|---|---|---|
| a All modelling was performed with QTools 2 Qsense software. Sauerbrey values were calculated from the 7th overtone. | ||
| Mass (ng cm−2) | 258 | 182 |
| Coverage (pmol cm−2) | 23 | 70 |
| Complete monolayer (pmol cm−2) | ≈24 | ≈83 |
| Coverage (%) | ≈96 | ≈84 |
The cysteine tagged Mms6 or Mms6 peptide was used to backfill a PEG-thiol patterned gold surface using conditions we have already developed and optimised.25 These surfaces were subject to a partial oxidation of ferrous hydroxide reaction with potassium hydroxide (POFHK) (Fig. 1, Scheme I), which precipitated MNPs. Once the reaction was complete and following cleaning, the surfaces were characterised by scanning electron microscopy (SEM), and the particles formed on both the surfaces and in the bulk solution were probed by X-ray diffraction (XRD) (Fig. 3). The interplanar distances of the particles produced are in close agreement to those corresponding to magnetite (Table 2), rather than the iron oxide maghemite that has a similar crystal structure. The (400) plane in particular, which can be used to distinguish between magnetite and maghemite, confirms the majority of the material is most likely to be magnetite.34 Diffraction peaks corresponding to gold are also present in surface biomineralisation data, obscuring the (222) magnetite peak. The bulk particles were visualised by transmission electron microscopy (TEM) and their dimensions measured using ImageJ35 software (Fig. 4). Grainsize analysis was also conducted for particles formed on both the protein and peptide patterned surfaces.
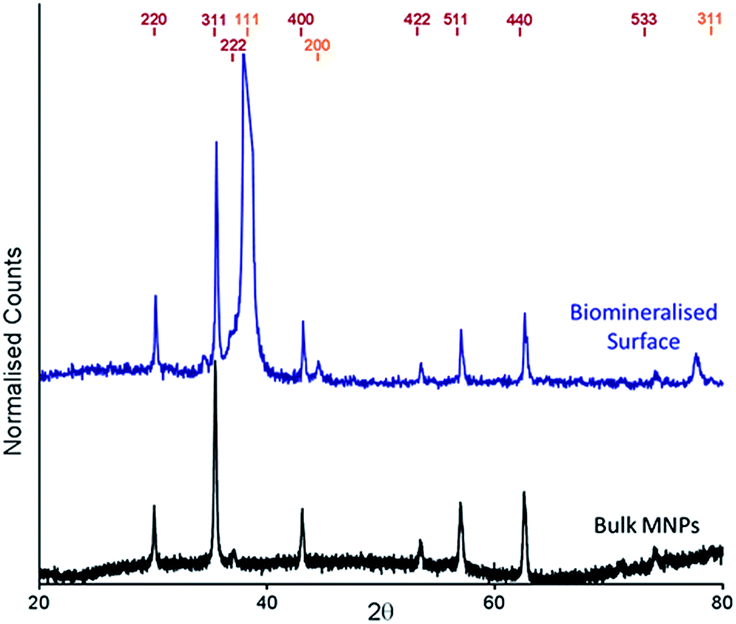 | ||
| Fig. 3 XRD spectra of POFHK (bulk) nanoparticles (black), and of a Mms6 (POFHK) surface. Each spectrum is offset for clarity and peak positions for magnetite (red) and gold (gold) are labelled. | ||
| Peak | Magnetite | Maghemite | POFHK(bulk) | Mms6(POFHK) |
|---|---|---|---|---|
| a Based on spectra from DIFFRAC Plus software.b Obscured by the Au (111) peak. | ||||
| (220) | 2.966 | 2.950 | 2.966 | 2.962 |
| (311) | 2.53 | 2.520 | 2.534 | 2.527 |
| (222) | 2.419 | 2.410 | 2.423 | —b |
| (400) | 2.096 | 2.080 | 2.097 | 2.097 |
| (422) | 1.712 | 1.700 | 1.718 | 1.711 |
| (511) | 1.614 | 1.610 | 1.615 | 1.614 |
| (440) | 1.483 | 1.480 | 1.483 | 1.483 |
| (533) | 1.279 | 1.270 | 1.280 | 1.276 |
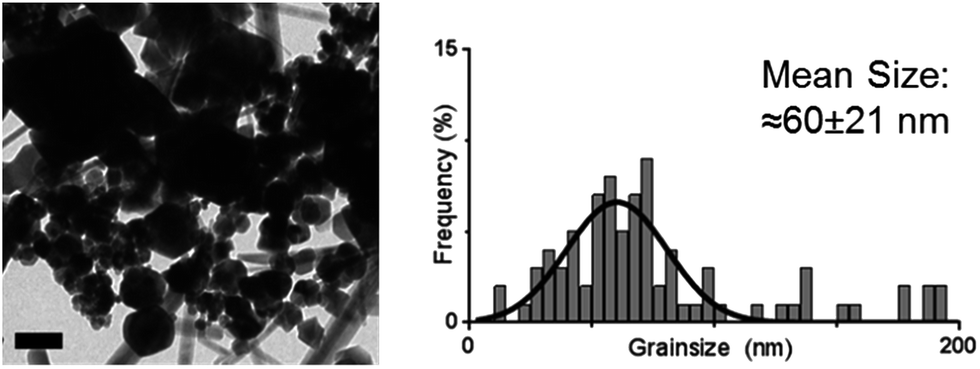 | ||
| Fig. 4 TEM image (left) and grainsize analysis of POFHK (bulk) nanoparticles. Scale bar is 100 nm. | ||
SEM of the Mms6 surface revealed clear, defined, dense patterns of magnetite nanoparticles in stripes corresponding to the areas covered by the protein. These patterns are consistent with our previous Mms6 surface biomineralisation experiments.25 When the peptide was used in place of the protein we observed a very different result. As shown in Fig. 5, the peptide patterns are not as clear or well defined as for the Mms6 protein surface templated particles. The peptide surface templated particles appear to be sparsely distributed on the surface. The extremely low amount of material observed is insufficient for diffraction analysis; although based on the XRD results from the Mms6 surface in the same reaction conditions we infer that the material is magnetite. The grainsize analysis of these particles also shows an interesting difference, (Fig. 4 & 5). The solution phase MNPs gives rise to a mean size of 60 nm, very close to the 65 nm size we observe on the peptide patterned surface, both with a similar broad distribution. By comparison, the Mms6 protein mediated particles are approximately 50% larger, with a mean size of 87 nm, and yet feature a much tighter size distribution. These data indicate that Mms6 is able to enhance both the size and homogeneity of the forming nanoparticles and also successfully anchor these particles to the surface. The peptide on the other hand appears to display particles with similar properties to the MNPs formed in the bulk solution, with no improvement in homogeneity, and with much less dense anchoring of the particles to the surface. It is possible that the peptide may be more susceptible to the destabilising conditions of the POFHK reaction, resulting in loss of function. If this is the case, it indicates that structure and assembly are necessary to the function of Mms6 rather than acidic C-terminal region sequence alone.
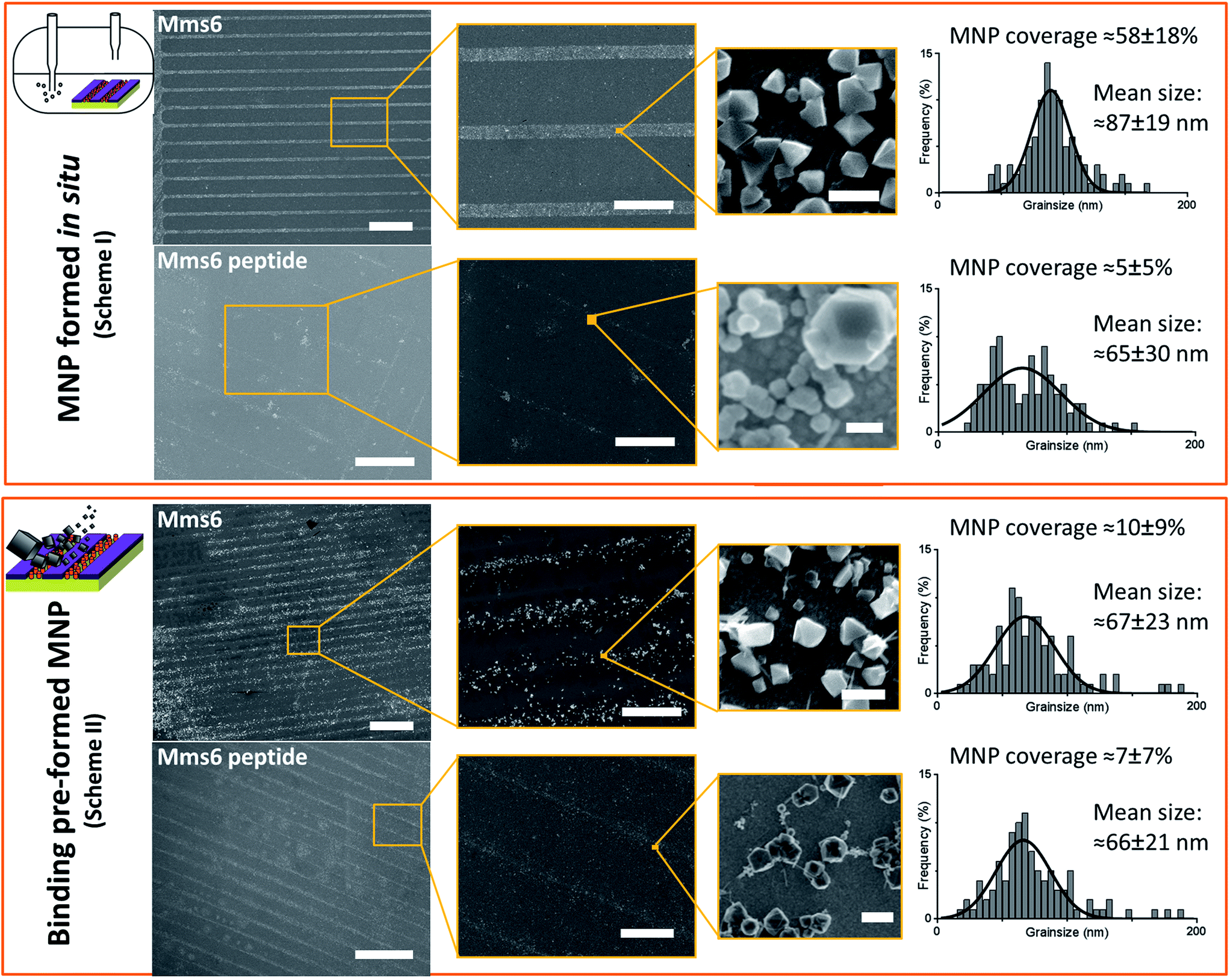 | ||
| Fig. 5 SEM analysis of the different surfaces at increasing magnification. Scale bars are 100 μm (left), 20 μm (centre), and 100 nm (right). MNP sizing histograms are shown with Gaussian fitting (GraphPad Prism). MNP coverage from 5 areas of biomolecule patterned regions is shown for each sample with standard deviation. | ||
To ascertain if the Mms6 protein or peptide was able to bind pre-made MNPs to the patterned surface, we modified the system. Rather than use the surfaces in an in situ biomineralisation reaction to produce particles, we simply took MNPs in water (prepared from a POFHK reaction), and applied them directly to a surface already patterned with either the Mms6 protein or peptide (Fig. 1, Scheme II). The resulting surfaces were washed, and analysed with SEM, and subsequent grainsizing was performed as before (Fig. 5). The main difference observed is between the two Mms6 surfaces (biomineralised and MNP binding), which revealed a less dense MNP pattern had been produced in the pre-formed MNP binding when compared to the in situ POFHK experiments; indicating that fewer MNP had been adsorbed. The protein also showed no selectivity towards binding larger MNPs, as the mean particle size from the grainsize analysis matched those of the applied bulk MNPs. SEM of the Mms6 peptide surface reveals no significant difference to that obtained from the control POFHK reaction, with sizes which again match those of the applied bulk MNPs. Taken together, these results help to build a picture of the differences between the protein and the peptide, and the mode of action of Mms6.
Intriguingly, if we compare the density of the MNPs on the Mms6 protein patterns resulting from the in situ biomineralisation to those formed from the addition of pre-made nanoparticles, we see it much reduced in the binding experiment when compared to the biomineralisation one. This is despite the biomineralisation surface being subject to much more extreme conditions of pH and heat, which suggests that the binding of MNPs by Mms6 may be enhanced when the MNPs are formed in the presence of the protein. We hypothesise that by binding iron ions, nucleating and stabilising the formation of the MNP, the C-terminal residues of Mms6 may mediate more contacts with the growing particle than if the MNP is supplied preformed. This may suggest that the strong attachment of Mms6 to magnetite is a by-product of its nucleating activity.
In this study, the Mms6 peptide appears to offer no effect on controlling the size or shape of the MNPs produced, and also sequesters nanoparticles with much lower density than the full Mms6 protein. Previous studies of an Mms6 C-terminal peptide in solution phase POFHK magnetite formation show modest particle size effects.22 A peptide with the additional glycine leucine repeat section displays greater activity.22 We considered that the shorter length of the peptide (when compared to the Mms6 protein) may mean it is not as accessible on the SAM patterned surface, which may limit its ability to function as fully as when free in solution. To test this, we prepared cys-pep surfaces with no SAM, thereby providing maximum accessibility to the peptides for both our process schemes. The surfaces, visualised by SEM (ESI Fig. 2 & 3†), revealed the same type of sparse particle deposition as before, showing that the peptides low activity is not due to masking by the SAM. Therefore, it may be that the Mms6 peptide is crowding itself, by packing more closely than is possible in the full length sequence. Alternatively, the shorter peptide may be more prone to destabilisation by heating than the full length Mms6 protein is.
The Mms6 peptide yielded sparse MNP coverage of the patterns under both experimental systems, and the apparent lack of any effect upon size or homogeneity of the surface bound nanoparticles suggests that this molecule exerts no apparent control over magnetite formation in this experimental system. The similarity between the pattern densities of the peptide resulting from Schemes I and II is suggestive of the peptide binding some particles weakly in both cases. It is possible that during the Scheme I POFHK reaction, the peptide may simply be binding to the particles produced in the bulk solution in a similar manner to the MNP binding by cys-pep in Scheme II. This mode of action would explain why both cys-pep patterned surfaces (biomineralised and MNP binding) look very similar.
Purified Mms6 forms spontaneous micelle-like structures; indicating that this protein has a natural propensity to aggregate.14,15,20 The Mms6 C-terminal peptide contains an abundance of acidic residues (which are considered an essential feature of Mms6 magnetite biomineralisation14) and previous analysis demonstrates some aggregation into oligomeric species of the range dimers to octamers.15 However, even with the same acidic residues and the locally high concentration brought about through the surface attachment, as well as any natural oligomerisation, the peptide appears to be unable to replicate the activity of the full length protein in our experiments. One important feature which is absent from the peptide is the distinctive glycine–leucine repeat motif (ESI Fig. 5–7†) which has been shown to be important in the oligomerisation and activity of Mms6 in previous studies.15,22 This type of low complexity repeating sequence is commonly associated with self-assembling proteins such as silk fibroins.36 We believe this motif could play a crucial role in the assembly of the complex; bringing about the correct packing and orientation of the proteins to facilitate iron ion coordination, binding, and nucleation of the magnetite nanoparticle. Molecular modelling of this sequence (ESI Fig. 5–7 and ESI† methods) suggests that the glycine and leucine residues in an α-helical conformation could produce regularly spaced interlocking knobs and holes along the length of the repeat motif. A parallel assembly of such helices would allow precise packing of multiple Mms6 molecules to generate a C-terminal surface of iron ion binding residues (aspartate and glutamate). This packing may give rise to an arrangement of these acidic residues that is able to support iron binding and crystallisation of magnetite, as opposed to the potentially uncontrolled surface packing of the peptide form of Mms6 used in our experiments (ESI Fig. 6†).
Using our biomimetic surface system as a mimic of the magnetosome membrane, we find that Mms6 is able to form nanoparticles which are different (in size and homogeneity) from the particles formed in a bulk solution, which is consistent with our previous studies. It should be noted that the ≈87 nm MNPs formed on our biomimetic surface are approximately twice the size of the 50 nm natural magnetosomes crystals. In previous studies where Mms6 was used in solution in a similar POFHK reaction, the particles were found to be approximately 50% smaller than control particles formed without protein.17 This is in direct contrast to the 50% size increase we see in our surface based experiment. One key consideration is the effect of the curvature present on the surface of Mms6 soluble micelles when free in solution, when compared to the immobilisation of Mms6 on a flat surface (Fig. 6). A planar arrangement of Mms6 may provide a greater expanse of the active acidic region, giving rise to increased nucleation and growth of larger crystals (Fig. 6b). The smaller convex surface present on the Mms6 micelles may provide a smaller nucleation surface and hence form smaller crystals (Fig. 6a). Neither the micelle form nor our surface experiment perfectly matches the concave assembly of Mms6 likely to be present on the interior face of the magnetosome (Fig. 6c). Further experiments could include enhancing our biomimetic system to better represent the curvature of the magnetosome interior. In addition, this biomimetic system provides a clear marker (larger MNP) of Mms6 activity in vitro. This could be exploited in future experiments to probe the effect of changes to the Mms6 sequence on MNP formation. This may help to further unlock the mode of action of Mms6 at the individual residue level.
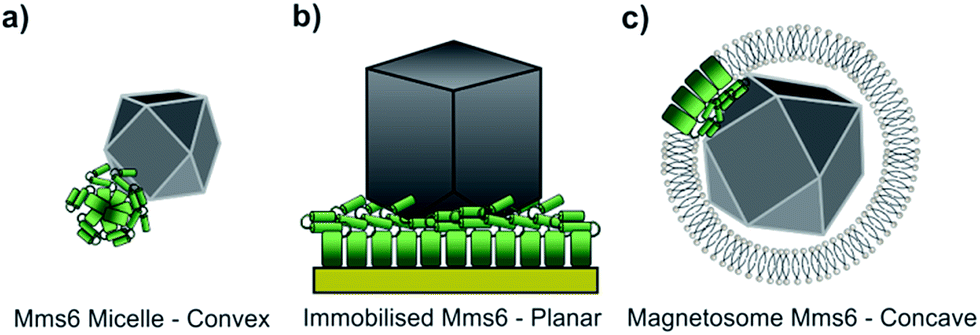 | ||
| Fig. 6 The assembly of Mms6 under different conditions (the N-terminal region of Mms6 is represented by a green rectangle and the iron binding C-terminal region by two green cylinders). (a) Mms6 in solution arranged into a micelle, with a convex surface interacting with a magnetite nanoparticle. (b) Surface immobilised Mms6, with a planar interaction with a magnetite crystal. (c) Mms6 within a magnetosome, presenting a concave surface that interacts with a magnetite particle. | ||
In summary, the results presented here suggest that Mms6 is a magnetite nucleation protein, where the assembled protein surface binds iron ions specifically to nucleate the formation of magnetite. Furthermore, in our biomimetic system it is not the C-terminal section alone, but the full length protein, which is required to provide the complete function of Mms6.
Acknowledgements
We would like to thank the LENNF facility at the University of Leeds and Stuart Micklethwaite for assistance with SEM, Professor Stephen Baldwin for the pTTQ18 based parent vector and the glycine, leucine modelling and Nik Reeves-McLaren for support with XRD. We also thank the BBSRC (BB/H005412/2) for funding this work, and the EPSRC for funding Scott Bird (CDT studentship (EP/J500458/1)) and Johanna Galloway (Post-Doctoral Prize Fellowship (EP/K503071/1)).Notes and references
- P. F. Lindley, Rep. Prog. Phys., 1996, 59, 867–933 CrossRef CAS.
- W. E. Winter, L. A. Bazydlo and N. S. Harris, Lab. Med., 2014, 45, 92–102 CrossRef.
- S. V. Torti and F. M. Torti, Nat. Rev. Cancer, 2013, 13, 342–355 CrossRef CAS.
- J. Collingwood and J. Dobson, J. Alzheimer's Dis., 2006, 10, 215–222 Search PubMed.
- J. Dobson, Ann. N. Y. Acad. Sci., 2004, 1012, 183–192 CrossRef CAS.
- R. Blakemore, Science, 1975, 190, 377–379 CrossRef CAS.
- D. A. Bazylinski, Chem. Geol., 1996, 132, 191–198 CrossRef CAS.
- D. A. Bazylinski and R. B. Frankel, Nat. Rev. Microbiol., 2004, 2, 217–230 CrossRef CAS.
- D. L. Balkwill, D. Maratea and R. P. Blakemore, J. Bacteriol., 1980, 141, 1399–1408 CAS.
- Y. A. Gorby, T. J. Beveridge and R. P. Blakemore, J. Bacteriol., 1988, 170, 834–841 CAS.
- R. B. Frankel, J. P. Zhang and D. A. Bazylinski, J. Geophys. Res.: Solid Earth, 1998, 103, 30601–30604 CrossRef.
- K. Grunberg, E. C. Muller, A. Otto, R. Reszka, D. Linder, M. Kube, R. Reinhardt and D. Schuler, Appl. Environ. Microbiol., 2004, 70, 1040–1050 CrossRef.
- A. Arakaki, J. Webb and T. Matsunaga, J. Biol. Chem., 2003, 278, 8745–8750 CrossRef CAS.
- L. Wang, T. Prozorov, P. E. Palo, X. Liu, D. Vaknin, R. Prozorov, S. Mallapragada and M. Nilsen-Hamilton, Biomacromolecules, 2012, 13, 98–105 CrossRef.
- S. R. Feng, L. J. Wang, P. Palo, X. P. Liu, S. K. Mallapragada and M. Nilsen-Hamilton, Int. J. Mol. Sci., 2013, 14, 14594–14606 CrossRef.
- M. Tanaka, E. Mazuyama, A. Arakaki and T. Matsunaga, J. Biol. Chem., 2011, 286, 6386–6392 CrossRef CAS.
- Y. Amemiya, A. Arakaki, S. S. Staniland, T. Tanaka and T. Matsunaga, Biomaterials, 2007, 28, 5381–5389 CrossRef CAS.
- T. Prozorov, S. K. Mallapragada, B. Narasimhan, L. J. Wang, P. Palo, M. Nilsen-Hamilton, T. J. Williams, D. A. Bazylinski, R. Prozorov and P. C. Canfield, Adv. Funct. Mater., 2007, 17, 951–957 CrossRef CAS.
- J. M. Galloway, A. Arakaki, F. Masuda, T. Tanaka, T. Matsunaga and S. S. Staniland, J. Mater. Chem., 2011, 21, 15244–15254 RSC.
- H. Zhang, X. Liu, S. Feng, W. Wang, K. Schmidt-Rohr, M. Akinc, M. Nilsen-Hamilton, D. Vaknin and S. Mallapragada, Langmuir, 2015, 31, 2818–2825 CrossRef CAS.
- S. Kashyap, T. J. Woehl, X. Liu, S. K. Mallapragada and T. Prozorov, ACS Nano, 2014, 8, 9097–9106 CrossRef CAS.
- A. Arakaki, F. Masuda, Y. Amemiya, T. Tanaka and T. Matsunaga, J. Colloid Interface Sci., 2010, 343, 65–70 CrossRef CAS.
- J. M. Galloway, J. P. Bramble, A. E. Rawlings, G. Burnell, S. D. Evans and S. S. Staniland, Small, 2012, 8, 204–208 CrossRef CAS.
- J. M. Galloway, J. P. Bramble, A. E. Rawlings, G. Burnell, S. D. Evans and S. S. Staniland, J. Nano Res., 2012, 17, 127–146 CrossRef CAS.
- S. M. Bird, J. M. Galloway, A. E. Rawlings, J. P. Bramble and S. S. Staniland, Nanoscale, 2015, 7, 7340–7351 RSC.
- Ł. Krzeminński, S. Cronin, L. Ndamba, G. W. Canters, T. J. Aartsma, S. D. Evans and L. J. Jeuken, J. Phys. Chem. B, 2011, 115, 12607–12614 CrossRef.
- M. V. Voinova, M. Rodahl, M. Jonson and B. Kasemo, Phys. Scr., 1999, 59, 391–396 CrossRef CAS.
- C. Zhou, J.-M. Friedt, A. Angelova, K.-H. Choi, W. Laureyn, F. Frederix, L. A. Francis, A. Campitelli, Y. Engelborghs and G. Borghs, Langmuir, 2004, 20, 5870–5878 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- J. M. Galloway, J. E. Talbot, K. Critchley, J. J. Miles and J. P. Bramble, Adv. Funct. Mater., 2015, 25, 4590–4600 CrossRef CAS.
- M. C. Dixon, J. Biomol. Tech., 2008, 19, 151–158 Search PubMed.
- G. Sauerbrey, Zeitschrift fur Physik, 1959, 155, 206–222 CrossRef CAS.
- D. Xu and Y. Zhang, Proteins, 2012, 80, 1715–1735 CrossRef CAS.
- J. A. R. Guivar, A. I. Martínez, A. O. Anaya, L. D. L. S. Valladares, L. L. Félix and A. B. Dominguez, Adv. Nanopart., 2014, 03, 114–121 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS.
- C. Z. Zhou, F. Confalonieri, M. Jacquet, R. Perasso, Z. G. Li and J. Janin, Proteins, 2001, 44, 119–122 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Including Mms6 protein and peptide sequences, additional QCM-D and SEM data and protein modelling. See DOI: 10.1039/c5ra16469a |
| ‡ Joint first authors (alphabetically). |
| This journal is © The Royal Society of Chemistry 2016 |