
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Impact of fluorine substitution upon the photovoltaic properties of benzothiadiazole-fluorene alternate copolymers†
Luke
Cartwright
a,
Ahmed
Iraqi
*a,
Yiwei
Zhang
b,
Tao
Wang
c and
David. G.
Lidzey
*b
aDepartment of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: a.iraqi@sheffield.ac.uk; Fax: +44 (0)114 222 9303; Tel: +44 (0)114 222 9566
bDepartment of Physics and Astronomy, University of Sheffield, S3 7RH, UK. E-mail: d.g.lidzey@sheffield.ac.uk; Fax: +44 (0)114 222 3555; Tel: +44 (0)114 222 3501
cSchool of Materials Science and Engineering, Wuhan University of Technology, Wuhan 430070, China
First published on 18th May 2015
Abstract
The effect of fluorine substitution on the ubiquitously-studied, moderate band gap polymer, poly(2,7-fluorene-alt-dithienylbenzothiadiazole) (PFO-DBT) has been investigated by substituting the hydrogen atoms at the 5,6-positions of benzothiadiazole to yield the polymer, PFO-DffBT. An analogous polymer, PFDo-DffBT, with longer chains on the 9H-positions of the fluorene moiety, was synthesised to rectify the low Mw of PFO-DffBT. In thin films, both fluorinated polymers display absorption bands with well-defined shoulders, giving optical band gaps of 1.91 and 1.89 eV, respectively. The optical band gap of PFDo-DBT is identical to that of PFDo-DffBT, despite the former having a significantly larger Mw. However, PFDo-DffBT displayed a blue-shifted λmax relative to its non-fluorinated counterpart. The HOMO levels of PFO-DffBT and PFDo-DffBT are lower than their non-fluorinated analogues, owing to the incorporation of electron withdrawing substituents on the benzothiadiazole moiety. The photovoltaic properties of all polymers were investigated by fabricating bulk heterojunction (BHJ) polymer solar cells using PC70BM as the electron acceptor. PFO-DffBT displayed the highest efficiency with a PCE of 4.4% despite having the lowest Mw of all polymers studied.
Introduction
Numerous energy resources are currently available, however, solar energy is believed to be the largest carbon-neutral energy source with the sun producing an estimated 3.86 × 1026 J of energy every second.1The first commercial crystalline silicon p–n junction solar cell was produced by Bell laboratories in 1954. The efficiency of such devices have improved remarkably, going from 6%, to modern era wafer-size single-junction crystalline silicon solar cells that have efficiencies of 25%.2,3 The current inorganic monocrystalline photovoltaic devices available are highly efficient at harvesting solar energy. However, they are fragile, have poor performance in low light intensities, their performance varies with temperature and they are produced using energy intensive manufacturing techniques. Thus, research into alternative, cost-efficient photovoltaic device that can efficiently harvest solar energy has resulted in research on so-called third generation PV devices (organic photovoltaic devices). These have potential advantages over their inorganic counterparts including reduced embodied energy, increased flexibility, abundant materials for fabrication and better operation at lower light intensity.3,4 Despite skeptics within the solar community who have challenged the viability of organic photovoltaic devices, the successful commercialization of organic light emitting diodes gives confidence that organic photovoltaics may complement traditional inorganic photovoltaic devices.5–13
The most commonly used architecture for the active layer of polymer solar cells is the bulk heterojunction (BHJ). A conjugated polymer (electron donor) and a fullerene derivative (electron acceptor) are blended together to form an interpenetrating network that forms the active layer of the BHJ cell. High efficiencies in polymer solar cells can be achieved by designing conjugated polymers with a low band gap, extended absorption in the visible and infrared region, high charge carrier mobilities, high absorption coefficients and high molecular weights. The length and flexibility of the side chains attached to the rigid polymer backbone are central in determining the solubility of the conjugated polymer.
Previous literature has shown that the easiest way to fulfil these criteria is to synthesise a conjugated polymer that comprises alternating donor–acceptor (D–A) repeat units along polymer chains.7,14,15 The internal charge transfer between the donor and acceptor moieties enables control of the band gap of the polymer.16 The HOMO and LUMO levels of D–A polymers can be finely tuned using various approaches. For example, a small band gap can be obtained when a more electron rich donor monomer is copolymerised with a more electron deficient acceptor monomer.17 Further adjustment of the HOMO and LUMO levels can be achieved through the introduction of electron withdrawing or electron donating substituents and the attachment of solubilising side chains.15,18,19
A growing body of literature now addresses the incorporation of substituents and the effect they have on the resulting polymer system.11,18–22 Arguably, the most interesting substituent is fluorine; the smallest electron withdrawing group with a Pauling electronegativity of 4.0.15 Li et al. was the first to design and synthesise a fluorine-functionalised benzothiadiazole-acceptor moiety that was polymerised with an electron rich benzodithiophene unit.20 An analogous, non-fluorinated polymer was also synthesised to help establish the effects of fluorine-substitution. The fluorinated polymer showed a dramatic increase in photovoltaic performance which was attributed to the lower optical band gap and deeper HOMO level. Other groups have reported the use of fluorinated-acceptors and have found a similar increase in performance.19,22–24 However, other groups have discovered that incorporation of the fluorinated benzothiadiazole unit does not always yield an improved photovoltaic performance.18,25
The objective of this work is to incorporate fluorine substituents into the commonly studied benzothiadiazole acceptor. This was then copolymerised with the electron rich 9,9-dioctylfluorene monomer flanked by two thienyl repeat units to afford a new copolymer poly[2,7-(9,9-dioctylfluorene)-alt-4,7-bis(thiophen-2-yl)-5,6-difluorobenzo-2,1,3-thiadiazole] (PFO-DffBT, Fig. 1). In view of the low molecular weight of PFO-DffBT larger solubilising alkyl chains were attached to the fluorene moiety to yield poly[2,7-(9,9-didodecylfluorene)-alt-4,7-bis(thiophen-2-yl)-5,6-difluorobenzo-2,1,3-thiadiazole] (PFDo-DffBT). To ascertain the effects of fluorination, its non-fluorinated analogue, poly[2,7-(9,9-didodecylfluorene)-alt-4,7-bis(thiophen-2-yl)benzo-2,1,3-thiadiazole] (PFDo-DBT), was also synthesised. The optical, thermal and electrochemical properties of these polymers were investigated and their performance in BHJ polymer solar cell devices were evaluated. We demonstrate that incorporation of fluorine blue-shifts the peak absorption wavelength (λmax). However, the optical band gaps of fluorinated polymers when cast into thin films were identical to their non-fluorinated analogues owing to improved molecular ordering. We fabricate photovoltaic devices from all polymers synthesized and optimize the solvent system and solvent additives used to cast the polymer![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :fullerene active-layer. We find that despite the fact that PFO-DffBT and PFDo-DffBT both display deeper HOMO levels relative to their non-fluorinated analogue, this does not apparently translate into a higher open circuit voltage (Voc). Nevertheless we show that OPVs based on PFO-DffBT, PFDo-DBT and PFDo-DffBT have power conversion efficiencies (PCEs) of 4.4, 2.4 and 3.6%, respectively.
:fullerene active-layer. We find that despite the fact that PFO-DffBT and PFDo-DffBT both display deeper HOMO levels relative to their non-fluorinated analogue, this does not apparently translate into a higher open circuit voltage (Voc). Nevertheless we show that OPVs based on PFO-DffBT, PFDo-DBT and PFDo-DffBT have power conversion efficiencies (PCEs) of 4.4, 2.4 and 3.6%, respectively.
 | ||
| Fig. 1 Structures of PFO-DffBT, PFDo-DBT and PFDo-DffBT. | ||
Results and discussion
Polymer synthesis
PFO-DffBT was prepared via Suzuki coupling using Pd(OAc)2 and tri(o-tolyl)phosphine as the catalyst and tetraethylammonium hydroxide as the base. The polymerisation proceeded rapidly, with large quantities of purple precipitate forming after 1 hour. Consequently, only a portion (20%) of the resulting polymer was soluble in common organic solvents and able to be processed into films for BHJ devices. The number-average molecular weight (Mn) and weight-average molecular weight (Mw) were estimated to be 6800 and of 9600 Da, respectively (Table 1). It is well documented that attaining a high Mw is imperative for photovoltaic performance as it facilitates the bicontinuous phase formation with fullerene derivatives and promotes efficient charge transportation.22 It was hypothesised that incorporating fluorine atoms on the benzothiadiazole moiety resulted in stronger π–π stacking and aggregation of polymer chains. Thus, the product precipitates out of solution quickly during the polymerisation, which limits the final molecular weight of the product. Attaching alkyl chains to the polymer backbones disrupts the π–π stacking. However, it was clear that larger alkyl chains needed to be attached to the donor-moieties in order to the overcome the aggregation that is brought about by incorporating fluorine onto the benzothiadiazole moiety.| Polymer | M n a (Da) | M w a (Da) | λ max solution (nm) | ε b (M−1 cm−1) | λ max film (nm) | E g opt c (eV) | HOMOd (eV) | LUMOe (eV) | E g elec f (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a Measurements conducted on the toluene fractions of polymers using a differential refractive index (DRI) detection method. b Absorption coefficient measured at λmax in CHCl3. c Optical energy gap determined from the onset of the absorption band in thin film. d HOMO position (vs. vacuum) determined from the onset of oxidation. e LUMO position (vs. vacuum) determined from the onset of reduction. f Electrochemical energy gap. | |||||||||
| PFO-DffBT | 6800 | 9600 | 518 | 41600 |
549 | 1.91 | −5.38 | −3.32 | 2.05 |
| PFDo-DBT | 20500 |
35700 |
548 | 40200 |
578 | 1.89 | −5.44 | −3.49 | 1.95 |
| PFDo-DffBT | 9700 | 15900 |
527 | 40600 |
556 | 1.89 | −5.51 | −3.48 | 2.03 |
Umeyama and coworkers demonstrated that the additional incorporation of solubilising 3-hexylthiophene repeat units yields higher molecular weight polymers. However, this negates the positive effects of fluorine substitution.18 Therefore, we have avoided this approach and consequently, redesigned the polymer to bear larger solubilising dodecyl chains on the fluorene moiety at the 9H-position. Previous work by Inganäs and coworkers demonstrated that this approach was successful in attaining higher molecular weight polymers.26 Both PFDo-DffBT and PFDo-DBT were hence prepared in this work using a similar method to that outlined above. Both polymerisations were conducted until precipitation of the polymers was first observed, which occurred around 48 hours after the start of polymerisations. Only small amounts of polymer precipitated out of solution in these reactions; a finding in contrast to the polymerisation of PFO-DffBT. PFDo-DffBT and its non-fluorinated analogue PFDo-DBT displayed excellent solubility in common organic solvents, permitting the formation of films for photovoltaic devices. GPC analysis estimated the Mn and Mw of PFDo-DBT to be 20500 and 35700 Da, respectively. In contrast, the Mn and Mw of PFDo-DffBT were estimated to be 9700 and 15900 Da, respectively (Table 1). The fluorinated analogue clearly displays a lower molecular weight. This data supports our earlier hypothesis that incorporation of fluorine on the benzothiadiazole moiety results in stronger π–π stacking and aggregation of polymer backbones, which limits the final molecular weight.
Optical properties
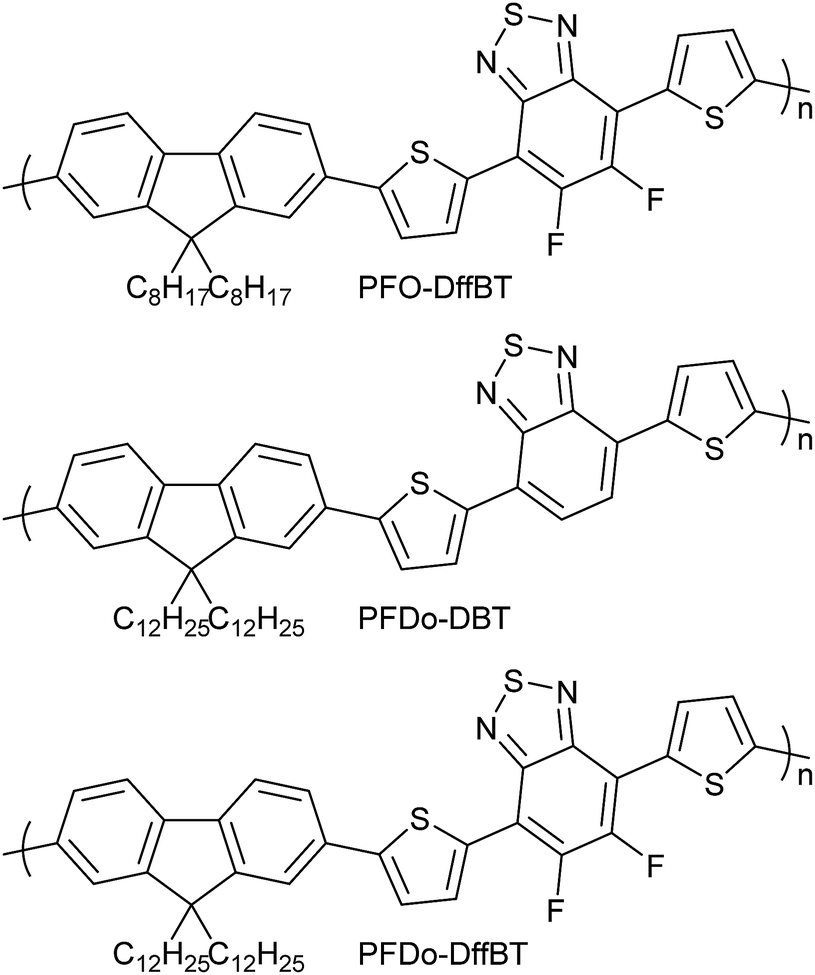
The optical properties of all polymers were investigated by UV-vis absorption spectroscopy on dilute chloroform solutions and drop-cast films cast on quartz substrates (Fig. 2). The optical properties of PFO-DffBT, PFDo-DffBT and PFDo-DBT are summarised in Table 1.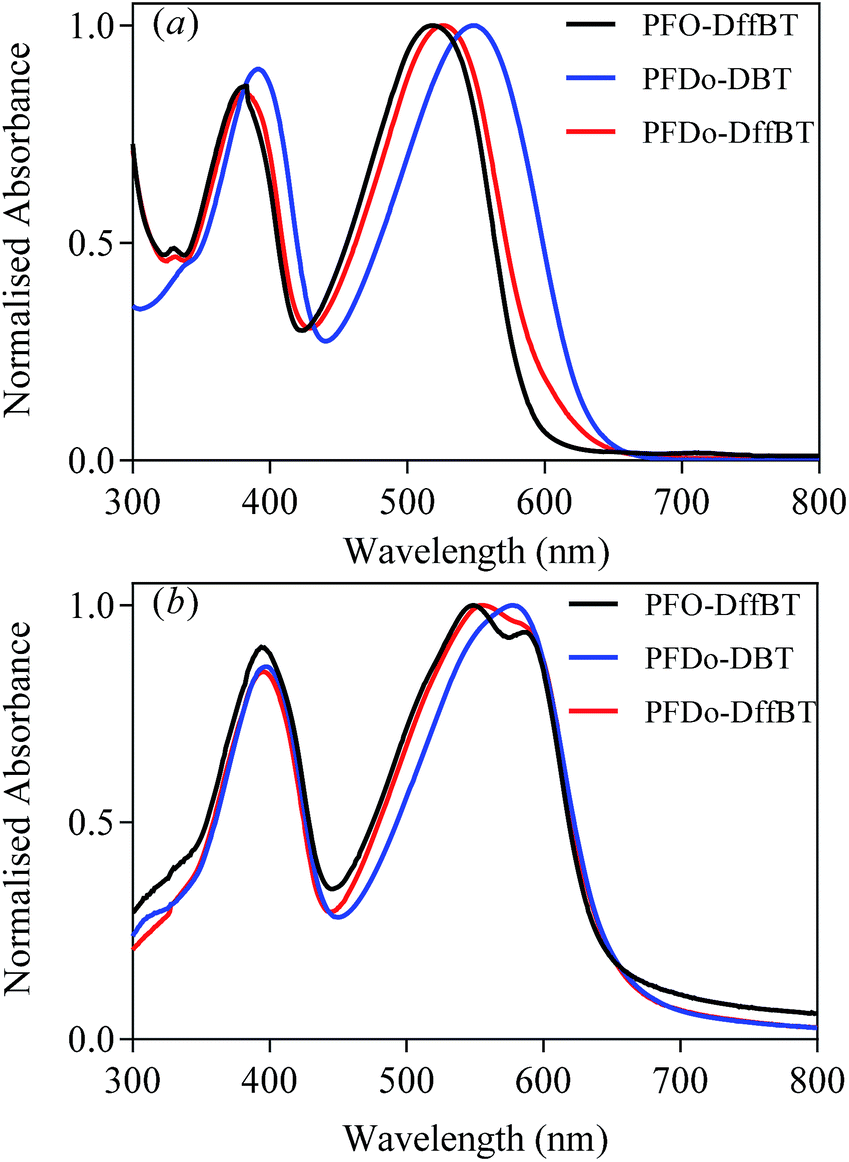 | ||
| Fig. 2 Normalised absorption spectra of PFO-DffBT, PFDo-DBT and PFDo-DffBT in; (a) chloroform solutions; and (b) thin films. | ||
Dilute solutions of PFO-DffBT revealed absorption bands in the visible region at 383 and 518 nm. When cast into a thin-film, the absorption spectra of PFO-DffBT displays red-shifted absorption maxima at 394 and 549 nm, relative to that in solution. The bathochromic shift that is observed can be ascribed to stronger π–π interchain stacking and a more coplanar structure in the solid state. Furthermore, PFO-DffBT displays a peak at ∼590 nm which we attribute to the purely electronic transition of the polymer; a result that indicates that inhomogeneous broadening within the molecular ensemble has been reduced as a result of improved molecular ordering in the solid state. We speculate that such enhanced molecular ordering is a direct result of interactions between fluorine atoms and components on adjacent aromatics. This yields a more planar polymer backbone which promotes π–π stacking in solid state. The optical band gap of PFO-DffBT was calculated to be 1.91 eV. The non-fluorinated analogue of this polymer, PFO-DBT, has previously been prepared by the Iraqi group and displays an absorption maximum at 549 nm in chloroform solution.27 When fabricated into a film, the absorption maximum was located at 592 nm; a wavelength red shifted relative to that of PFO-DffBT.27 This phenomenon has been reported by previous groups.18,19,22,25,28 However, PFO-DBT did not display any improved resolution in solid state indicating that the material has a higher degree of structural and electronic disorder.
PFDo-DBT, displayed an optical band gap of 1.89 eV and absorption maxima at 397 and 578 nm in films. Clearly, the introduction of longer alkyl chains on the fluorene moiety facilitates the formation of higher molecular weights, lowering the band gap and increasing the λmax of the resulting polymer when compared to PFO-DffBT. The absorption maxima of PFDo-DffBT (at 556 nm), is red shifted relative to that of PFO-DffBT but blue shifted relative to that of PFDo-DBT. However, the absorption spectra of PFDo-DffBT, like PFO-DffBT, is broader and clearly displays a peak at 595 nm, which is again indicative of increased ordering in the solid state. Interestingly this peak is completely absent in PFDo-DBT suggesting that fluorination of the benzothiadiazole moiety does not hinder backbone planarization and in actuality improves polymer-chain stacking in the solid state. Previous literature has reported the same phenomenon.18,19,25 We speculate that the non-covalent interaction between fluorine atoms and components on adjacent aromatics are responsible for this phenomenon. It is possible that the high planarity of PFDo-DffBT may introduce efficient interchain interactions, thereby resulting in an optical band gap that is identical to that of PFDo-DBT (1.89 eV), despite the lower molecular weight and blue-shifted absorption maxima.
All polymers displayed similar absorption coefficients suggesting that neither the incorporation of fluorine substituents nor the change of alkyl groups on the fluorene moiety has any significant effect on the light harvesting qualities of the polymer.
Electrochemical properties
The HOMO and LUMO energy levels (vs. vacuum) of the polymers were determined from the onsets of oxidation and reduction (Table 1). The onsets were determined via cyclic voltammetry measurements, which were conducted on drop-cast polymer films in acetonitrile with tetrabutylammonium perchlorate as the electrolyte (Fig. 3). PFO-DffBT displayed a HOMO level of −5.38 eV, which is deeper than its non-fluorinated analogue, PFO-DBT.27 This is consistent with literature concerning the effects of fluorine substitution and the impact it has on the HOMO levels of the resulting polymer.18–22,29 It should be noted that the limited solubility of PFO-DffBT hindered the formation of films on the working electrode. Consequently, such thin films degraded after several sweeps. It is possible therefore that the results obtained for the energy levels of PFO-DffBT do not fully represent their true value. Indeed, we speculate that the true HOMO level may be deeper still. The LUMO level of PFO-DffBT was estimated to be −3.32 eV. This corresponds to an electrochemical band gap of 2.05 eV for PFO-DffBT.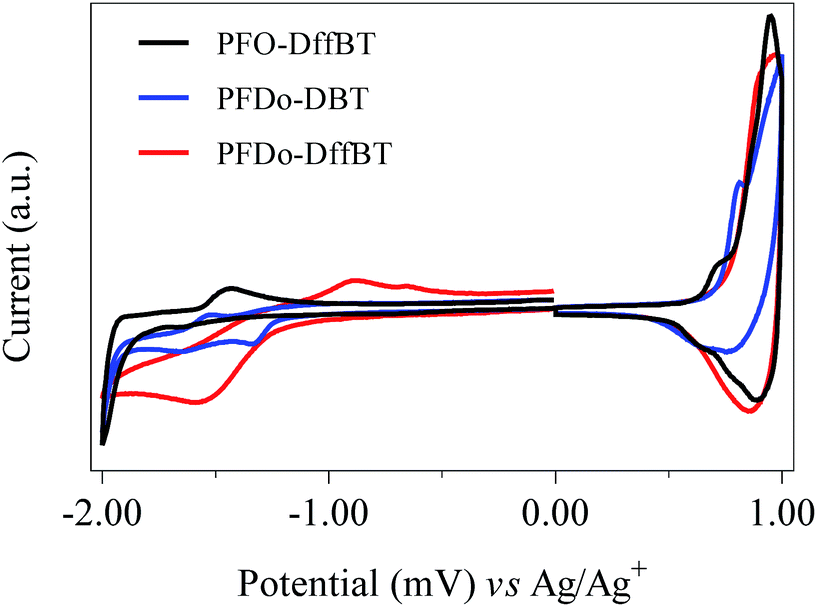 | ||
| Fig. 3 Cyclic voltammograms of PFO-DffBT, PFDo-DBT and PFDo-DffBT on platinum disc electrodes (area 0.031 cm2) at a scan rate of 100 mV s−1 in acetonitrile/tetrabutyl ammonium perchlorate (0.1 mol dm−3). | ||
The onset potentials of the first oxidation wave of PFDo-DBT and PFDo-DffBT appeared at 0.72 and 0.79 V vs. Ag/Ag+, corresponding to HOMO energy levels of −5.44 and −5.51 eV respectively. The onset potentials of the first reduction wave of PFDo-DBT and PFDo-DffBT appeared at −1.23 and −1.24 V, corresponding to LUMO energy levels of −3.49 and −3.48 eV, respectively. This corresponds to an electrochemical band gap of 1.95 and 2.03 eV for PFDo-DBT and PFDo-DffBT, respectively. Incorporation of fluorine does not significantly alter the LUMO levels of the resulting polymer. However, it does lower the HOMO level of the resulting polymer which is consistent with previous literature.18–22,29 The lower HOMO levels of PFO-DffBT and PFDo-DffBT should result in better oxidative stability in ambient conditions and yield a higher open-circuit voltage (Voc) in photovoltaic devices.
Thermal properties and XRD studies
The thermal stabilities of the polymers were investigated by thermogravimetric analysis (TGA) (Fig. 4). All polymers were determined to be thermally stable with decomposition temperatures (5% weight loss) higher than 400 °C in a nitrogen atmosphere. The initial onset of decomposition temperatures for PFO-DffBT, PFDo-DffBT and PFDo-DBT were estimated to be 417, 432 and 419 °C, respectively. All of these initial weight loss peaks can be attributed to loss of alkyl chains from the fluorene donor moiety. The results indicate that the fluorinated polymer, PFDo-DffBT, undergoes decomposition at a slightly higher temperature relative to its non-fluorinated analogue. Unfortunately, no comparison can be drawn between PFO-DffBT and PFO-DBT owing to a lack of thermal analysis reported. Li and Iyer also demonstrated a marginal increase in decomposition temperature, indicating that this phenomenon occurs more generally.20,29 | ||
| Fig. 4 TGA plots of PFO-DffBT, PFDo-DBT, and PFDo-DffBT with a heating rate of 10 °C min−1 under an inert atmosphere of nitrogen. | ||
Powder X-ray diffraction (XRD) studies were conducted on the three polymers (Fig. 5). The broad features of the PFDo-DBT are consistent with an incoherent scatter from an amorphous solid. The presence of several sharp peaks in the XRD of PFO-DffBT and PFDo-DffBT suggest the polymers adopt a microcrystalline arrangement in the solid state. PFO-DffBT and PFDo-DffBT display peaks at 2θ values of 6.5 and 5.0, respectively. This corresponds to distances of ∼13 Å for PFO-DffBT and ∼16 Å for PFDo-DffBT. Previous literature reports have shown that this value corresponds to the distance between polymer main chains, where alkyl chains on the fluorene moiety self-organise into an interdigitated manner.30,31 A sharp, narrow peak appeared at 2θ values of 25.32 and 25.65 for PFO-DffBT and PFDo-DffBT, respectively, corresponding to small coplanar stacking distances of 3.52 and 3.48 Å, respectively. It is assumed that the smaller stacking distances are a result of enhanced π–π stacking and intermolecular interactions brought about by the incorporation of fluorine. The XRD of PFDo-DBT does not show any peak in this area. Evidently, incorporation of fluorine improves the packing of polymer chains by promoting stronger coplanar interactions. This further suggests that PFDo-DffBT has improved organisation in solid state compared to PFDo-DBT; a finding consistent with the reduced inhomogeneous broadening observed in the solid-state absorption spectra.
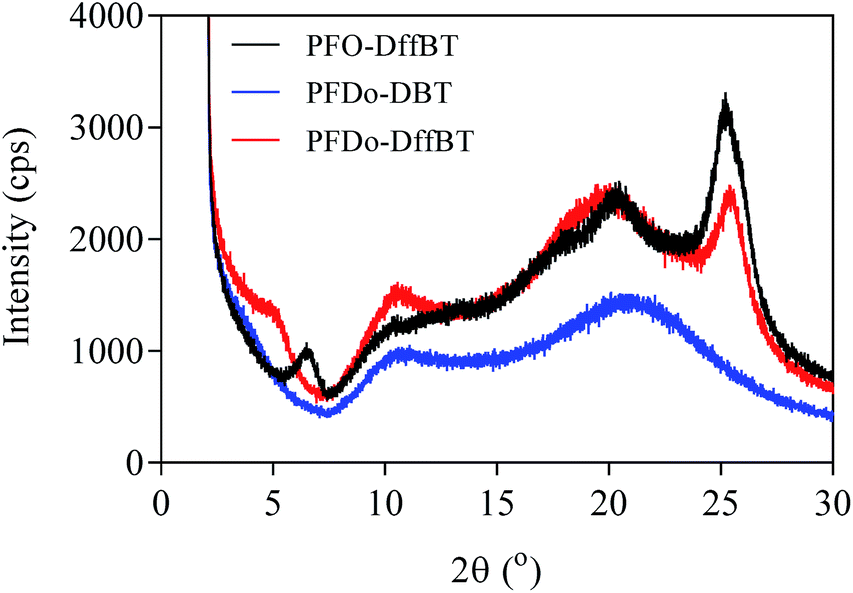 | ||
| Fig. 5 Powder X-ray diffraction (XRD) patterns of PFO-DffBT, PFDo-DBT and PFDo-DffBT. | ||
Photovoltaic properties
Photovoltaic measurements were made on a series of glass/ITO/PEDOT:PSS/polymer:PC70BM/Ca/Al devices using blends of polymers:PC70BM in weight ratios from 1:1 to 1:4. A detailed device-fabrication process is described in the Experimental section. Photovoltaic measurements revealed that devices with the active layer cast from 1:4 polymer:PC70BM (w/w ratio) gave the optimum performance. The influence of solvents used to cast the active layer was then investigated (Table 2). Chloroform was found as the best solvent for PFDo-DBT whilst chlorobenzene with a 3.0% diiodooctane (DIO) additive yielded the best results for PFO-DffBT and PFDo-DffBT. It is known that the addition of DIO decreases the size of fullerene domains and facilitates the formation of a bicontinous interpenetrating donor:acceptor network.32 The J–V characteristic curves for the best performing devices are shown in Fig. 6. Measurements of the external quantum efficiency (EQE) were made, with the integrated photocurrent being consistent with the short circuit current density (Jsc) values quoted here.
:PC70BM/Ca/Al BHJ polymer solar cells under a simulated photovoltaic light with 1000 W m−2 illumination (AM 1.5). PCE values given represent the highest and average values obtained
| Polymer | Polymer:PC70BMa (w/w) |
Solvent | J sc (mA cm−2) | V oc (V) | FF | PCE (%) |
|---|---|---|---|---|---|---|
|
a Polymer:PC70BM weight ratio.
b CF = chloroform.
c CB = chlorobenzene.
d CB + DIO = chlorobenzene + diiodooctane (3% additive).
|
||||||
| PFO-DffBT | 1:4 |
CFb | 4.16 | 0.92 | 58.4 | 2.2 (2.2 ± 0.1) |
| 1:4 |
CBc | 5.00 | 0.96 | 55.4 | 2.7 (2.5 ± 0.1) | |
| 1:4 |
CB + DIOd | 7.97 | 0.95 | 58.3 | 4.4 (4.2 ± 0.2) | |
| PFDo-DBT | 1:4 |
CFb | 4.16 | 0.97 | 59.7 | 2.4 (2.4 ± 0.1) |
| 1:4 |
CBc | 3.75 | 0.99 | 48.2 | 1.8 (1.8 ± 0.05) | |
| 1:4 |
CB + DIOd | 2.47 | 0.88 | 54.1 | 1.2 (1.1 ± 0.2) | |
| PFDo-DffBT | 1:4 |
CFb | 3.49 | 0.82 | 50.2 | 1.4 (1.4 ± 0.1) |
| 1:4 |
CBc | 6.69 | 0.88 | 58.3 | 3.4 (3.3 ± 0.2) | |
| 1:4 |
CB + DIOd | 8.06 | 0.86 | 51.3 | 3.6 (3.4 ± 0.1) | |
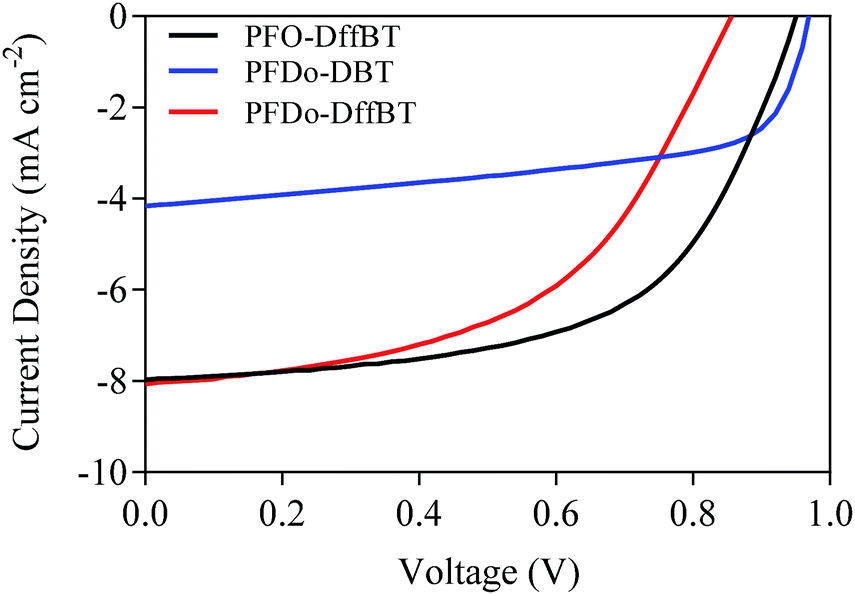 | ||
| Fig. 6 J–V characteristic curves of photovoltaic devices fabricated from PFO-DffBT, PFDo-DBT and PFDo-DffBT. | ||
A comparison of the performance of the best devices from the polymers indicates that all three materials exhibit moderate efficiencies with PCEs of 4.4, 2.4 and 3.6% for PFO-DffBT, PFDo-DBT and PFDo-DffBT, respectively. Optimised photovoltaic devices based on PFO-DffBT demonstrated an open-circuit voltage (Voc) of 0.95 V, a Jsc of 7.97 mA cm−2 and a fill-factor (FF) of 58.3%. In contrast, PFO-DBT synthesised by Iraqi et al., demonstrated a Voc of 1.03 V, a Jsc of 9.66 mA cm−2 and a FF of 54.7%.27 The polymer displayed a moderate PCE of 5.4%, which is higher than the fluorinated-analogue synthesised within this report. The optimised photovoltaic device based on PFDo-DBT demonstrated a Voc of 0.97 V, a Jsc of 4.16 mA cm−2 and a FF of 59.7%. In contrast, optimised devices fabricated the fluorinated polymer PFDo-DffBT demonstrated a Voc of 0.86 V, Jsc of 8.06 mA cm−2 and a FF of 51.3%.
PFO-DffBT demonstrated the best performance in this series of polymers, despite it having the lowest molecular weight. We speculate that the shorter alkyl chains on the fluorene moiety facilitate the packing of polymer chains in the solid state, resulting in a material having improved molecular order and thus enhanced charge-carrier mobility resulting in higher PCE values. However, further studies are required to elucidate whether this is the case. In spite of this, PFO-DffBT still displays lower Voc, Jsc and PCE values relative to its non-fluorinated analogue. Additionally, PFDo-DffBT displayed lower Voc values when compared to its non-fluorinated analogue, PFDo-DBT. It is well documented that the Voc is closely related to the energy difference between the HOMO of the donor and the LUMO of the acceptor.33 Interestingly, the lower HOMO levels of PFO-DffBT and PFDo-DffBT did not translate into higher Voc values. Previous groups have found that the discrepancy can be explained in terms of the morphology of the blended active layer.18,20,34 The fluorinated polymer PFDo-DffBT provided a better PCE than that of PFDo-DBT when the best devices were compared. This was chiefly as a result of the better Jsc values of the fluorinated polymer.
Atomic force microscopy (AFM) images
To explore the origin of the varied photovoltaic performances, we have used atomic force microscopy (AFM) in tapping mode to image the surface morphologies of polymer:PC70BM blends thin films (as shown in Fig. 7). It can be seen that both PFO-DffBT:PC70BM and PFDo-DffBT:PC70BM blends display films characterized by morphology that we interpret as fine-scale phase separation (and possibly in PFO-DffBT:PC70BM crystallization), a result consistent with improved exciton dissociation and photocharge generation. Previous work suggests that the lower surface energy of fluorinated polymers drives such materials to the surface during film-casting (so-called surface-enrichment); a process that can result in an unfavourable geometry for charge extraction and the accumulation of space charge within the device.20 Such a process is consistent with the reduced Voc observed in PFO-DffBT:PC70BM based devices, although further work is necessary to characterize the morphology of such films in a direction normal to the substrate. We find that PFO–PFDo-DffBT:PC70BM blends display a distinctive morphology having features that are coarser compared to those observed in blends based on PFDo-DBT:PC70BM. In contrast, PFDo-DBT:PC70BM blend films are characterized by a series of significantly larger (micron-sized) features that we speculate may be fullerene-rich. Phase-separation at such micron length-scales invariably results in inefficient exciton dissociation; a result consistent with the low Jsc and FF associated with photovoltaic devices fabricated from PFDo-DBT.
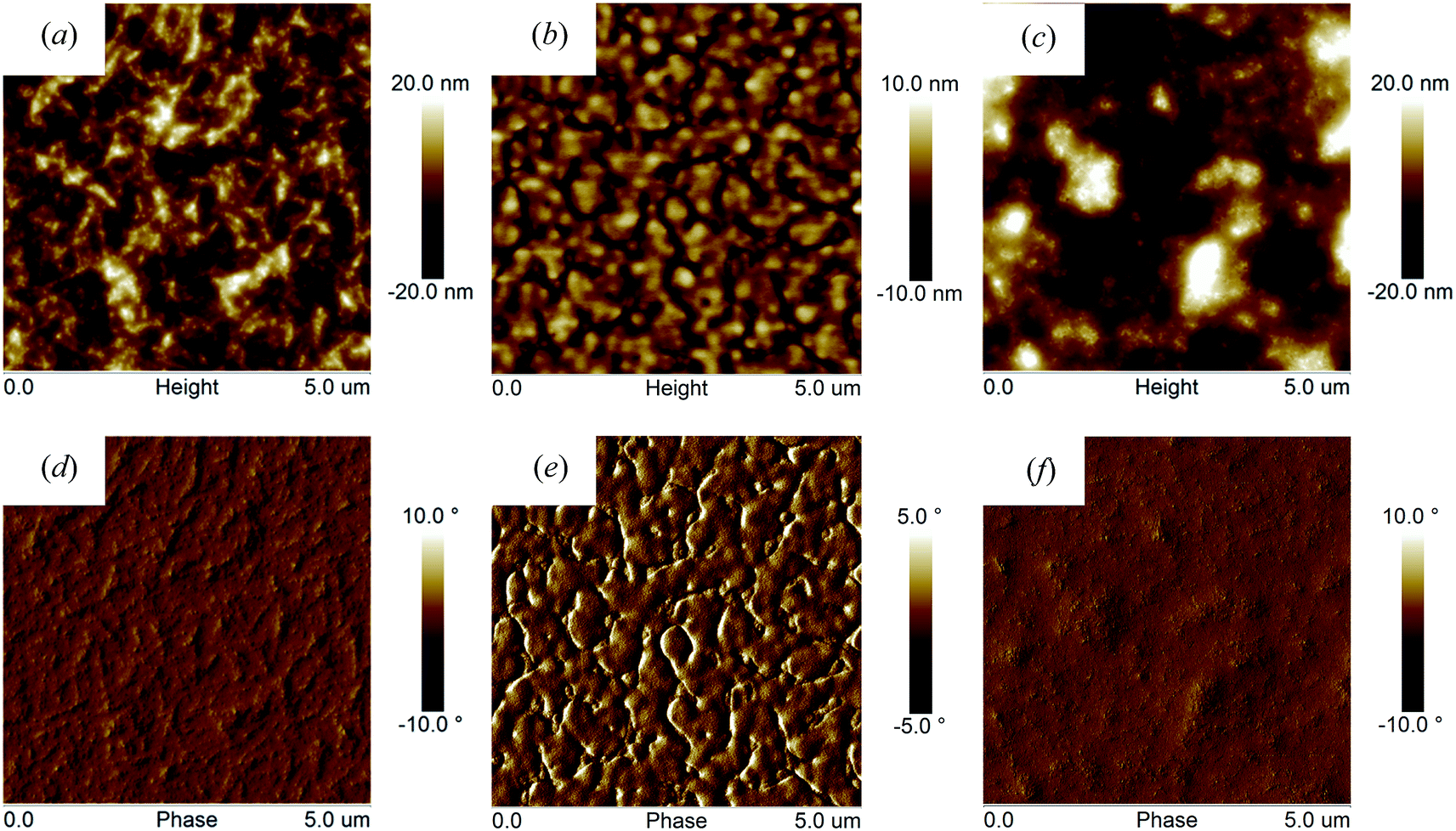 | ||
| Fig. 7 Tapping-mode atomic force microscopy topography (a, b and c) and phase images (d, e and f) of; PFO-DffBT:PC70BM (1:4 w/w, 3% DIO) (a and d); PFDo-DBT:PC70BM (1:4 w/w) (b and e); and PFDo-DffBT:PC70BM (1:4 w/w, 3% DIO) (c and f). | ||
Conclusions
The effect of fluorine substitution on the molecular, optical, electrochemical and photovoltaic properties of the well-studied poly(2,7-fluorene-alt-dithienylbenzothiadiazole) has been investigated. A fluorinated benzothiadiazole acceptor-moiety was synthesised and polymerised with 9,9-dioctlyfluorene units to obtain PFO-DffBT. The low molecular weight of PFO-DffBT resulted in a blue-shifted absorption maxima and a larger optical band gap relative to its non-fluorinated analogue PFO-DBT. The fluorinated benzothiadiazole unit was then polymerised with fluorene units that were functionalized with larger dodecyl chains (PFDo-DffBT). A non-fluorinated analogue, PFDo-DBT was also synthesised to help understand the effects of fluorination. UV-visible spectroscopy revealed that incorporation of fluorine results in a blue-shifted absorption maxima. However, the absorption band of PFDo-DffBT, relative to PFDo-DBT, is wider and displays electronic and vibrational structure, a result that we interpret in terms of improved molecular ordering in PFDo-DffBT. We find that the band gap of PFDo-DffBT is identical to that of PFDo-DBT (1.89 eV) although it displayed a deeper HOMO level relative to its non-fluorinated analogue. The LUMO levels of the polymers are however identical. BHJ polymer solar cell devices fabricated from PFO-DffBT:PC70BM, PFDo-DffBT:PC70BM and PFDo-DBT:PC70BM gave efficiencies of 4.4, 3.6 and 2.4%, respectively. The improved PCE values for the fluorinated polymers mainly result from increased Jsc values. However, the Voc of devices made from PFO-DffBT and PFDo-DffBT were lower than devices made from non-fluorinated analogues despite both fluorinated analogues displaying deeper HOMO levels relative to their non-fluorinated analogues. We speculate that this may be attributed to the segregation of fluorinated polymer to the surface of the active layer. We find that when formed into a polymer-fullerene blend, films based on the fluorinated polymers are characterized by fine length-scale features; an observation consistent with improved exciton dissociation and charge-generation.
Experimental
Materials
5,6-Difluoro-4,7-bis(5-bromo-4,2-thienyl)-2,1,3-benzothiadiazole was prepared according to previous literature procedures.22,35 4,7-Bis(5-bromo-2-thienyl)-2,1,3-benzothiadiazole was prepared according to literature procedures.10 9,9-Didodecylfluorene-2,7-diboronic acid (97%) and 9,9-dioctylfluorene-2,7-diboronic acid bis(1,3-propanediol) ester (97%) were purchased from Sigma Aldrich and used as received. PC70BM (95%) was purchased from Ossila Ltd. All chemicals and solvents, with the exception of those stated below, were of reagent grade quality, purchased commercially and used without further purification unless otherwise stated. Toluene was dried and distilled over sodium under an inert argon atmosphere. Acetonitrile (high performance liquid chromatography (HPLC) grade) was dried and distilled over phosphorous pentoxide under an inert argon atmosphere, then stored over molecular sieves, 3 Å.Measurements
1H and 13C nuclear magnetic resonance (NMR) spectra were recorded on either Bruker AV 250 (250 MHz), Bruker AV 400 (400 MHz) or Bruker Avance III HD 500 (500 MHz) spectrometers at room temperature using chloroform-d (CDCl3) or at 100 °C using 1,2-dideutrotetrachloroethane as the solvent. NMR spectra were recorded in parts per million (ppm) relative to tetramethysilane (δH 0.00). Coupling constants are given in Hertz (Hz). Carbon, nitrogen and sulphur elemental analysis was performed on a Perkin Elmer 2400 series 11 CHNS/O analyser. Analysis of halides was undertaken using the Schöniger flask combustion method. GPC analysis was conducted on polymer solutions in 1,2,4-trichlorobenzene at 140 °C. Polymer samples were spiked with toluene as a reference. GPC curves were obtained using a Viscotek GPCmax VE 2001 GPC solvent/sample module and a Waters 410 Differential Refractometer, which was calibrated using a series of narrow polystyrene standards (Polymer Laboratories). TGA's were obtained using a Perkin Elmer TGA-1 Thermogravimetric Analyser at a scan rate of 10 °C min−1 under an inert nitrogen atmosphere. Powder X-ray diffraction samples were recorded on a Bruker D8 advance diffractometer with a CuKα radiation source (1.5418 Å, rated as 1.6 kW). The scanning angle was conducted over the range 2–30°. UV-visible absorption spectra were recorded at ambient temperature using a Hitachi U-2010 Double Beam UV/visible Spectrophotometer. Polymer solutions were made using chloroform (spectrophotometric grade) and measured using quartz cuvettes (path length = 1 × 10−2 m). Thin films, used for absorption spectra, were prepared by drop casting solutions onto quartz plates using 1 mg cm−3 polymer solutions that were made using chloroform (HPLC grade). AFM measurements were made using a Veeco Dimension 3100, operating in tapping mode. Cyclic voltammograms were recorded using a Princeton Applied Research Model 263A Potentiostat/Galvanostat. A three electrode system was employed comprising a Pt disc (area = 3.14 × 10−2 cm2), platinum wire and Ag/AgCl as the working electrode, counter electrode and reference electrode, respectively, in a tetrabutylammonium perchlorate acetonitrile solution (0.1 mol dm−3). Measurements were conducted on polymer films that were prepared by drop casting 1.0 mm3 of polymer solution (1 mg cm−2 in chloroform (HPLC grade)). In accordance with IUPAC's recommendations, ferrocene was employed as a reference redox system.36Fabrication and testing of BHJ polymer solar cells
The polymers and PC70BM were dissolved separately in either CF, CB or CB with 3% DIO, and were then put on a hotplate held at 70 °C overnight with stirring to allow dissolution. The solutions were then mixed at different polymer:fullerene blending ratios before spin casting. Photovoltaic devices were fabricated onto pre-patterned ITO glass substrates (20 Ohms per square) that were supplied by Ossila Limited. The ITO/glass substrates were first cleaned by sonication in dilute NaOH followed by IPA. A 30 nm thick PEDOT:PSS layer was spin-coated onto the ITO substrates. These were then transferred to a hot-plate held at a temperature of 120 °C for 5 min before being transferred to a nitrogen glove-box. All active layers were spin cast onto the glass/ITO/PEDOT:PSS substrate, the devices were then transferred into a thermal evaporator for deposition of OPV cathode (5 nm of calcium capped by a 100 nm of aluminium evaporated at a base pressure of ∼10−7 mbar). The cathode was deposited through a shadow-mask, producing a series of independent pixels. Devices were finally encapsulated using a glass slide and epoxy glue before testing. PCEs were determined using a Newport 92251A-1000 AM 1.5 solar simulator. An NREL calibrated silicon cell was used to calibrate the power output to 100 mW cm−2 at 25 °C. An aperture mask having an area of 2.06 mm2 was placed over the devices to define the test area. At least two devices were prepared for each sample to give 12 pixels, each reporting an independent J–V curve. EQE values were determined over the wavelength range of interest by comparing the photocurrent of the OPV cell to a reference silicon photodiode having a known spectral response.
Poly[2,7-(9,9-dioctylfluorene)-alt-4,7-bis(thiophen-2-yl)-5,6-difluorobenzo-2,1,3-thiadiazole] (PFO-DffBT)
5,6-Difluoro-4,7-bis(5-bromo-4,2-thienyl)-2,1,3-benzothiadiazole (0.478 g, 0.9676 mmol) and 9,9-dioctylfluorene-2,7-diboronic acid bis(1,3-propanediol) ester (0.540 g, 0.9676 mmol) were added to a 100 cm3 single neck round bottom flask and placed under argon using standard Schlenk link techniques. Dry toluene (21 cm3), followed by tetraethyl ammonium hydroxide (20% wt, 7.0 cm3, degassed) were added and the system was degassed again. To this solution Pd(OAc)2 (8.0 mg) and tri(o-tolyl)phosphine (21.7 mg) were added, degassed and heated to 90 °C for 1 hour. The mixture was cooled to room temperature and bromobenzene (0.1 cm3, 0.94 mmol) was added. The mixture was degassed and heated at 90 °C for 2 hours. The mixture was cooled to room temperature and phenylboronic acid (150 mg, 1.23 mmol) was added, degassed and heated to 90 °C for 3 hours. The mixture was poured into degassed methanol (500 cm3) and stirred overnight. The mixture was then filtered through a membrane filter. The solids were cleaned using Soxhlet extraction with solvents in the order; methanol (250 cm3), acetone (250 cm3), hexane (250 cm3), toluene (250 cm3), chloroform (250 cm3) and chlorobenzene (250 cm3). The toluene, chloroform and chlorobenzene fractions were concentrated (∼50 cm3) and then poured into degassed methanol (500 cm3). The resulting mixtures were stirred overnight and the polymers collected by filtration as purple powders. Toluene (143 mg, 20%), chloroform (3 mg, 0.4%) and chlorobenzene (110 mg, 16%). GPC: toluene fraction, Mn = 6800 g mol−1, Mw = 9600 g mol−1, PDI = 1.41; chloroform fraction Mn = 8800 g mol−1, Mw = 11900 g mol−1, PDI = 1.35; chlorobenzene fraction, Mn = 11900 g mol−1, Mw = 15400 g mol−1, PDI = 1.29. 1H NMR (toluene fraction) (500 MHz, C2D2Cl4, 100 °C) (δH/ppm) 8.31 (m, 2H), 7.71 (m, 6H), 7.52 (m, 2H), 2.08 (br, 4H), 1.5–0.95 (m, 24H), 0.71 (m, 6H). 19F NMR (toluene fraction) (500 MHz, C2D2Cl4, 100 °C) (δF/ppm) −127.97. Anal. calcd for C43H44F2N2S3: C, 71.43; H, 6.13; N, 3.87; S, 13.30. Found: C, 72.25; H, 6.26; N 3.54; S, 11.8.
Poly[2,7-(9,9-didodecylfluorene)-alt-4,7-bis(thiophen-2-yl)-benzo-2,1,3-thiadiazole (PFDo-DBT)
4,7-Bis(5-bromo-2-thienyl)-2,1,3-benzothiadiazole (0.443 g, 0.9676 mmol) and 9,9-didodecylfluorene-2,7-diboronic acid (0.571 g, 0.9676 mmol) were added to a 100 cm3 single neck round bottom flask and placed under argon using standard Schlenk link techniques. Dry toluene (21 cm3), followed by tetraethyl ammonium hydroxide (20% wt, 7.0 cm3, degassed) were added and the system was degassed again. To this solution Pd(OAc)2 (8.0 mg) and tri(o-tolyl)phosphine (21.7 mg) were added, degassed and heated to 90 °C for 24 hours. The mixture was cooled to room temperature and bromobenzene (0.1 cm3, 0.94 mmol) was added. The mixture was degassed and heated at 90 °C for 2 hours. The mixture was cooled to room temperature and phenylboronic acid (150 mg, 1.23 mmol) was added, degassed and heated to 90 °C for 3 hours. After cooling to room temperature, the mixture was partially dissolved in CHCl3 (300 cm3) and to this solution ammonia (200 cm3, 35%) was added, heated at reflux for 3 hours and then cooled to room temperature. The organic layer was separated. To the organic phase was added ethylenediaminetetraacetic acid disodium salt dihydrate (300 mg) and stirred overnight. The mixture was washed with distilled water (2 × 200 cm3). The organic phase was separated, concentrated to about 50 cm3, poured into degassed methanol:water (10:1, 500 cm3) and stirred overnight. The mixture was then filtered through a membrane filter and collected. The solids were cleaned using Soxhlet extraction with solvents in the order; methanol (250 cm3), acetone (250 cm3), hexane (250 cm3), toluene (250 cm3) and chlorobenzene (250 cm3). The toluene and chlorobenzene fractions were concentrated (∼50 cm3) and then poured into degassed methanol (500 cm3). The resulting mixtures were stirred overnight and the polymers collected by filtration as purple powders. Toluene (184 mg, 24%), and chlorobenzene (26 mg, 3%). GPC: toluene fraction, Mn = 20500 g mol−1, Mw = 35700 g mol−1, PDI = 1.74; chlorobenzene fraction, Mn = 20200 g mol−1, Mw = 40500 g mol−1, PDI = 2.00. 1H NMR (toluene fraction) (500 MHz, C2D2Cl4, 100 °C) (δH/ppm) 8.14 (m, 2H), 7.91 (m, 2H), 7.69 (m, 6H), 7.47 (m, 2H) 2.08 (br, 4H), 1.41 (s, 8H) 1.16 (br, 32H), 0.82 (m, 6H). Anal. calcd for C51H62N2S3: C, 76.64; H, 7.82; N, 3.51; S, 12.03. Found: C, 75.36; H, 7.77; N, 3.16; S, 11.00.
Poly[2,7-(9,9-didodecylfluorene)-alt-4,7-bis(thiophen-2-yl)-5,6-difluorobenzo-2,1,3-thiadiazole] (PFDo-DffBT)
5,6-Difluoro-4,7-bis(5-bromo-4,2-thienyl)-2,1,3-benzothiadiazole (0.478 g, 0.9676 mmol) and 9,9-didodecylfluorene-2,7-diboronic acid (0.571 g, 0.9676 mmol) were added to a 100 cm3 single neck round bottom flask and placed under argon using standard Schlenk link techniques. Dry toluene (21 cm3), followed by tetraethyl ammonium hydroxide (20% wt, 7.0 cm3, degassed) were added and the system was degassed again. To this solution Pd(OAc)2 (8.0 mg) and tri(o-tolyl)phosphine (21.7 mg) were added, degassed and heated to 90 °C for 48 hours. The mixture was cooled to room temperature and bromobenzene (0.1 cm3, 0.94 mmol) was added. The mixture was degassed and heated at 90 °C for 2 hours. The mixture was cooled to room temperature and phenylboronic acid (150 mg, 1.23 mmol) was added, degassed and heated to 90 °C for 3 hours. After cooling to room temperature, the mixture was dissolved in CHCl3 (300 cm3) and to this solution ammonia (200 cm3, 35%) was added, heated at reflux for 3 hours and then cooled to room temperature. The organic layer was separated. To the organic phase was added ethylenediaminetetraacetic acid disodium salt dihydrate (300 mg) and stirred overnight. The mixture was washed with distilled water (2 × 200 cm3). The organic phase was separated, concentrated to about 50 cm3, poured into degassed methanol (500 cm3) and stirred overnight. The mixture was then filtered through a membrane filter. The solids were cleaned using Soxhlet extraction with solvents in the order; methanol (250 cm3), acetone (250 cm3), hexane (250 cm3), toluene (250 cm3) and chlorobenzene (250 cm3). The toluene and chlorobenzene fractions were concentrated (∼50 cm3) and then poured into degassed methanol (500 cm3). The resulting mixtures were stirred overnight and the polymers collected by filtration as purple powders. Toluene (213 mg, 26%), and chlorobenzene (173 mg, 21%). GPC: toluene fraction, Mn = 9700 g mol−1, Mw = 15900 g mol−1, PDI = 1.64; chlorobenzene fraction, Mn = 16100 g mol−1, Mw = 23700 g mol−1, PDI = 1.47. 1H NMR (toluene fraction) (500 MHz, C2D2Cl4, 100 °C) (δH/ppm) 8.37 (m, 2H), 7.71 (m, 6H), 7.52 (m, 2H), 2.09 (br, 4H), 1.40 (s, 4H), 1.15 (m, 36H), 0.82 (m, 6H). 19F NMR (toluene fraction) (500 MHz, C2D2Cl4, 100 °C) (δF/ppm) −127.32, −127.94. Anal. calcd for C51H60F2N2S3: C, 73.34; H, 7.24; N, 3.35; S, 11.52. Found: C, 74.05; H, 7.30; N, 2.92; S, 10.81.
Acknowledgements
We would like to acknowledge EPSRC for financial support of this work via research grants EP/J017361/1 and EP/I028641/1. YZ thanks the University of Sheffield for the award of a scholarship.References
- P. L. T. Boudreault, A. Najari and M. Leclerc, Chem. Mater., 2011, 23, 456–469 CrossRef CAS.
- E. Wang, L. Wang, L. Lan, C. Luo, W. Zhuang, J. Peng and Y. Cao, Appl. Phys. Lett., 2008, 92, 5–7 CrossRef.
- G. Dennler, M. C. Scharber and C. J. Brabec, Adv. Mater., 2009, 21, 1323–1338 CrossRef CAS.
- J. Y. Kim, K. Lee, N. E. Coates, D. Moses, T. Nguyen, M. Dante and A. J. Heeger, Science, 2007, 222–226 CrossRef CAS PubMed.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed.
- Y. Liang and L. Yu, Acc. Chem. Res., 2010, 43, 1227–1236 CrossRef CAS PubMed.
- S. H. Park, A. Roy, S. Beaupre, S. Cho, N. Coates, J. S. Moon, D. Moses, M. Leclerc, K. Lee and A. J. Heeger, Nat. Photonics, 2009, 3, 297–302 CrossRef CAS.
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lussem and K. Leo, Nature., 2009, 459, 234–238 CrossRef CAS PubMed.
- Z. Tan, S. Li, F. Wang, D. Qian, J. Lin, J. Hou and Y. Li, Sci. Rep., 2014, 4, 4691 CrossRef PubMed.
- H. Yi, S. Al-Faifi, A. Iraqi, D. C. Watters, J. Kingsley and D. G. Lidzey, J. Mater. Chem., 2011, 21, 13649–13656 RSC.
- J. You, L. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C.-C. Chen, J. Gao, G. Li and Y. Yang, Nat Commun., 2013, 4, 1446 CrossRef PubMed.
- T. Umeyama and H. Imahori, J. Mater. Chem. A, 2014, 2, 11545–11560 RSC.
- H.-J. Jhuo, P.-N. Yeh, S.-H. Liao, Y.-L. Li, Y.-S. Cheng and S.-A. Chen, J. Chin. Chem. Soc., 2014, 61, 115–126 CrossRef CAS.
- C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2011, 133, 10062–10065 CrossRef CAS PubMed.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- Q. T. Zhang and J. M. Tour, J. Am. Chem. Soc., 1998, 120, 5355–5362 CrossRef CAS.
- H. Zhou, L. Yang, S. Stoneking and W. You, ACS Appl. Mater. Interfaces, 2010, 2, 1377–1383 Search PubMed.
- T. Umeyama, Y. Watanabe, E. Douvogianni and H. Imahori, J. Phys. Chem. C, 2013, 117, 21148–21157 CrossRef CAS.
- Y. Zhang, S.-C. Chien, K.-S. Chen, H.-L. Yip, Y. Sun, J. a. Davies, F.-C. Chen and A. K.-Y. Jen, Chem. Commun., 2011, 47, 11026–11028 RSC.
- Z. Li, J. Lu, S.-C. Tse, J. Zhou, X. Du, Y. Tao and J. Ding, J. Mater. Chem., 2011, 21, 3226–3233 RSC.
- S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, J. Kniepert, S. Inal, P. Pingel, K. Fostiropoulos, N. Koch and D. Neher, J. Am. Chem. Soc., 2012, 134, 14932–14944 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- J. Min, Z.-G. Zhang, S. Zhang and Y. Li, Chem. Mater., 2012, 24, 3247–3254 CrossRef CAS.
- Z. Fei, M. Shahid, N. Yaacobi-Gross, S. Rossbauer, H. Zhong, S. E. Watkins, T. D. Anthopoulos and M. Heeney, Chem. Commun., 2012, 48, 11130–11132 RSC.
- J. Kim, M. H. Yun, G. H. Kim, J. Lee, S. M. Lee, S. J. Ko, Y. Kim, G. K. Dutta, M. Moon, S. Y. Park, D. S. Kim, J. Y. Kim and C. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 7523–7534 Search PubMed.
- O. Inganäs, M. Svensson, F. Zhang, a. Gadisa, N. K. Persson, X. Wang and M. R. Andersson, Appl. Phys. A: Mater. Sci. Process., 2004, 79, 31–35 CrossRef.
- A. A. B. Alghamdi, D. C. Watters, H. Yi, S. Al-Faifi, M. S. Almeataq, D. Coles, J. Kingsley, D. G. Lidzey and A. Iraqi, J. Mater. Chem. A., 2013, 1, 5165–5171 RSC.
- H. J. Son, W. Wang, T. Xu, Y. Liang, Y. Wu, G. Li and L. Yu, J. Am. Chem. Soc., 2011, 133, 1885–1894 CrossRef CAS PubMed.
- A. Iyer, J. Bjorgaard, T. Anderson and M. E. Ko, Macromol. Rapid Commun., 2012, 45, 6380–6389 CrossRef CAS.
- S. H. Chen, A. C. Su and S. A. Chen, J. Phys. Chem. B, 2005, 10067–10072 CrossRef CAS PubMed.
- M. Grell, D. D. C. Bradley, G. Ungar, J. Hill and K. S. Whitehead, Macromolecules, 1999, 32, 5810–5817 CrossRef CAS.
- A. Zusan, B. Gieseking, M. Zerson, V. Dyakonov, R. Magerle and C. Deibel, Sci. Rep., 2015, 5, 8286 CrossRef CAS PubMed.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- B. Qi and J. Wang, J. Mater. Chem., 2012, 24315–24325 RSC.
- N. Cho, K. Song, J. K. Lee and J. Ko, Chem.–Eur. J., 2012, 18, 11433–11439 CrossRef CAS PubMed.
- G. Gritzner, Pure Appl. Chem., 1990, 62, 1839–1858 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR spectra of the polymers. See DOI: 10.1039/c5ra06076a |
| This journal is © The Royal Society of Chemistry 2015 |