
The behaviour of β-triketimine cobalt complexes in the polymerization of isoprene†
Mohammed N. Alnajrani‡
* and
Francis S. Mair*
Organic Materials Innovation Centre, School of Chemistry, The University of Manchester, Brunswick Street, Manchester M13 9PL, UK. E-mail: mair@manchester.ac.uk
First published on 18th May 2015
Abstract
Four new β-triketimine cobalt complexes [(LCo-μ-Br)2][BArF]2 where L = N,N′,N′′-triaryl-β-diketimine were synthesised and then characterized by single-crystal X-ray diffraction, MALDI-MS and elemental analysis. In combination with three previously reported, they provided a series of seven subtly different complexes which were screened for activity in the catalysis of isoprene polymerization: the structure of the ligand (L) had strong effects on activity and stereoselectivity. The produced polyisoprene contained a mixture of cis-1,4, trans-1,4 and 3,4-enchained monomers; 4,1-errors followed 3,4 errors. The highest percentage of cis-1,4 content (80%) was obtained with complex 5 where two electron-withdrawing fluorine substituents occupied the 2- and 6-positions of two of the imino-aryl rings. This polymer was obtained in 98.5% conversion. Both cis-1,4 content and activity decreased when electron donor substituents were present on the aryl rings, so that complex 1, with three methyl substituents in positions 2, 4 and 6 gave the lowest cis-1,4 content (73.6%), with a conversion of only 58.5%, under identical conditions. The temperature of the polymerization, the type of aluminium co-catalyst used, and the Al/Co ratio strongly affected the activity and microstructure of the polyisoprene produced. Ethylaluminiumsesquichloride was the most active of a range of organoaluminiums screened. A kinetic study using complex 6 as catalyst demonstrated that the polymerization was first-order in monomer, and that approximately 12% of cobalt formed active centers. The combination of high molecular weight (>105) with moderately high activity at conveniently accessible temperatures to give predominantly cis-1,4-polyisoprene but with 3,4 units to promote efficient crosslinking is potentially attractive, and has not previously been attained with 3d elements.
Introduction
The stereospecific polymerization of 1,3-diene monomers has offered an interesting challenge to many academic and industrial research groups because of the numerous applications for polydienes in the rubber industry, where the various forms of monomer enchainment possible lead to starkly different materials properties which select for a range of end-uses.1 The notion that addressing these issues is part of the ‘green chemistry’ agenda2 may be superficially challenged by the current supply of high-cis polyisoprene from renewable tree (Hevea brasiliensis), rather than petrochemical, sources: natural rubber. However, there is still a current market for synthetic polyisoprene, since it offers product consistency, lack of odour, lack of allergenic proteins, reproducible molecular weight and molecular weight distribution, and easy initial processing/compounding, all of which are problems with natural rubber.3,4 Furthermore, supply of natural rubber is exposed to the risk of crop failure if the South American Leaf Blight fungus (Microcyclus ulei) migrates to current rubber-producing areas in the Asia-Pacific region from its home in South America, where it has already almost destroyed commercial production.5 Moreover, production of natural rubber consumes large quantities of anti-fungals and insecticides, such as arsenic trioxide; replacement of primary or secondary forest with Hevea brasiliensis monoculture can have strongly negative local environmental effects, and can reduce the total carbon biomass held in the land.6 Finally, the environmental footprint of synthetically engineered rubbers can be significantly offset by the lower fuel consumption of vehicles fitted with low-rolling-resistance tyres produced from synthetic rubbers.7,8 Consequently, there is a strong justification for further research to improve the catalysis of synthetic diene polymerization. Historically and currently, Ziegler–Natta catalysts have often been used in the polymerization of 1,3-dienes;9–11 however, molecular catalysts have also met with wide interest. This field of diene polymerization, both of butadiene and isoprene, has received much academic study, which has been recently reviewed.12,13 The key modes of enchainment of polyisoprenes are shown in Chart 1. A series of different catalytic systems have been found to be efficient in controlling such microstructure; ligands co-ordinated to the active metal centre were found to affect the form (syn or anti) of the putative allyl intermediate of the diene polymerization, and hence the conformation of the remaining double bond in the produced polymer, and also the activity of the propagating centre during the polymerization.12,13 | ||
| Chart 1 Various polymerization modes available. | ||
These effects can be modified by changing the steric and electronic properties of the ligands. While industrial catalysis has tended to move towards lanthanide (Yb) catalysts for high-cis polydienes, cognisance of the medium-term competing uses of these elements for their magnetic and photochemical properties combined with their limited mineral reserves has re-focused academic attention on the first-row d-block elements due to their greater long-term availability;14 while earlier examples using these elements were far inferior to lanthanides in the degrees of activity and/or stereocontrol offered,9,15–20 ligand design offers an opportunity to tailor the properties of the base elements (Co, Ni, Fe), so that, even if they cannot surpass the remarkably high degrees of cis-1,4-selectivity of isoprene enchainment offered by some lanthanide catalysts of over 96%,21–27 they may still have their activity tailored to produce alternative potentially useful polymers unknown in Nature. It is now possible to obtain high trans-1,4 polymer,16,28–30 high 3,4-isotactic polymer31–33 and high 3,4 atactic or syndiotactic polymer,34 mostly with lanthanides, but it is also possible to obtain unique cis-1,4-alt-3,4 polyisoprene with cobalt phosphine complexes.35 Here, we report our results on cobalt complexes of N,N,N facially capping ligands, recently employed to give high activity and selectivity in polymerization of butadiene,36 and their performance in the polymerization of isoprene.
Results and discussion
Synthesis and characterization of β-triketimine cobalt complexes
A series of modified β-triketimine ligands with substituents varied from bulky electron-donating (alkyl) to less-bulky electron-withdrawing (fluoro) substituents were synthesised and then fully characterized by NMR, IR, elemental analyses and electrospray mass spectrometry. These ligands exhibited extensive tautomerism in solution, as reported for earlier examples,37 but as previously found when metal-bound,36,37 the seven examples all adopted fac-triketimine ligated forms: β-triketimine cobalt complexes were prepared from the reaction of these ligands with cobalt bromide in the presence of NaBArF, Scheme 1.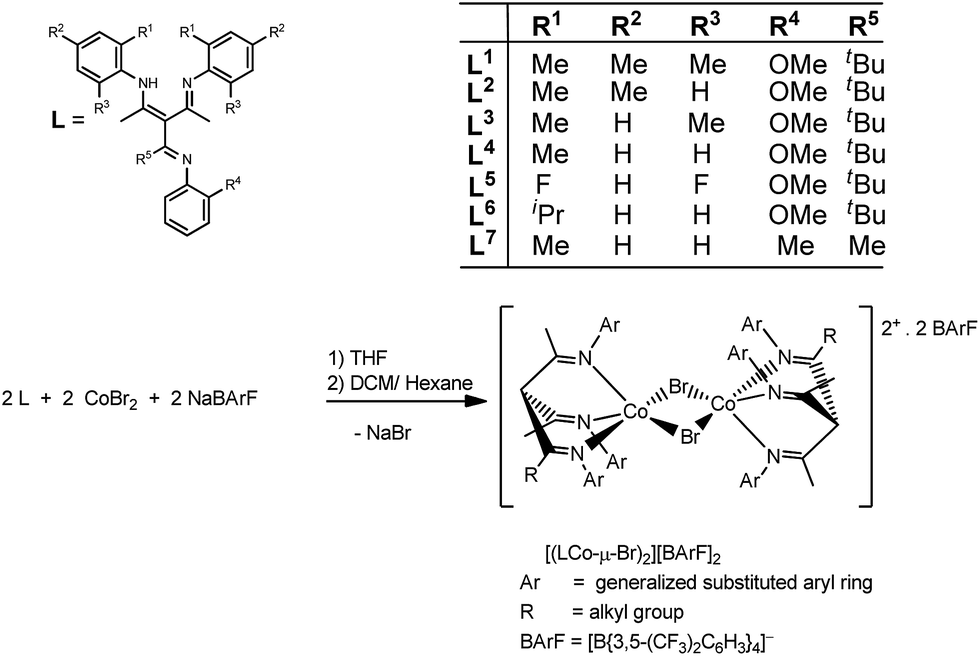 | ||
| Scheme 1 Definition of ligands and synthesis route of complexes 1–7. | ||
The complexes were then characterized by elemental analyses, MALDI mass spectrometry and single crystal X-ray diffraction. These complexes were all found to be five-coordinate centrosymmetric dimeric bromide-bridged species [(LCo-μ-Br)2][BArF]2. The structures of complexes 1, 2 and 4 (nomenclature: [(L1Co-μ-Br)2][BArF]2 = 1; [(L2Co-μ-Br)2][BArF]2 = 2; etc.) have been previously reported,36 where the co-ordination geometry at the cobalt centre was described as distorted square-pyramidal: one nitrogen atom (N2) occupied the axial site, whilst the other two nitrogen atoms (N1 and N3) and two bromine atoms (Br and Br′) occupied basal sites. Complexes 1–7 are broadly isostructural. The angles between the axial site (N2) and the other nitrogen atoms are less than 90° while they are higher than 90° between N2 and bromine atoms, indicating the axial site was bent toward N1 and N3. Fig. 1 shows the structure of the dication of 5, whereas Fig. 2 shows that of 6. Other complexes are broadly similar in structure (figures and tabulated data are in ESI†). There were no close borate anion–cation interactions. The cobalt atoms lay in the direction of N1 in the axial site, since the angles between N1–Co–Br and N3–CoBr′ were less than 180° i.e. it lay proud of the square plane, as is common in such geometry. Key angles and bond lengths are listed in Table 1.
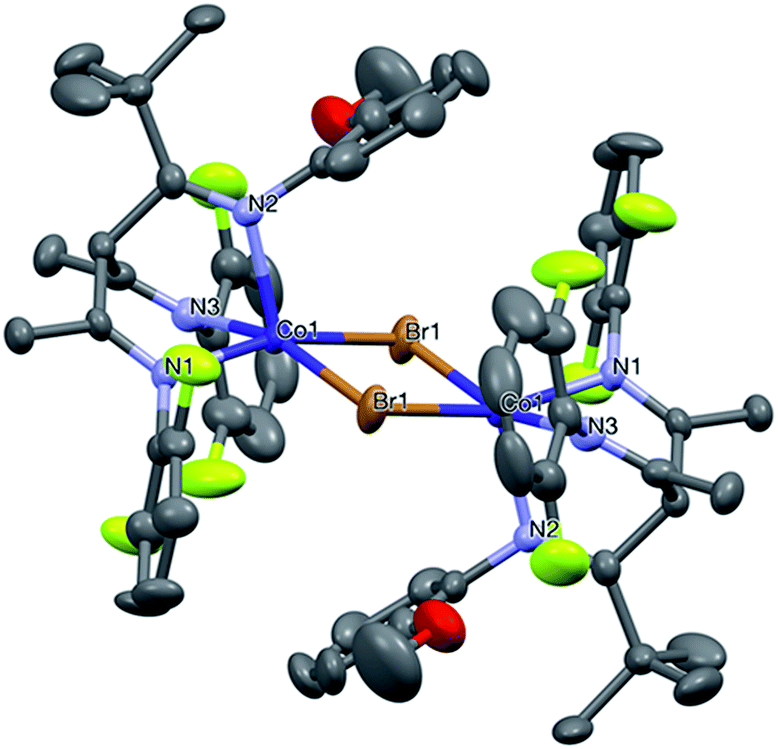 | ||
| Fig. 1 Structure of the dicationic unit in 5, with all hydrogen atoms and BArF ions omitted. | ||
 | ||
| Fig. 2 Structure of the dicationic unit in 6, with all hydrogen atoms and BArF ions omitted. | ||
| 3 | 5 | 6 | 7 | |
|---|---|---|---|---|
| Co–N1 | 2.160(3) | 2.118(6) | 2.114(3) | 2.078(6) |
| Co–N2 | 2.079(4) | 2.082(5) | 2.072(3) | 2.067(4) |
| Co–N3 | 2.134(3) | 2.114(5) | 2.138(3) | 2.152(5) |
| Co–Br | 2.4990(7) | 2.4997(11) | 2.4899(6) | 2.421(4) |
| Co–Br′ | 2.4778(7) | 2.5070(11) | 2.55099(7) | 2.488(4) |
| N2–Co–N1 | 87.71(13) | 85.3(2) | 87.14(11) | 88.54(19) |
| N2–Co–N3 | 88.82(14) | 86.6(2) | 86.05(14) | 85.56(18) |
| N2–Co–Br | 97.88(10) | 110.90(16) | 118.92(8) | 124.44(19) |
| N2–Co–Br′ | 122.45(10) | 110.27(15) | 104.51(8) | 100.92(18) |
| N1–Co–Br | 174.13(10) | 163.16(16) | 153.29(9) | 146.29(19) |
| N3–Co–Br′ | 148.12(10) | 162.67(16) | 169.41(12) | 173.26(17) |
Interestingly, in the cases of 5 and 6, the methoxyaryl group adopted the axial position (Fig. 1 and 2), whereas in 1–4,36 it adopted one of the basal positions. There is no discernible pattern either in the Co–N lengths, indicating that they are subject to several subtle steric and packing effects, except to note that the mean Co–N length is significantly greater for 3 than for all other examples, underlining the effect of the two ortho methyl substituents on the steric demand of the imines.
The degree of distortion of the square pyramids is quite varied, being smallest for the 2,6-difluoro-substituted 5 (τ = 0.008, near-perfect square planar),38 but largest for 3 (τ = 0.432, almost half way between square planar and trigonal bipyramidal), amongst the bulkiest of 1–7. However, catalyst 1 possessed a closely similar degree of ortho bulk to that of catalyst 3, the only difference between them being a p-methyl substituent on two aryl rings, and yet with τ = 0.03, was among the least-distorted. Thus, ortho bulk alone does not explain the data. It may be a function of crystal packing forces. Such details become academic, since these pre-catalysts react with alkylaluminium species, thus exchanging their bridging bromides for alkyl groups, most probably reverting to monomers in solution on the route to becoming catalytically active sites.
The steric demand of the substituents is measurable by considering the average N–N separation in the ligand, as a measure of its bite-size. For 5, this is 2.858(3) Å, whereas for 3 (Me2 in place of difluoro, and replacement of tBu imine substituent by Me) it is 2.924(3) Å. Mono-substituted aryls in 6 and 7 have intermediate values {2.877(7), 2.902(6)}Å. There are few other clear trends in the data for 1–7, though the modest structural differences do result in variations in both activity and selectivity of polymerization (vide infra).
Polymerization of isoprene
β-Triketimine cobalt complexes 1–7 were tested in the polymerization of isoprene. The polymerizations were carried out in chlorobenzene using diethylaluminium chloride (DEAC) as co-catalyst at 35 °C. The results are summarized in Table 2.| Complex | Conversion (%) | Mwb (g mol−1) × 105 | Mnb (g mol−1) × 105 | Đb | Microstructurec % | |||
|---|---|---|---|---|---|---|---|---|
| Cis-1,4 | Trans-1,4 | 1,2 | 3,4 | |||||
| a Conditions: catalyst: 5 μmol; co-catalyst: DEAC; Al/Co: 150; isoprene: 5 mL; solvent: 30 mL chlorobenzene; T: 35 °C; time: 24 h.b Determined by GPC.c Determined by 1H and 13C NMR. | ||||||||
| 1 | 58.5 | 7.2 | 2.8 | 2.57 | 73.6 | 1.0 | 0 | 26.4 |
| 2 | 63.0 | 7.4 | 2.8 | 2.64 | 74.4 | 0.6 | 0 | 25.0 |
| 3 | 66.9 | 8.3 | 2.5 | 3.32 | 74.8 | 0.5 | 0 | 24.7 |
| 4 | 75.3 | 8.8 | 2.6 | 3.38 | 76.8 | 0 | 0 | 23.2 |
| 5 | 98.5 | 6.7 | 2.0 | 3.35 | 80.0 | 0 | 0 | 20.0 |
| 6 | 100 | 9.1 | 2.8 | 3.25 | 76.0 | 0 | 0 | 24.0 |
| 7 | 88.2 | 9.0 | 2.8 | 3.21 | 74.5 | 0.5 | 0 | 25.0 |
Ligand effects
Firstly, it is to be noted that in contrast to the case for butadiene,1,36 strong differences between differently ligated cobalt complexes are found. Cobalt carboxylates are poorly selective for cis-1,4 polymer, giving ratios below 50%, with significant trans-1,4 as well as 3,4 enchainments.19 In contrast, phosphine-ligated complexes give exactly 50% ratio in the form of a perfectly alternating 3,4/cis-1,4 polymer.35 Nitrogen-ligated examples are rare, the only examples in the open literature being N,N,N meridional chelates from He, Sun and co-workers, the most active example of which is shown as 8, below.39,40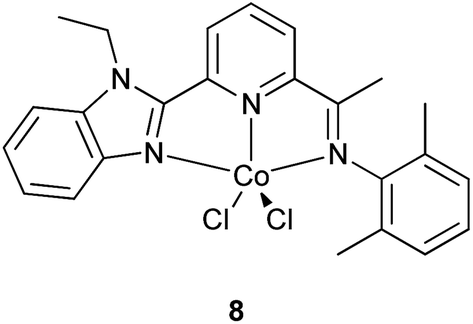
Our results with 1–7 mirror those obtained with 8: higher degrees of cis-1,4 enchainment than previously observed with cobalt are attained for both classes of tridentate nitrogen ligands, fac or mer. While our estimates of selectivity, based on a comparison of CH3 integrals in the 13C(1H) NMR spectra (see ESI†)35,41 range from 74 to 80%, whereas 8 and analogous examples gave values claimed up to 90%, there are some irregularities in the spectral assignments leading to these high values.§ This makes a comparison of 1–7 with 8 and its analogues on the basis of selectivity somewhat challenging, but it is clear that weight-average relative molar masses Mw obtained with 8 were typically in the range 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000,39 whereas 1–7 gave Mw of approximately 700000–900000 as determined by GPC. This difference is not unexpected, since catalysts 8 are more active under their most favoured conditions, and polymerization times are shorter.39,40 However, activity comparisons are marred also by solvent and aluminium effects; the high activities were attained with ethylaluminiumsequichloride, EASC (vide infra for study of aluminium effects). It seems likely that both activities and chain transfer rates were higher in these circumstances than for 1–7.
000–50000,39 whereas 1–7 gave Mw of approximately 700000–900000 as determined by GPC. This difference is not unexpected, since catalysts 8 are more active under their most favoured conditions, and polymerization times are shorter.39,40 However, activity comparisons are marred also by solvent and aluminium effects; the high activities were attained with ethylaluminiumsequichloride, EASC (vide infra for study of aluminium effects). It seems likely that both activities and chain transfer rates were higher in these circumstances than for 1–7.
We can, however make a direct comparison within the set 1–7, which shows that all catalysts are fairly selective for cis-1,4 enchainment of isoprene, with 1,2-linkages undetectable, and trans-1,4-linkages at very low levels, while maintaining significant levels of 3,4 enchained monomers which can act as points for crosslinking at later processing, as for 8.39,40 An interesting outcome of the detailed analysis of 13C NMR spectra of the polymers was the observance of two environments for 3,4 units of almost equal abundance, which we assign as cis-1,4-3,4-cis-1,4 triads, and cis-1,4-3,4-cis-4,1 triads, i.e. that the chain end resulting from a 3,4 mis-insertion is poorly selective for head-to-tail vs. head-to-head enchainment of the next monomer. Furthermore, there was no evidence for clustering of the 3,4 units, which seem statistically spread throughout the polymer.
It is notable that complexes bearing di- or trimethyl-phenyl groups (i.e. 1–3) were less active than those with single alkyls or fluoro-substituents (4–7). It is also notable that the least-active complexes produced the largest traces of trans-1,4-linkages. A comparison of 4 and 7 shows that the presence of the methoxyaryl, present in all but 7, does not in fact offer advantage in polyisoprene production, with 7 outperforming 4 in terms of activity and Mw, but with marginally lower selectivity for cis-1,4 enchainment, though all values of this parameter were in a rather narrow range around 75%, excepting the fluorinated 5, which attained 80% cis-selectivity with high activity and high molecular weight. The most active was 6, followed by 5 then 7. In general the presence of two ortho-substituents is disfavoured, unless they be fluorine atoms. The fact that 6 outperforms 4 shows that it is not a simple effect either of bulk or electron donicity, but rather that asymmetry in the disposition of bulk seems to offer advantage. The fact that all of 1–7 are cations, partnered by weakly co-ordinating BArF anions,42 is almost certainly a helpful feature, obviating the need for separation of an aluminium-based anion from the active centre. Advantage of electron-withdrawing substituents has been seen before in polymerization of butadiene by cobalt catalysts43 and can be rationalised here as being due to a more electrophilic metal demanding more electron-density from the co-ordinated isoprene thus facilitating its attack by polymeryl chain-end.
Kinetic analysis
The behaviour of the polymerization over time was studied for the most active catalyst, 6. This kinetic study used the 6/DEAC catalyst system (Al/Co = 150) at 35 °C, and monitored conversion of monomer to polymer as a function of time. The results are summarized in Table 3. Plotting In([IP]0/[IP]t) against polymerization time, where [IP]0 is the initial isoprene monomer concentration and [IP]t is isoprene monomer concentration at time t, gave a linear relationship (Fig. 3). Therefore, the polymerization of isoprene catalysed by β-triketimine cobalt complexes is first order in isoprene, i.e. Rp = kp[C*][IP] where Rp is the propagation rate, kp is the rate constant of propagation, [C*] is the concentration of active centres. This is in accord with prior studies on other metals.2,16,17,20,44 However, there was no impact on the stereoregularity of the polymer; it remained almost constant for the entire polymerization time. The molecular weight increased with polymerization time, reaching a value of 3.3 × 105 g mol−1 after one hour, and 9.1 × 105 g mol−1 after 24 hours at full conversion. It was noticed that the value of dispersity Đ increased with polymerization time even though the molecular weight was increasing almost linearly with time. At high conversions, there was a significant increase in the viscosity of the solution of the polymerization; as monomer dwindled and polymer became abundant, so grew the possibility of crosslinking reactions through reaction of main chain alkenes of 1,4-enchained units, or, more likely, side-chain alkenes of 3,4-enchained units, thus accelerating the climb in viscosity.45| Catalyst | Time (h) | Conversion (%) | [Isoprene] (M) | Mwb (g mol−1) × 105 | Mnb (g mol−1) × 105 | Polymer yield (g) | Đb | Number of chains × 1018 | Number of chains per Co |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: catalyst: 5 μmol; co-catalyst: DEAC; Al/Co: 150; isoprene: 5 mL; solvent: 30 mL chlorobenzene; temperature: 35 °C.b Determined by GPC. | |||||||||
| 6 | 0 | 0 | 1.667 | — | — | — | — | — | — |
| 6 | 1 | 13.0 | 1.450 | 3.3 | 1.6 | 0.443 | 2.06 | 1.66 | 0.55 |
| 6 | 2 | 50.3 | 0.833 | 4.4 | 1.9 | 1.703 | 2.32 | 5.40 | 1.79 |
| 6 | 4 | 67.9 | 0.535 | 4.8 | 1.9 | 2.312 | 2.53 | 7.33 | 2.43 |
| 6 | 6 | 77.5 | 0.375 | 6.2 | 2.2 | 2.639 | 2.82 | 7.22 | 2.40 |
| 6 | 24 | 100 | 0 | 9.1 | 2.8 | 3.405 | 3.25 | 7.32 | 2.43 |
 | ||
| Fig. 3 ln([IP]0/[IP]t) against polymerization time. | ||
The data (Table 3) would fit a situation where chain transfer was slow (hence, Mn increased with time and conversion), but not zero (hence, dispersity Đ increased with conversion). In fact, the number of chains (obtained by dividing the polymer yield by Mn and multiplying by Avogadro's number) variation with conversion gives a linear plot (see ESI;† R2 = 0.9995) where points from high conversion are excluded from the fit. At high conversion, chain transfer to polymer events reduce the number of chains, and chain transfer to monomer is inhibited by the dwindling of monomer, while at conversions less than 70%, chain transfers to monomer and/or aluminium dominate. The line can be extrapolated to zero conversion to give a number of active centres,46 equal to the number of chains before any chain transfer has occurred, which in this case is 0.3 × 1018 chains, which represents a 12%-efficient initiation in terms of cobalt. Taken together with the linear first-order kinetic plot, which indicates that the number of active centres remains at steady state throughout this polymerization, these facts suggest that in common with metallocenes and other organoaluminium-activated polymerization catalysts,47,48 there is a pre-equilibrium between closely associated but inactive transition metal-aluminium species and dissociated, active catalyst forms, and that in this case the equilibrium constant for the equilibrium between dormant and active site, Kinit = 0.11. Furthermore, the steady state of active centres indicates that at 35 °C any catalyst death events are balanced by new initiation events. Further discussion of the detail of all possible chain transfer events is available in the ESI.†
Organoaluminium effects
In order to attempt to distinguish the possibilities of chain transfer to monomer vs. chain transfer to aluminium, a series of polymerizations were carried out where [Al] was the varied parameter: the Al:Co ratios of 25 to 800 were probed, for both catalysts 2 and 7, keeping at the same initial monomer:Co ratio of 10000 (Table 4). In neither case was there any clear relationship between [Al] and Mn. The data appear to suggest that the chain transfer events are zero-order in Al. This is in stark contrast to most group 4 cases, where strong dependence of Mn on [Al] is normally found,48 as also with lanthanides and isoprene,49 and also even on our own results with closely similar Co complexes with butadiene where Mn decreased as [Al] increased,36 though these, as for most group 4 and lanthanide work, used MAO or R3Al rather than DEAC. If zero order, either there is no aluminium in the chain transfer process, i.e. chain transfer to monomer dominates, or, there is an already-saturated pre-equilibrium of associated Co–Al species even at a ratio as low as 25. If chain transfer to monomer was dominant, then some dependence of rate on state of conversion (i.e. on monomer concentration) might be expected, though the very high initial concentration of isoprene (10000:1 isoprene:Co) might mask this dependence until conversion had significantly dropped the isoprene concentration. No such drop in the rate of chain transfer was apparent until conversion reached beyond 70%. The slowing of the increase in no. of chains at that point was most likely a result of chain transfer to monomer being replaced by chain transfer to polymer, given that large increases in viscosity and the presence of insoluble gel fractions became an issue at high conversions. So, neither [Al] nor [isoprene] showed a clear influence on the rate of chain transfers over the concentration ranges studied, which however remained constant from 0 to 70% conversion before slowing. Furthermore, the Mn values were too high to make viable the spectroscopic identification of end-groups, which might distinguish these cases. We are unable to distinguish the cases of chain transfer to aluminium from transfer to monomer, though if the former dominates, it must imply a saturated pre-equilibrium of Co–Al contact species. Such a hypothesis, closely analogous to the situation proven for zirconium metallocenes,48,50 might also explain the fact of the similar ratio of active centres to metals present in those zirconium studies to that shown by our data. The case at hand differs from zirconocene-catalysed ethylene polymerization, however, in two key respects. Firstly, there are copious quantities of chloride ligands in the polymerization medium in the cobalt case (via DEAC or EASC); secondly, the allyl chain end will selectively π-co-ordinate to Co;51 the more charge-dense and polarizing Al σ-co-ordinates to allyls.52 Both of these factors in concert make it likely that any bridging between Co and Al would be likely to occur via chloride, rather than via polymeryl, and so reduce the opportunity for chain transfer to Al to occur. However, the most interesting observation to come from the study of [Al] was the dependence of stereoselectivity on ratio: at ratios of 25:1 Al:Co and at 35 °C, trans contents of 59% (2) and 52% (7) were found, both of which dropped to zero once the ratio approached 200:1. The overall activity also improved, but at the low Al level activity was still appreciable at this temperature, with around 24% conversion after 3 h. This rose to 88% conversion over the same timeframe at the favoured level of 150:1, before dropping again at very high conversions, though selectivity levels stabilised at around 75% cis, as [Al] was raised. Such an effect can only be explained by invoking notions of contact species between the catalyst and the organoaluminium, almost certainly via bridging chlorides. An equilibrium between a dormant such species and a looser, active pair, which becomes saturated in the associated state at high [Al], could explain the tail-off in activity and stabilization of selectivity, but does little to explain the switch in selectivity upon variation of [Al]. Similar effects have been observed before,2,19,53 but a clear explanation is still awaited.
| Catalyst | Al/Co | Conversion (%) | Mwb (g mol−1) × 105 | Mnb (g mol−1) × 105 | Đb | Microstructurec | |||
|---|---|---|---|---|---|---|---|---|---|
| Cis-1,4 | Trans-1,4 | 1,2 | 3,4 | ||||||
| a Conditions: catalyst: 5 μmol; co-catalyst: DEAC; isoprene: 5 mL; solvent: 30 mL chlorobenzene; time: 3 h; temperature: 35 °C.b Determined by GPC.c Determined by 1H and 13C NMR. | |||||||||
| 2 | 25 | 23.6 | 2.4 | 1.2 | 2.00 | 26.7 | 59.2 | 0 | 14.1 |
| 50 | 27.6 | 4.9 | 1.9 | 2.58 | 68.0 | 12.0 | 0 | 20.0 | |
| 100 | 52.9 | 5.0 | 2.2 | 2.27 | 73.0 | 1.5 | 0 | 25.5 | |
| 150 | 63.0 | 7.4 | 2.8 | 2.64 | 74.4 | 0.6 | 0 | 25.0 | |
| 200 | 49.1 | 4.6 | 1.9 | 2.42 | 74.0 | 0 | 0 | 26.0 | |
| 400 | 30.0 | 4.0 | 1.5 | 2.67 | 73.0 | 0 | 0 | 27.0 | |
| 800 | 27.8 | 3.4 | 1.2 | 2.83 | 73.3 | 0 | 0 | 26.7 | |
| 7 | 25 | 24.5 | 2.6 | 1.1 | 2.36 | 34.3 | 52.1 | 0 | 13.6 |
| 50 | 36.5 | 3.4 | 1.5 | 2.27 | 64.5 | 16.8 | 0 | 18.7 | |
| 100 | 77.4 | 9.6 | 4.3 | 2.23 | 76.9 | 0 | 0 | 23.1 | |
| 150 | 88.2 | 11.3 | 5.1 | 2.22 | 74.1 | 0 | 0 | 25.9 | |
| 200 | 73.5 | 10.0 | 4.5 | 2.22 | 74.3 | 0 | 0 | 25.7 | |
| 400 | 44.4 | 6.9 | 2.6 | 2.65 | 76.0 | 0 | 0 | 24.0 | |
| 800 | 26.2 | 5.3 | 2.0 | 2.65 | 74.0 | 2.5 | 0 | 23.5 | |
While on the subject of the effects of Al, an examination of the literature in this area19,39,40 led to the trialling of ethylaluminiumsequichloride (EASC) as a catalyst in place of DEAC. Other organoaluminium species were also trialled (triethylaluminium (TEA), trimethylaluminium (TMA), triisobutylaluminium (TIBA) and methylaluminoxane (MAO), in the range 150–500 Al:Co); all were found to be inactive. Chloride ligands appear essential, and the greater number of these in EASC over DEAC results in a significant improvement (approximately tenfold) in activity in trials with 6, (Table 5) where EASC produced not only 100% conversion in 4 h, but also improved selectivity, to 80.5% cis-1,4 polyisoprene.
| Catalyst | Al/Co | Time (h) | Conversion (%) | Mwb (g mol−1) × 105 | Mnb (g mol−1) × 105 | Đb | Microstructurec | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Cis-1,4 | Trans-1,4 | 1,2 | 3,4 | |||||||
| a Conditions: catalyst: 5 μmol; isoprene: 5 mL; solvent: 30 mL chlorobenzene.b Determined by GPC.c Determined by 1H and 13C NMR. | ||||||||||
| DEAC | 150 | 4 | 67.9 | 4.8 | 1.9 | 2.53 | 78.3 | 0.4 | 0 | 21.3 |
| EASC | 150 | 4 | 100 | 4.4 | 0.8 | 5.50 | 80.5 | 1.7 | 0 | 17.8 |
So to conclude, the polymerization seems to have rapid initiation, and some chain transfer to monomer, which is overtaken by chain transfer to polymer at high conversions, leading to some crosslinking at conversions nearing 100%, and selectivities depend modestly on ligand substituents, but more strongly on [Al].
Polymer microstructure
In our previous study on butadiene,36 there was no significant impact of the complex structure on the microstructure of the obtained polymer, which was predominantly cis-1,4-polybutadine (>97%). However, the introduction of the methyl group to the structure of isoprene causes a difference in both steric and electronic properties of the two double bonds of the diene. The unsubstituted double bond will have a weaker σ-bond but stronger π back-bond compared to the substituted double bond in isoprene, and is also less sterically encumbered, and more susceptible to attack by a nucleophile. The cobalt active centre can be η4-or η2-co-ordinated; in the case of η2 it can be 1,2 or 3,4 co-ordinated, and for η4 can be s-cis or s-trans co-ordinated.12 Chart 2 shows the possibilities of isoprene co-ordination at the active centre, and the type of enchainment most likely to result.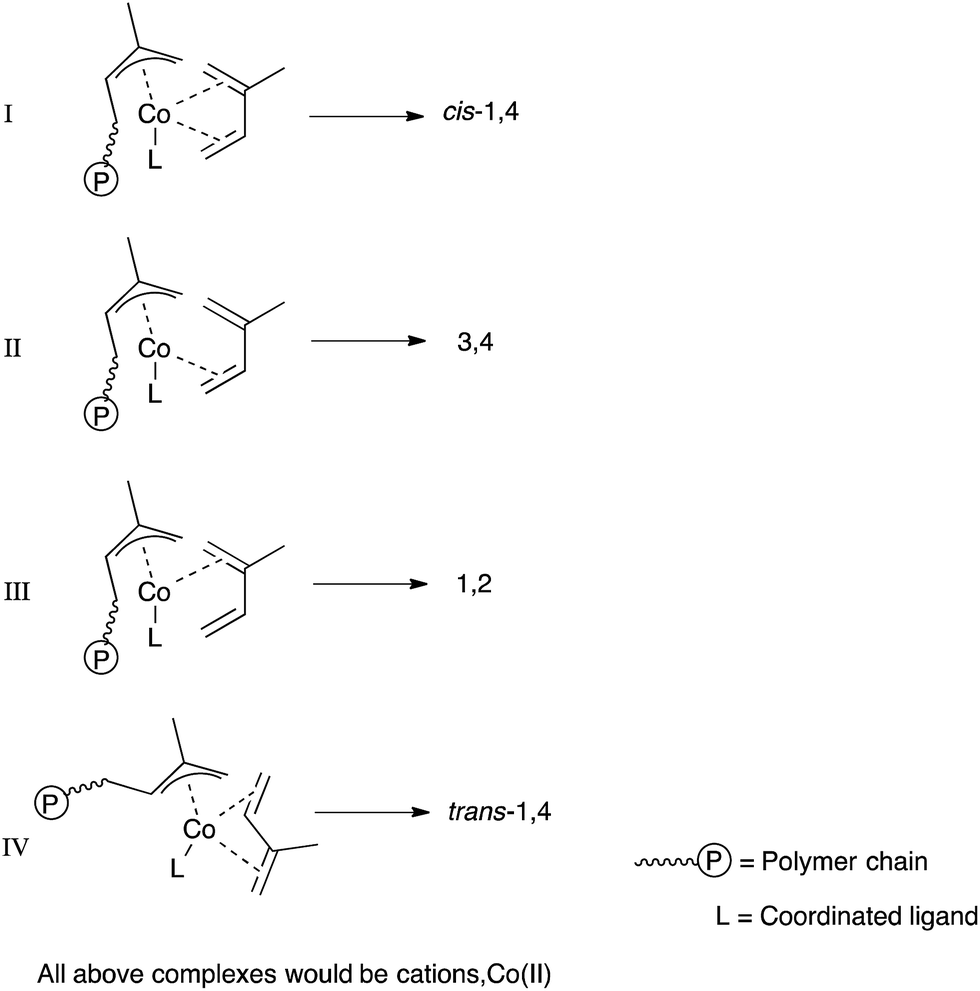 | ||
| Chart 2 Various polymerization modes available. | ||
If isoprene was bound as in III then 1,2-polyisoprene is likely; if as in II then 3,4-polyisoprene is likely (see also Chart 1). In the case of s-cis-η4 co-ordination, I, the produced polymer would be expected to be cis-1,4 if no isomerization of the diene occurs while it is in the expected anti allyl form of the active chain end, while trans-1,4 is possible where isomerization of the allyl to the more stable syn form precedes attack of the chain end on a further monomer, or where the s-trans form reacts directly without isomerization (IV).12 The temperature dependence data for polymerizations using 3, 6 and 7 are listed in Table 6. It can be noted that expected Arrhenius increase in rate with temperature operates up to around 70 °C, but then there is a decrease. This may partly be due to outgassing of monomer from the reaction solution, but also is likely to indicate catalyst de-activation at high temperatures.
| Catalyst | T (°C) | Conversion (%) | Mwb (g mol−1) × 105 | Mnb (g mol−1) × 105 | Đb | Microstructurec | |||
|---|---|---|---|---|---|---|---|---|---|
| Cis-1,4 | Trans-1,4 | 1,2 | 3,4 | ||||||
| a Conditions: catalyst: 5 μmol; co-catalyst: DEAC; Al/Co: 150; isoprene: 5 mL; solvent: 30 mL chlorobenzene time: 3 h.b Determined by GPC.c Determined by 1H and 13C NMR. | |||||||||
| 3 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 20 | 1.8 | 2.7 | 1.8 | 1.50 | 27.5 | 60.6 | 0 | 11.9 | |
| 35 | 14.2 | 3.1 | 1.2 | 2.58 | 73.8 | 4.7 | 0 | 21.5 | |
| 70 | 38.3 | 3.2 | 1.1 | 2.91 | 72.0 | 1.3 | 0 | 26.7 | |
| 100 | 32.4 | 1.9 | 0.6 | 3.17 | 71.9 | 1.9 | 0 | 26.2 | |
| 6 | 0 | Trace | 3.1 | 1.8 | 1.72 | 41.0 | 46.4 | 0 | 12.6 |
| 20 | 4.1 | 3.4 | 1.9 | 1.79 | 58.8 | 24.9 | 0 | 16.3 | |
| 35 | 70.6 | 5.2 | 2.2 | 2.36 | 76.0 | 0 | 0 | 24.0 | |
| 70 | 94.1 | 4.1 | 1.1 | 3.73 | 76.3 | 0 | 0 | 23.7 | |
| 100 | 78.5 | 2.0 | 0.5 | 4.00 | 76.0 | 0 | 0 | 24.0 | |
| 7 | 0 | Trace | 1.7 | 1.2 | 1.42 | 16.4 | 75.5 | 0 | 7.1 |
| 20 | 1.3 | 3.0 | 1.3 | 2.31 | 70.1 | 10.3 | 0 | 19.6 | |
| 35 | 44.1 | 4.9 | 2.1 | 2.33 | 75.8 | 0 | 0 | 24.2 | |
| 70 | 70.6 | 3.7 | 1.4 | 2.64 | 74.3 | 0 | 0 | 25.7 | |
More intriguing is the effect of temperature on microstructure. The thermodynamic aspects of diene polymerizations are fairly well-understood:12 (1) trans-1,4 polymer is the thermodynamic product; cis-1,4 is a kinetic product. (2) The parent allyl also is more stable in the syn (transoid) form than the anti (cisoid) form. Also, for the free dienes themselves, s-trans forms are more stable than s-cis forms. The situation is less-clear cut in η4-ligated forms: there are far more examples of η4-s-cis-co-ordinated dienes than of s-trans-co-ordinated ones, and there are more examples of cis-selective polymerizations than of trans-selective ones.54 However, in computational studies of some lanthanide and iron systems, it was found that the s-trans-bound form of isoprene or butadiene was in fact energetically preferred, but had a higher barrier to propagation than the s-cis-forms.55 Thus, while the thermodynamics is not in doubt, the kinetic pathways can be quite varied. Hence, the less-stable and less-abundant form of allyl (anti), arrived at via a relatively low barrier to isomerization, is responsible for the majority of polymerization events in some cases. With such a complicated potential surface to negotiate, it is reasonable to expect significant effects from catalyst ligand, temperature, co-catalyst ratio and co-catalyst type, and these are all found.
At the lowest temperatures, trans-1,4 (the thermodynamic product) predominates; paradoxically, this is replaced by cis-1,4 (the kinetic product) at higher temperatures. It should be noted that the productivity of 6 at 0 °C was very low, and elsewhere at low conversions, more trans product was seen. However, if it is accepted that all polymer is produced by the catalysts at hand, rather than by some parallel radical mechanism operating at a low level and only detected when other polymerization is slow, then it is possible to explain the observations with the following hypothesis: at low temperature, s-trans-co-ordinated diene is preferred, as for iron and lanthanides as recently shown computationally.55 The rate of monomer enchainment is slow, but some polymerization is seen. The rate of isomerization from syn to the more reactive anti allyl is also slow, and hence relatively low amounts of cis-enchainment result. As temperature rises, more isomerization can occur, and as this happens, the more reactive anti form starts to dominate in the products. Whether trans-co-ordination and addition is followed by syn-anti-allyl isomerization, which given the high electron count at the metal would be expected to be facile, or whether cis-co-ordination competes at higher temperatures, it is not possible to say at present. However, we can summarise our findings in the form of Chart 3 if we assume cis-co-ordination is at least partly responsible for the high levels of cis-polymer we find.
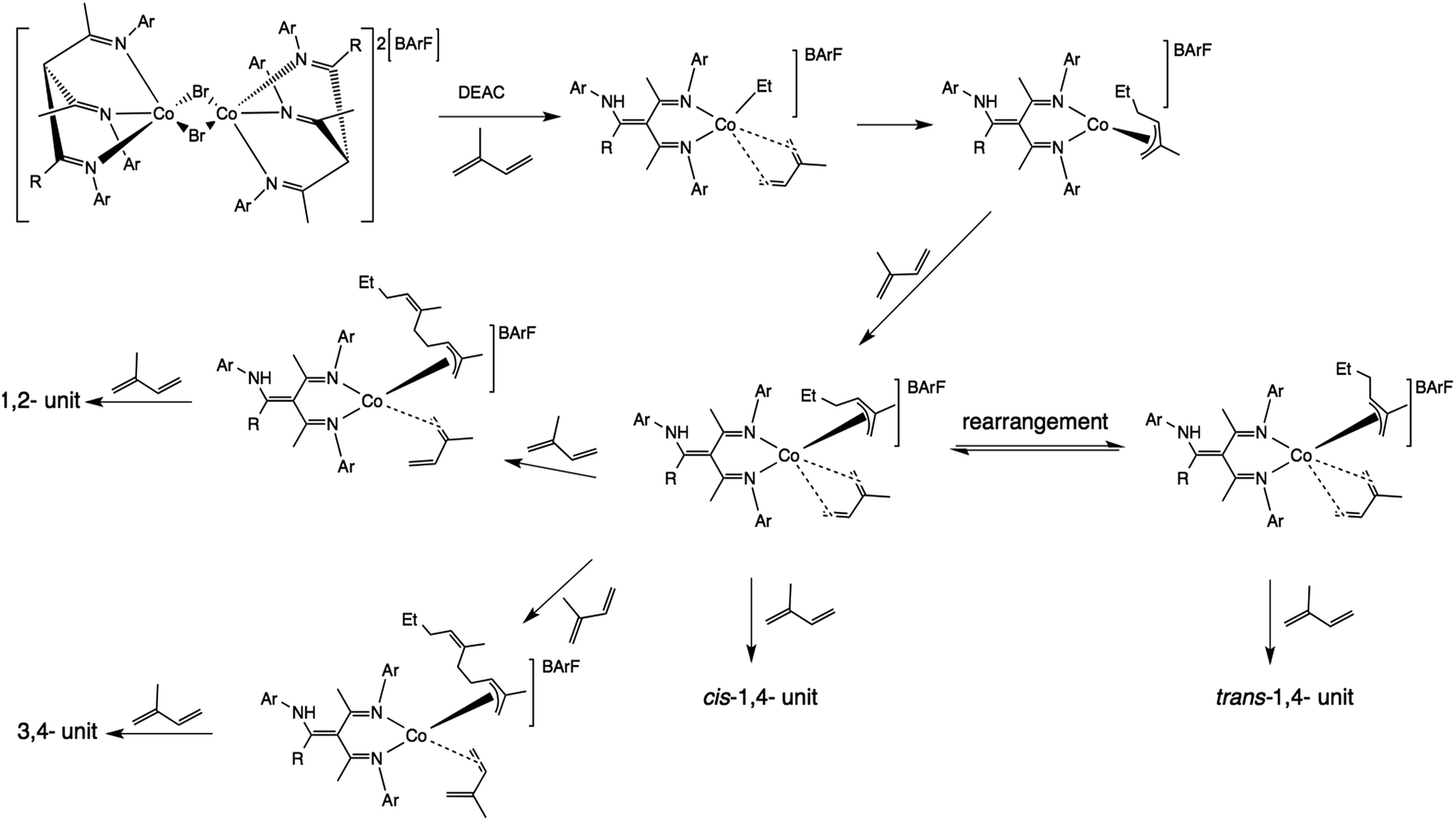 | ||
| Chart 3 Mechanism for polymerization of isoprene using 1–7. | ||
We now turn to the changes in the precatalysts 1–7 needed in order to become competent catalysts. Firstly, the dimers must break, since the bromides would be rapidly replaced by ethyl groups from DEAC or EASC, which are much less-efficient bridging ligands. Secondly, if a 4-electron isoprene and a 4-electron π-allyl (ionic count) is to be accommodated around d7 Co(II), then if the triketimine remained tridentate it would bring the electron count at the metal to 21. It seems likely that the triketimine might revert to bidentate enamine-imine form (19-electron), in order to relieve this stress. This then makes the triketimines ambidentate ligands with labile donors, the efficacy of which has previously been seen for butadiene polymerization.36,56 This might also explain the relatively modest (12%) ratio of active centres; dormant centres might include those with tridentate imine ligands. It remains an open question whether the NH in such bidentate tautomers would become metallated by the aluminium reagent, and whether this is a mode of activation or of de-activation. It is also possible to posit that the significant levels of 3,4 polymer obtained are a result of bidentate co-ordination of the isoprene facilitated by tridentate re-co-ordination of the triketimines. If this is the case, then such re-co-ordination must be transient, since clustering of 3,4 units was not apparent in the NMR data.
Finally, the purpose of the research was to find rubbers to challenge natural rubber in a range of applications, and so the thermal behaviour becomes relevant: the rubbers made by 1–7 at 35 °C are indeed elastic materials, with Tg values of approximately −32 °C as determined by the peak in tanδ from a DMTA analysis run in tension mode. Similar analysis of a commercial Nd high-cis polyisoprene gave Tg of −48 °C and for natural rubber, −50 °C. The higher Tg for polymers derived from 1–7 is to be expected from the impediment to relaxation offered by the statistically distributed 3,4 units protruding from the main chain.57 Given the difficulties encountered in preparation of regular films by melt pressing of some samples, it is likely also that some crosslinking had already occurred, which is known to raise tanδ Tg values.58 It is also the case that in tests of fully vulcanised rubber blended samples, the polyisoprenes from 1–7 gave rubbers with properties indistinguishable from natural rubber-containing analogues by DMTA, both in terms of tanδ and storage modulus.59 The DSC behaviour was more complex, with all polymers showing two to three broad endotherms (see ESI†), at higher temperatures than for natural rubber, in a manner similar to that reported for polymers derived from 8. The presence of the 3,4 units clearly has a profound influence on the phase behaviour of the materials. It would be of interest to investigate how crosslinked samples behave at higher strains, using such techniques as have recently been applied to probe the important phenomenon of strain induced crystallization.60 Further materials characterization of the rubbers and their compounded formulations will be the subject of a future paper.59
Catalyst performance comparison
We conclude our discussion with an attempt to place our activity and selectivity results in proper context (see ESI† for tabulated activity and selectivity comparisons). It becomes clear that catalysts 5–7 are moderately active; much more active than cationic61 or anionic polymerizations,62 more active that nickel salts,20 more active than some lanthanum and lanthanide catalysts,63 but less active than most.33,64 The activities are broadly in line with most other Co complexes, once the superior effect of EASC over DEAC is taken into account, which is to say, in the range of hundreds to a few thousands of mol IP mol−1 catalyst h−1.15–17,35,39,40A few cases stand out from this range: cobalt octoate with EASC reached a value of 43650 mol IP mol−1 Co h−1, but at the expense of selectivity (only 45% cis 1,4), and molecular weight (Mn only 3.7 × 104).19 In fact, it is clear that within the field of cobalt, it is possible to obtain a wide range of selectivities, from alternating 3,4/1,4-cis,35 to mixtures of all three main types of enchainment (e.g. 3,4/cis-1,4/trans-1,4 of 46/43/9%)19 to those more strongly biased to 3,4 (e.g. 57%).65 Most commonly with Co, cis-1,4 is favoured, from the aforementioned very modest levels of 45%,19 through 50%,35 a respectable 66% attained with silica-supported CoCl2 (with very high activity, though no Mn data are reported),18 to 75% attained with very low activity from [cobalt(dimethylglyoxime)],15 to our own values ranging from 74 to 80%, up to the highest yet-reported selectivities for cobalt in isoprene polymerization, ranging from 70% even up to 96% claimed (though not persuasively evidenced by 13C NMR spectroscopy),41 with catalysts similar to ours (8), from He, Sun and co-workers.39,40 However, their meridional N3 ligands, dosed as neutral dichloride complexes rather than as bromide BArF salts, while giving good activities, had much lower Mn than achieved by our facial N3-ligated catalysts. Hence, the rate of chain transfer, as well as the rate of polymerization, was lower in our case. This allows with 5–7 a combination of reasonable cis-1,4 selectivity (up to 80%) with high molecular weight and satisfactory activity. Though higher activities and selectivities have been reported for Co, no other case of which we are aware provides this optimum combination.
The market, where not served by natural rubber, has historically been served by traditional Zeigler–Natta catalysts based on Ti; these give cis-1,4 values in excess of 90%, and respectable activities and Mn values.9,10 For high-value applications where colour, odour or protein content of natural rubber may be an issue, such as in surgical gloves, anionically polymerized isoprene has been used, though low temperatures are required to keep cis-1,4 selectivity high with the organolithium initiators used.62
The most recent work in the area has been dominated by investigation of Rare Earths, which do indeed offer higher cis selectivity and activity.13,64 However, there is a long-term problem with use of lanthanides for this purpose.2 As discussed in our previous paper on butadiene,36 there are strongly competing uses for the unique magnetic and optical properties of the lanthanides, and it is strongly preferable to use base metals where possible. In fact, the most abundant transition metal, iron, would be ideal, and has some precedent. Some time ago Fe was shown to provide very high activity of 82400 mol IP mol−1 Fe h−1 but a moderate Mn (1.5 × 104) atactic 3,4-polymer in 85% selectivity with [(bipy)2FeEt2], or a lower selectivity (67%) but higher Mn (1.4 × 106)17 with [(bipy)FeCl2], in both cases activated by methylaluminoxane.66 More recently, replacement of one pyridyl group with a bulky imine has presented intriguing results. If the imine was aliphatic, a trans-1,4 polyisoprene (92% selective) was obtained with good activity (1000 mol IP mol−1 Fe h−1) and moderately good Mn (1.7 × 105) if sufficient monomer was used, while if an aromatic imine was used, then cis-1,4 polymer was obtained, in selectivities which ranged from 66 to 86%, with the balance in all cased being predominantly 3,4 polymer, i.e. comparable with those polymers reported here.67 However, in order to attain a good activity (1250 mol IP mol−1 Fe h−1) and a good cis-1,4 selectivity (83%), and sufficient Mn (23 × 105) it was necessary to use cryogenic temperatures (−78 °C).67 Our catalysts 5–7, then, are the only 3d transition metal complexes capable of producing high Mn polyisoprene of >75% cis-1,4 content at industrially preferable ambient or above-ambient temperature with satisfactory activity. Consequently, if our activities can be improved through increases in the proportion of cobalt which is active, we may yet find 3d-metal catalysts to make polymers to replace natural rubber, so as to protect against the economic and commercial effects of a catastrophic attack on the South-East Asia-Pacific rubber plantations by South American leaf blight,5 and protect scarce supplies of lanthanides by substituting more readily and widely available metals,68 as well as give the advantages of processing and reproducibility attendant to synthetic rubbers.3,4 While iron remains the best catalytic candidate for long-term supply, it has not yet reached the levels of activity and molecular weight at industrially relevant ambient or above-ambient temperatures, to yield 75–80% cis-1,4 rubbers, that we have attained with our cobalt catalysts 5–7. Supply of cobalt, as a byproduct of copper and nickel production, is less under immediate threat than that of lanthandides.68 Hence, we consider that our results contained herein, reporting good activity, moderately good 1,4 selectivity and high molecular weight for cobalt complexes of triketimines, may be of interest for applications where a modest degree of 3,4 enchained polymer, facilitative of efficient vulcanization, could offer advantage. Indeed, patents underline the improved wet-skid resistance of tyres with 3,4-content,7 while low rolling resistance of cis-1,4 polymer remains a primary concern to reduce vehicular fuel emissions.8
Conclusions
Cobalt β-triketimine complexes catalyse the polymerization of isoprene at 35 °C to give cis-1,4 polymer (74–80%) with the balance being 3,4-enchained, thus offering sites for crosslinking. At lower temperatures the catalysts are less-active but more selective for trans-1,4 polymer. Low ratios of DEAC co-catalyst also favoured trans. EASC gave higher activities than DEAC. At high conversions the catalysts are prone to crosslinking, broadening molecular weight distributions (Đ > 3) and generating multimodal features in the distribution. At low conversions molecular weight distribution is monomodal. Only 12% of catalyst is active at any moment, but chain transfer does occur to some extent, though not at sufficient rate to prevent attainment of high Mn, which can reach up to 9 × 105. Polymerization is first-order in monomer. The polymers produced have a higher Tg than natural rubber due to the 3,4 units.Experimental
General considerations
All procedures were carried out under an atmosphere of nitrogen. The ligands, CoBr2 and NaBArF were stored in an argon-filled glovebox before they were transferred into Schlenk tubes. Nitrogen gas was dried by passage through a column of phosphorus pentoxide supported on vermiculite. The solvents toluene, hexane, diethyl ether and tetrahydrofuran (THF) were distilled from sodium/benzophenone, while 2,6-dimethyl aniline, 2,6-difluoro aniline, dichloromethane (DCM), chlorobenzene and triethyl amine were distilled from calcium hydride. Isoprene, acetylacetone, nBuLi, CoBr2, 2,6-dimethylaniline, 2,6-difluoroaniline, acetyl chloride, 2,6-di-tert-butyl-4-methylphenol, trimethylaluminium, triethylaluminium, triisobutylaluminium and methylaluminoxane were purchased from Sigma-Aldrich while p-toluenesulfonic acid and ethylaluminum sesquichloride were purchased from Acros Organics. NaBArF,42 tBuCONH-2-OCH3C6H4,69 MeCONH(2-MeC6H4),70 tBuCClN-2-OCH3C6H4,37 the β-diketimines (2,6-Me2C6H3)NHCMeCHCMeN(2,6-Me2C6H3) {HBDK2,6Me2}71 (2-MeC6H4)NHCMeCHCMeN(2-MeC6H4) {HBDK2Me}72 and (2,6-F2C6H3)NHCMeCHCMeN(2,6-F2C6H3) {HBDK2,6F2}73 triketimine HC{MeCN(2-iPrC6H4)}2tBuCN(2-MeOC6H4)37 and complexes 1, 2 and 4 (ref. 36) were synthesised according to literature procedures. A Bruker 400 MHz spectrometer was used in order to record the NMR spectra, using CDCl3 as solvent. For quantitative 13C{1H} NMR spectra for assessment of selectivity, an extended time-delay of 6 s was used, and [Cr(acac)3] was added to accelerate relaxation. Assignments were by analogy to the literature.35,41 Infrared spectra were recorded on a Bruker Alpha-p ATR FTIR spectrometer, using OPUS 6.5 software. The mass spectra were recorded using the electrospray (ES) or MALDI techniques, with acetonitrile as solvent. Gel permeation chromatography (GPC) was used to measure Mn, Mw and dispersity Đ. GPC was carried out at 35 °C using a PL 2MB500A column in THF at a flow rate of 1 cm3 min−1; 100 μL was injected using a Viscotek VE2001 GPC equipped with a Viscotek VE5880 Refractive Index detector. A universal calibration was employed. X-ray diffraction (XRD) was used to measure the structure of single crystals. The data for 3 were collected on an Oxford SMART CCD diffractometer with Mo K-α X-ray source (λ = 0.71073 Å) at 150 K, while the data for 5 and 7 were collected on a Bruker Super Nova diffractometer with Mo K-α X-ray source (λ = 0.71073 Å) at 150 K; the data for 6 were collected on Bruker-Prospector diffractometer with Cu K-α X-ray source (λ = 1.54184 Å) at 150 K. The collected data were solved using the SUPERFLIP74 program and refined by the SHELX-97 (ref. 75) and OLEX2 (ref. 76) programs.Synthesis of imidoyl chlorides
Synthesis and characterization of β-triketimines
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CH3), 1.70 (6H, s, o-CH3), 2.06 (6H, s, o-CH3), 3.65 (3H, s, OCH3), 6.49–6.95 (10H, aromatic CH), 13.50 (1H, s, NH). 13C{1H} NMR (400 MHz, CDCl3): peaks due to CH3: δ 17.76 (o-CH3),18.22 (o-CH3), 20.14 (2 NC–CH3), 30.64 (C(CH3)3), 55.26 (OCH3); peaks due to CH: 110.96, 119.82, 120.14, 124.03, 124.34, 127.78, 127.84 (aromatic CH); peaks due to aliphatic C: 42.11 (C(CH3)3), 131.69 (CC), 159.05 (NH–C–CH3), 182.73 (NH–CC), peaks due to aromatic C: 131.79, 133.33, 141.10, 143.11, 150.39. Elemental analysis, calculated for C33H41N3O (%): C, 79.96; H, 8.34; N, 8.48. Found: C, 79.91; H, 8.32; N, 8.39. Mass spectrometry (ESI+) m/z: 496 [MH]+.C–CH3), 3.78 (3H, s, OCH3), 6.57 (1H, br dd, 3JF–H = 7.6 Hz, 4JH–H = 1.8 Hz, OMe-aryl, C6–H), ca. 6.75 (1H, C3–H) and 6.82 (1H, C4–H), br, overlapped with 6.79, 4H, dd. 3JF–H = 8.3 Hz, 3JH–H = 7.6 Hz, difluoro-aryl, meta-CH; 6.94, 2H, tt, 3JH–H = 8.5 Hz, 4JF–H = 6.7 Hz, difluoro-aryl, p-CH, overlapped with 6.98, 1H, OMe-aryl, C5–H, 13.26 (1H, s, NH). 13C{1H} NMR, 125.77 MHz, CDCl3, 35 °C: NMR at room temperature was broad. Recorded at 35 °C, resolved at least 3 and sometimes 4 separate environments for each nucleus, indicating tautomers. Major: 84% N,N′-bis-(o-difluorophenyl)-enamine-imine with pendant non-conjugated tBuC = N(o-anisyl), minor: 11% N-o-difluorophenyl-N′-(o-anisyl)-enamine-imine with pendant non-conjugated MeC = N(o-difluorophenyl) and 5% β-triketimine. All peaks were singlets unless stated otherwise. δ 20.52 (major), 19.38, 18.95 (minor) MeCN; 30.57 (major), 29.04, 27.99 (minor) Me3C; 42.72 (major), 42.30, 41.84 (minor) Me3C; 55.35 (major), 55.23, 55.45 (minor) OMe; 93.6 (minor) HC(RC = NAryl)3, β-triketimine; 106.01 (major), 108.48 (minor) CCNAryl; 111.4 (major, overlapped), 110.53 (minor) o-anisyl C5; 111.51, dd, 3JC–F = 17.5 Hz, 5JC–F = 5.6 Hz (major) 110.86, dd, 3JC–F = 15.0 Hz, 5JC–F = 6.4 Hz (minor) o-difluorophenyl C5/C3; 115.11 anisyl C3; 121.88 (major), t, 2JC–F = 16.4 Hz, 116.19 (minor), t, 2JC–F = 16.8 Hz, o-difluorophenyl ipso C–N; 124.98 (major), 116.6, 127.85, 127.33 (minor) t, 4JC–F = 8.4 Hz, 9.0 Hz, 9.5 Hz, 9.4 Hz respectively, o-difluorophenyl C4; 120.53 with shoulder(major + minor), 118.56 (minor), anisyl C4; 119.53 (broad, major + minor) 121.10 (minor), anisyl C6; 124.68 (major) anisyl C3; 136.07 (major) anisyl ipso C–N; 150.47 (major) 149.76 (minor) anisyl C–OMe. 156.27 (major) 152.27, 158.05, 158.34, (minor), dd, 1JC–F = 247.9 Hz, 4JC–F = 4.5 Hz, 1JC–F = 240.2 Hz, 4JC–F = 7.6 Hz, 1JC–F = 248.6 Hz, 4JC–F = 9.9 Hz, 1JC–F = 250.5 Hz respectively, o-difluorophenyl C2/C6; 161.54 (major), 159.84, 161.77 (minor) MeC = N; 194.70 tBuC = N. 19F NMR (470.65 MHz, 35 °C, externally referenced to CFCl3): δ −120.80, dd, apparent t, 3JF–H = 6.8 Hz (major); −118.42, dd, 3JF–H = 11.3 Hz, 4JF–H = 6.1 Hz. Elemental analysis, calculated for C35H45N3O (%): C, 68.09; H, 5.71; N, 8.21. Found: C, 67.97; H, 5.75; N, 8.23. Mass spectrometry (ESI+) m/z: 512.4 [MH]+.N), and other minor peaks (s) in this area, indicating tautomers. Further minor peaks: 4.68 (s, α-HC triketimine), 6.68 (d, aryl CH), 6.74 (t, aryl CH), major tautomer: 6.84 (1H, broad t,3 J = 6 Hz, aryl CH) 7.12–7.24 (3H broad m, overlapping, aromatic CH × 2), 13.72 (1H, s, NH). 13C NMR (125.78 MHz, CDCl3, 25 °C): d 18.28, 18.71, 24.27 (3 × minor, CH3), 18.42 (major, CH3), 110.39 (CCN), 118.27, 117.97, 117.70, 124.01 (4 × aryl o-CH), 123.43, 126.31, 130.50 (aryl m- and p-CH), 131.05 (aryl C–CH3), 143.76 (aryl C–N), 158.02 (CN conjugated), 171.72 (CN isolated). Elemental analysis, calculated for C28H31N3 (%): C, 82.11; H, 7.63; N, 10.26. Found: C, 81.87; H, 7.58; N, 10.27. Mass spectrometry (ESI+) m/z: 410.4 [MH]+.
C–CH3), 1.70 (6H, s, o-CH3), 2.06 (6H, s, o-CH3), 3.65 (3H, s, OCH3), 6.49–6.95 (10H, aromatic CH), 13.50 (1H, s, NH). 13C{1H} NMR (400 MHz, CDCl3): peaks due to CH3: δ 17.76 (o-CH3),18.22 (o-CH3), 20.14 (2 NC–CH3), 30.64 (C(CH3)3), 55.26 (OCH3); peaks due to CH: 110.96, 119.82, 120.14, 124.03, 124.34, 127.78, 127.84 (aromatic CH); peaks due to aliphatic C: 42.11 (C(CH3)3), 131.69 (CC), 159.05 (NH–C–CH3), 182.73 (NH–CC), peaks due to aromatic C: 131.79, 133.33, 141.10, 143.11, 150.39. Elemental analysis, calculated for C33H41N3O (%): C, 79.96; H, 8.34; N, 8.48. Found: C, 79.91; H, 8.32; N, 8.39. Mass spectrometry (ESI+) m/z: 496 [MH]+.C–CH3), 3.78 (3H, s, OCH3), 6.57 (1H, br dd, 3JF–H = 7.6 Hz, 4JH–H = 1.8 Hz, OMe-aryl, C6–H), ca. 6.75 (1H, C3–H) and 6.82 (1H, C4–H), br, overlapped with 6.79, 4H, dd. 3JF–H = 8.3 Hz, 3JH–H = 7.6 Hz, difluoro-aryl, meta-CH; 6.94, 2H, tt, 3JH–H = 8.5 Hz, 4JF–H = 6.7 Hz, difluoro-aryl, p-CH, overlapped with 6.98, 1H, OMe-aryl, C5–H, 13.26 (1H, s, NH). 13C{1H} NMR, 125.77 MHz, CDCl3, 35 °C: NMR at room temperature was broad. Recorded at 35 °C, resolved at least 3 and sometimes 4 separate environments for each nucleus, indicating tautomers. Major: 84% N,N′-bis-(o-difluorophenyl)-enamine-imine with pendant non-conjugated tBuC = N(o-anisyl), minor: 11% N-o-difluorophenyl-N′-(o-anisyl)-enamine-imine with pendant non-conjugated MeC = N(o-difluorophenyl) and 5% β-triketimine. All peaks were singlets unless stated otherwise. δ 20.52 (major), 19.38, 18.95 (minor) MeCN; 30.57 (major), 29.04, 27.99 (minor) Me3C; 42.72 (major), 42.30, 41.84 (minor) Me3C; 55.35 (major), 55.23, 55.45 (minor) OMe; 93.6 (minor) HC(RC = NAryl)3, β-triketimine; 106.01 (major), 108.48 (minor) CCNAryl; 111.4 (major, overlapped), 110.53 (minor) o-anisyl C5; 111.51, dd, 3JC–F = 17.5 Hz, 5JC–F = 5.6 Hz (major) 110.86, dd, 3JC–F = 15.0 Hz, 5JC–F = 6.4 Hz (minor) o-difluorophenyl C5/C3; 115.11 anisyl C3; 121.88 (major), t, 2JC–F = 16.4 Hz, 116.19 (minor), t, 2JC–F = 16.8 Hz, o-difluorophenyl ipso C–N; 124.98 (major), 116.6, 127.85, 127.33 (minor) t, 4JC–F = 8.4 Hz, 9.0 Hz, 9.5 Hz, 9.4 Hz respectively, o-difluorophenyl C4; 120.53 with shoulder(major + minor), 118.56 (minor), anisyl C4; 119.53 (broad, major + minor) 121.10 (minor), anisyl C6; 124.68 (major) anisyl C3; 136.07 (major) anisyl ipso C–N; 150.47 (major) 149.76 (minor) anisyl C–OMe. 156.27 (major) 152.27, 158.05, 158.34, (minor), dd, 1JC–F = 247.9 Hz, 4JC–F = 4.5 Hz, 1JC–F = 240.2 Hz, 4JC–F = 7.6 Hz, 1JC–F = 248.6 Hz, 4JC–F = 9.9 Hz, 1JC–F = 250.5 Hz respectively, o-difluorophenyl C2/C6; 161.54 (major), 159.84, 161.77 (minor) MeC = N; 194.70 tBuC = N. 19F NMR (470.65 MHz, 35 °C, externally referenced to CFCl3): δ −120.80, dd, apparent t, 3JF–H = 6.8 Hz (major); −118.42, dd, 3JF–H = 11.3 Hz, 4JF–H = 6.1 Hz. Elemental analysis, calculated for C35H45N3O (%): C, 68.09; H, 5.71; N, 8.21. Found: C, 67.97; H, 5.75; N, 8.23. Mass spectrometry (ESI+) m/z: 512.4 [MH]+.N), and other minor peaks (s) in this area, indicating tautomers. Further minor peaks: 4.68 (s, α-HC triketimine), 6.68 (d, aryl CH), 6.74 (t, aryl CH), major tautomer: 6.84 (1H, broad t,3 J = 6 Hz, aryl CH) 7.12–7.24 (3H broad m, overlapping, aromatic CH × 2), 13.72 (1H, s, NH). 13C NMR (125.78 MHz, CDCl3, 25 °C): d 18.28, 18.71, 24.27 (3 × minor, CH3), 18.42 (major, CH3), 110.39 (CCN), 118.27, 117.97, 117.70, 124.01 (4 × aryl o-CH), 123.43, 126.31, 130.50 (aryl m- and p-CH), 131.05 (aryl C–CH3), 143.76 (aryl C–N), 158.02 (CN conjugated), 171.72 (CN isolated). Elemental analysis, calculated for C28H31N3 (%): C, 82.11; H, 7.63; N, 10.26. Found: C, 81.87; H, 7.58; N, 10.27. Mass spectrometry (ESI+) m/z: 410.4 [MH]+.Synthesis and characterization of β-triketimine cobalt complexes
Polymerization of isoprene
In the glovebox, the desired amount of catalyst 1–7 was transferred into a Schlenk tube, to which 10 cm3 of solvent (chlorobenzene) was added after removal from the glovebox and attachment to a N2/vacuum double manifold. A two neck round bottom flask was connected to a condenser which was fitted to the flask on a N2/vacuum double manifold. When the desired concentration of the catalyst (5 μmol) was added, the desired amount of the alkylating reagent (EASC or Et2AlCl) was added. The solution was left stirring for five minutes. Thereafter, isoprene (5 cm3, 0.05 mol) was added; the polymerization was carried out for 24 h unless otherwise stated. The polymerization was terminated by pouring the polymer solution into acidified methanol (10% HCl conc.) containing 2,6-di-tert-butyl-4-methylphenol as antioxidant. The mixture was left stirring 12 hours. The polymer was filtered, washed with methanol and then dried under vacuum for 24 hours at room temperature. Each polymerization was run in triplicate.Dynamic mechanical thermal analysis
Sheets of polymer (1 mm thick) were first pressed on a melt-press at 100 °C and then allowed to cool. Test pieces were cut (25 × 3 × 1 mm), at least 6 for each polymer. The polymer strips were clamped in the jaws of a Tritec 200 DMA running in tension mode (gap between clamps: 15 mm). At least 6 replicates were used for each sample. The storage and loss moduli E′, E′′ and tanδ were recorded during temperature scanning from −100 °C to 100 °C at a heating rate of 2 °C min−1, while the test-pieces were tensioned at a frequency of 1 Hz. In addition to research samples, samples of Natural Rubber and of commercial Nd high-cis polyisoprene were measured.
Differential scanning calorimetry
Heat flows were recorded by differential scanning calorimetry (DSC), using a Perkin Elmer Diamond DSC. Measurements were repeated three times for each sample, which was weighed (5–10 mg) and sealed in an aluminium hermetic pan. The experiment used the heating–cooling–heating program with a temperature scan from −80 °C to 100 °C. This upper limit was chosen to avoid crosslinking.The samples were heated from −80 °C to 100 °C with a heating rate of 10 °C min−1, held at 100 °C for 5 min, and then cooled from 100 °C to −80 °C at a rate of 10 °C min−1, kept at −80 °C for 5 min, and then heated from −80 °C to 100 °C at the heating rate of 10 °C min−1. The second heating data were recorded as a function of temperature and are presented as ESI† (endothermic events up).
Acknowledgements
The King Abdul Aziz City for Science and Technology (KACST) is acknowledged for provision of a studentship to MNA. Mr Martin Jennings of the University of Manchester School of Chemistry Analytical Service is acknowledged for provision of elemental analyses. The practical assistance of Mr Miao Jiang in collection of thermal characterization data is gratefully acknowledged.Notes and references
- R. Cariou, J. J. Chirinos, V. C. Gibson, G. Jacobsen, A. K. Tomov, G. J. P. Britovsek and A. J. P. White, Dalton Trans., 2010, 39, 9039 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- B. Krutzer, M. Ros, J. Smit and W. de Jong, Rubber World, 2012, 247(18), 31 Search PubMed.
- N. Nakajima, Science and Practice of Rubber Mixing, RAPRA Technology Ltd, Shawbury, Shropshire, UK, 2000 Search PubMed.
- Protection against South American leaf blight of rubber in Asia and the Pacific region, FAO Regional Office for Asia and the Pacific, 2011.
- J. Qiu, Nature, 2009, 457, 246 CrossRef CAS PubMed.
- J. Wolpers, H. B. Fuchs, C. Herrmann and W. Hellermann, EP456902A1, 1991.
- J. D. Massie II, W. L. Hsu, A. F. Halasa and P. H. Sandstrom, US5356997A, 1994.
- G. Saltman, W. E. Gibbs and J. Lal, J. Am. Chem. Soc., 1958, 80, 5615 CrossRef.
- G. Natta, L. Porri, A. Mazzei and D. Moreno, Chim. Ind., 1959, 41, 398 CAS.
- R. N. Kovalevskaya, E. I. Tinyakova and B. A. Dolgoplosk, Vysokomol. Soedin., 1962, 4, 1338 CAS.
- G. Ricci, A. Sommazzi, F. Masi, M. Ricci, A. Boglia and G. Leone, Coord. Chem. Rev., 2010, 254, 661 CrossRef CAS PubMed.
- Z. Zhang, D. Cui, B. Wang, B. Liu and Y. Yang, Struct. Bonding, 2010, 137, 49 CAS.
- E. Nakamura and K. Sato, Nat. Mater., 2011, 10, 158 CrossRef CAS PubMed.
- H. Noguchi and S. Kambara, J. Polym. Sci., Part B: Polym. Lett., 1964, 6, 593 CrossRef PubMed.
- G. Delheye and F. Dawans, Makromol. Chem., 1966, 98, 164 CrossRef CAS PubMed.
- F. Dawans and P. Teyssie, Makromol. Chem., 1967, 109, 68 CrossRef CAS PubMed.
- K. Soga, K. Yamamoto and S.-I. Chen, Polym. Bull., 1981, 5, 1 CrossRef CAS.
- V. A. Yakovlev, I. F. Gavrilenko, N. N. Glebova and G. N. Bondarenko, Polym. Sci., Ser. B, 2014, 56, 31 CrossRef CAS.
- U. Gebauer, S. Engelmann and K. Gehrke, Acta Polym., 1989, 40, 341 CrossRef CAS PubMed.
- A. Fischbach, F. Perdih, E. Herdtweck and R. Anwander, Organometallics, 2006, 25, 1626 CrossRef CAS.
- A. Fischbach, M. G. Klimpel, M. Widenmeyer, E. Herdtweck, W. Scherer and R. Anwander, Angew. Chem., Int. Ed., 2004, 43, 2234 CrossRef CAS PubMed.
- M. Zimmermann, N. A. Froeystein, A. Fischbach, P. Sirsch, H. M. Dietrich, K. W. Toernroos, E. Herdtweck and R. Anwander, Chem.–Eur. J., 2007, 13, 8784 CrossRef CAS PubMed.
- L. Zhang, T. Suzuki, Y. Luo, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2007, 46, 1909 CrossRef CAS PubMed.
- G.-L. Li, C.-Y. Ren, W.-M. Dong, L.-S. Jiang, X.-Q. Zhang and F.-S. Wang, Chin. J. Polym. Sci., 2010, 28, 157 CrossRef CAS.
- J. Zhang, Z. Hao, W. Gao, L. Xin, L. Zhang and Y. Mu, Chem.–Asian J., 2013, 8, 2079 CrossRef CAS PubMed.
- G. Zhang, Y. Wei, L. Guo, X. Zhu, S. Wang, S. Zhou and X. Mu, Chem.–Eur. J., 2014, 21, 2519 CrossRef PubMed.
- M. Zimmermann, K. W. Toernroos, H. Sitzmann and R. Anwander, Chem.–Eur. J., 2008, 14, 7266 CrossRef CAS PubMed.
- M. Zimmermann, K. W. Toernroos and R. Anwander, Angew. Chem., Int. Ed., 2008, 47, 775 CrossRef CAS PubMed.
- H. Liu, J. He, Z. Liu, Z. Lin, G. Du, S. Zhang and X. Li, Macromolecules, 2013, 46, 3257 CrossRef CAS.
- L. Zhang, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2005, 127, 14562 CrossRef CAS PubMed.
- L. Zhang, M. Nishiura, M. Yuki, Y. Luo and Z. Hou, Angew. Chem., Int. Ed., 2008, 47, 2642 CrossRef CAS PubMed.
- B. Liu, L. Li, G. Sun, J. Liu, M. Wang, S. Li and D. Cui, Macromolecules, 2014, 47, 4971 CrossRef CAS.
- C. Yao, D. Liu, P. Li, C. Wu, S. Li, B. Liu and D. Cui, Organometallics, 2014, 33, 684 CrossRef CAS.
- G. Ricci, G. Leone, A. Boglia, A. C. Boccia and L. Zetta, Macromolecules, 2009, 42, 9263 CrossRef CAS.
- M. N. Alnajrani and F. S. Mair, Dalton Trans., 2014, 43, 15727 RSC.
- D. Barnes, G. L. Brown, M. Brownhill, I. German, C. J. Herbert, A. Jolleys, A. R. Kennedy, B. Liu, K. McBride, F. S. Mair, R. G. Pritchard, A. Sanders and J. E. Warren, Eur. J. Inorg. Chem., 2009, 9, 1219 CrossRef PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- A. He, G. Wang, W. Zhao, X. Jiang, W. Yao and W.-H. Sun, Polym. Int., 2013, 62, 1758 CrossRef CAS PubMed.
- G. Wang, X. Jiang, W. Zhao, W.-H. Sun, W. Yao and A. He, J. Appl. Polym. Sci., 2014, 131, 39703 Search PubMed.
- A. S. Khachaturov, E. R. Dolinskaya, L. K. Prozeno, E. L. Abramenko and V. A. Kormer, Polymer, 1977, 18, 871 CrossRef CAS; V. A. Rozentsvet, A. S. Khachaturov and V. P. Ivanova, Polym. Sci., Ser. A, 2009, 51, 870 CrossRef; N. Mahiyanov, I. G. Akhmetov and A. M. Vagizov, Polym. Sci., Ser. A, 2012, 54, 942 CrossRef; Y. Tanaka, H. Sato, A. Ogura and I. Nagoya, J. Polym. Sci., Polym. Chem. Ed., 1976, 14, 73 CrossRef PubMed; A. J. Brandolini and D. D. Hills, NMR spectra of Polymers and Polymer Additives, Marcel Dekker, New York, 2000 Search PubMed.
- M. Brookhart, B. Grant and A. F. Volpe Jr, Organometallics, 1992, 11, 3920 CrossRef CAS.
- H. Liu, X. Jia, F. Wang, Q. Dai, B. Wang, J. Bi, C. Zhang, L. Zhao, C. Bai, Y. Hu and X. Zhang, Dalton Trans., 2013, 42, 13723 RSC; D. Gong, B. Wang, H. Cai, X. Zhang and L. Jiang, J. Organomet. Chem., 2011, 696, 1584 CrossRef CAS PubMed.
- V. K. M. Riches, Makromol. Chem., 1967, 103, 175 CrossRef PubMed.
- P. J. Flory, J. Am. Chem. Soc., 1947, 69, 2893 CrossRef CAS.
- D. G. Boucher, I. W. Parsons and R. N. Haward, Makromol. Chem., 1974, 175, 3461 CrossRef CAS PubMed.
- F. Vaultier, V. Monteil, R. Spitz, J. Thuillez and C. Boisson, Polym. Chem., 2012, 3, 1490 RSC.
- F. Ghiotto, C. Pateraki, J. R. Severn, N. Friederichs and M. Bochmann, Dalton Trans., 2013, 42, 9040 RSC.
- Z. Jian, D. Cui, Z. Hou and X. Li, Chem. Commun., 2010, 46, 3022 RSC; Y. Pan, T. Xu, G.-W. Yang, K. Jin and X.-B. Lu, Inorg. Chem., 2013, 52, 2802 CrossRef CAS PubMed.
- G. Fan and J.-Y. Dong, J. Mol. Catal. A: Chem., 2005, 236, 246 CrossRef CAS PubMed; F. Song, R. D. Cameron and M. Bochmann, J. Am. Chem. Soc., 2003, 125, 7641 CrossRef PubMed.
- P. Grossheimann and P. W. Jolly, Inorg. Chim. Acta, 1998, 270, 60 CrossRef.
- A. Stefani and P. Pino, Helv. Chim. Acta, 1972, 55, 1110 CrossRef CAS PubMed; Z. Peng, T. D. Blümke, P. Mayer and P. Knochel, Angew. Chem., Int. Ed. Engl., 2010, 49, 45 Search PubMed.
- L. N. Jende, C. O. Hollfelder, C. Maichle-Mossmer and R. Anwander, Organometallics, 2015, 34, 32 CrossRef CAS.
- C. Elschenbroich, Organometallics, Wiley, Weinheim, 3rd edn, 2006 Search PubMed.
- S. Tobisch, Can. J. Chem., 2009, 87, 1392 CrossRef CAS PubMed; L. Perrin, F. Bonnet, M. Visseaux and L. Maron, Chem. Commun., 2010, 46, 2965 RSC; X. Kang, Y. Luo, G. Zhou, X. Wang, X. Yu, Z. Hou and J. Qu, Macromolecules, 2014, 47, 4596 CrossRef.
- D. Gong, B. Wang, X. Jia and X. Zhang, Dalton Trans., 2014, 43, 4169 RSC.
- M. Baboo, M. Dixit, K. Sharma and N. S. Saxena, Polym. Bull., 2011, 66, 661 CrossRef CAS PubMed.
- S. Mitra, S. Chattopadhyay and A. K. Bhowmick, J. Appl. Polym. Sci., 2008, 107, 2755 CrossRef CAS PubMed.
- M. Jiang, M. N. Alnajrani and F. S. Mair, to be published.
- S. Toki, J. Che, L. Rong, B. S. Hsiao, S. Amnuaypornsi, A. Nimpaiboon and J. Sakdapipanich, Macromolecules, 2013, 46, 5283 CrossRef CAS; Y. Ren, S. Zhao, Q. Yao, Q. Li, X. Zhang and L. Zhang, RSC Adv., 2015, 5, 11317 RSC.
- S. Ouardad, T. Lebarbé, A. Deffieux and F. Peruch, Polym. Chem., 2013, 4, 407 RSC; V. A. Rozentsvet, V. G. Kozlov, E. F. Ziganshina, N. P. Boreiko and S. V. Kostjuk, Polym. Int., 2013, 62, 817 CrossRef CAS PubMed.
- K. Ratkanthwar, N. Hadjichristidis, S. Lee, T. Chang, Z. Pudukulathan and D. Vlassopoulos, Polym. Chem., 2013, 4, 5645 RSC; K. Ratkanthwar, N. Hadjichristidis and Z. Pudukulathan, Chem. J., 2013, 3, 90 Search PubMed.
- L. Wang, D. Liu and D. Cui, Organometallics, 2012, 41, 2367 Search PubMed.
- See, for example: M. J. Vitorino, P. Zinck and M. Visseaux, Eur. Polym. J., 2012, 48, 1289 CrossRef CAS PubMed; N. Martins, F. Bonnet and M. Visseaux, Polymer, 2014, 55, 5013 CrossRef PubMed ; and further examples cited in ESI.†.
- G. Ricci, A. Forni, A. Boglia, T. Motta, G. Zannoni, M. Canetti and F. Bertini, Macromolecules, 2005, 38, 1064 CrossRef CAS.
- C. Bazzini, A. Giarrusso and L. Porri, Macromol. Rapid Commun., 2002, 23, 922 CrossRef CAS; C. Bazzini, A. Giarrusso, L. Porri, B. Pirozzi and R. Napolitano, Polymer, 2004, 45, 2871 CrossRef PubMed.
- J. Raynaud, J. Y. Wu and T. Ritter, Angew. Chem., Int. Ed., 2012, 124, 11975 CrossRef PubMed.
- E. A. Brande and G. B. Brook, Smithills Metals Reference Book, Elsevier, Amsterdam, 1998 Search PubMed.
- G. W. Gribble and F. P. Bousquet, Tetrahedron, 1971, 27, 3785 CrossRef CAS.
- B. S. Kim, C. Jang, D. J. Lee and S. W. Youn, Chem.–Asian J., 2010, 5, 2336 CrossRef CAS PubMed.
- G. Xie and C. Qian, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 211 CrossRef CAS PubMed.
- R. Jiao, M. Xue, X. Shen, Y. Zhang, Y. Yao and Q. Shen, Eur. J. Inorg. Chem., 2010, 2523 CrossRef CAS PubMed.
- K. Yliheikkilä, K. Axenov, M. T. Räisänen, M. Klinga, M. P. Lankinen, M. Kettunen, M. Leskelä and T. Repo, Organometallics, 2007, 26, 980 CrossRef.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- M. F. Lappert and A. J. Oliver, J. Chem. Soc., Dalton Trans., 1974, 65 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Extended discussion of kinetic analysis, polymerization activity comparisons, polymer characterization data, and crystallographic information. CCDC 1055722, 1055723, 1055593 and 1055579. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra06792h |
| ‡ Current address: King Abdulaziz City for Science and Technology, PO Box 6086, Riyadh 11442, Kingdom of Saudi Arabia. E-mail: mnajrani@kacst.edu.sa. |
| § Notwithstanding the fact that reported 13C NMR spectra for a claimed 90% cis sample39 had main peaks at shifts of 22.7, 33.5 and 46.5 ppm, high-cis 1,4 polyisoprene more typically has shifts of 23, 26 and 32 ppm. Our own data from an industrial high-cis sample correspond with those, as reported in standard ref. 35 and 41. Only very weak peaks at 26 ppm were present in the spectra reported in ref. 39, and a clear explanation for the strong peak at 46.5, which in our data treatment we ascribe to a 3,4-enchained unit on the basis of analogy to authentic samples containing such units,65 was absent. Data presented for a sample of claimed 75% cis content were in closer accord with our own. See ESI† for fully assigned spectra of some examples of polymers prepared from 1–7. |
| This journal is © The Royal Society of Chemistry 2015 |