
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, chiroptical and SHG properties of polarizable push–pull dyes built on π-extended binaphthyls†
Carmine
Coluccini
a,
Marco
Caricato
a,
Elena
Cariati
bc,
Stefania
Righetto
b,
Alessandra
Forni
c and
Dario
Pasini
 *ad
*ad
aDepartment of Chemistry, University of Pavia, Viale Taramelli 10, 27100 Pavia, Italy. E-mail: dario.pasini@unipv.it
bDipartimento di Chimica dell'Università degli Studi di Milano, UdR dell'INSTM, via Golgi 19, 20133 Milano, Italy
cIstituto di Scienze e Tecnologie Molecolari – Consiglio Nazionale delle Ricerche (ISTM-CNR), Università degli Studi di Milano, UdR dell'INSTM, via C. Golgi 19, 20133 Milano, Italy
dINSTM Research Unit, University of Pavia, Italy
First published on 19th February 2015
Abstract
We report on new enantiopure binaphthyl derivatives in which electron-donating and electron-withdrawing substituents are placed in direct conjugation, to create active push–pull dyes for NLO applications. The dyes, unprecedentedly, extend their π-bridge from the 3,3′ positions of the binaphthyl units, and incorporate as acceptors pyridine units, possessing a coordinating nitrogen atom useful for further supramolecular polarization of the chiral dyes. The π-bridge is constructed by the sequential attachment of phenylenevinylene units to the enantiopure binaphthyl derivatives through Horner–Wadsworth–Emmons olefination, which proceed with high stereoselectivity, affording stereodefined chiral dyes. The polarization of the terminal pyridine units by means of labile complexation with Pd2+ ions has been demonstrated using both optical and chiroptical methods. The polarization by protonation can be made reversible in solution and solid state by exposure to ammonia vapors, as shown by absorption and emission spectroscopies. NLO properties, as determined by EFISH generation measurements in solution, are significant for the bisprotonated species when compared to previously reported binaphthyl substrates. TDDFT calculations show that the hyperpolarizability tensor contribution is responsible for enhancing SHG values upon protonation up to one order of magnitude, highlighting the potential of such ortho related, axially-chiral push–pull dyes for functional NLO applications.
Introduction
Conjugated organic materials are currently used or being developed for a number of technological applications. Amongst these, well-defined π-extended organic oligomers have been the subject of increasing attention in the last few decades, either in solution or in bulk. Their utility as model structures for the understanding of the solution and bulk scale properties of conjugated polymers should also be stressed.1 One of the most prominent examples of the application of conjugated organic molecules is in the field of second harmonic generation (SHG),2 a nonlinear optical property which is at the foundation for advanced technologies in materials science and biological imaging. SHG is forbidden in the presence of a centrosymmetric material; chiral organic molecules, by definition noncentrosymmetric, have thus been widely studied as materials capable of SHG.2Chirality can play a fundamental role in tuning the properties of nanoscale structures, and of matter in general; in addition, nanostructuring via self-assembly of the chiral organic dyes has been shown to have pronounced effects and amplifications of their SHG response.3 Amongst known atropoisomeric chiral compounds, binaphthyls play a preeminent role, but applications of these compounds in the field of nanosciences are recent, and not yet fully explored.4 Typical efficient organic π-conjugated chromophores for SHG are “push–pull” in nature, bearing both electron-withdrawing and electron-donating organic functionalities in direct conjugation with each other, by means of an extended conjugated bridge with delocalized π-electrons.
Binaphthyl compounds have been exploited for the realization of second-order nonlinear optical (NLO) materials, having the combined advantages of being chromophores, and carrying the required element of chirality for bulk anisotropy.5 In fact, several organic molecular modules have been reported in which electron donating substituents in the 2,2′ positions of the binaphthyl skeleton (dialkoxy substituents, such as in compound 1 (ref. 6) in Fig. 1, or dialkylamino substituents, such as in compound 2 (ref. 7) in Fig. 1) have their counterpart in the electron-withdrawing substituents placed at the most electrophilic 6,6′ position (Fig. 1, top). Pyridines, being electron-poor heterocycles, can be ideal “pull” end moieties, when in direct π-conjugation with other “push” functionalities, for the preparation of efficient SHG chromophores: in fact, as an additional feature, they can be coordinated to their nitrogen atom, and their “pull” electronic character is in this case further enhanced.8 This coordination can be either irreversible (covalent functionalization, as in compound 2) or reversible, by means of weak coordination with a metal cation, protonation, supramolecular complexation through halogen bonding.9 The reversible protonation of the pyridinic moiety has recently resulted in interesting switchable emissive and non linear optical (NLO) materials.10 In addition to compound 2, several other pyridine-binaphthyl π-conjugated systems have been reported, branching out from the 3,3′ or 6,6′-positions of the binaphthyl skeleton, for elegant applications in the field of metal–organic frameworks, or metallamacrocycles.11
 | ||
| Fig. 1 Known, representative examples of functionalized binaphthyls for SHG (top), and design of novel 3,3′-binaphthyl disubstituted synthons displaying reversible coordination at the pyridine nitrogen atoms. | ||
The substitution pattern can be amongst the various factors, which include weak intramolecular interactions, buttressing effects by sterically hindered substituents in vicinal positions, and at the supramolecular level, packing effects in crystals, which can have a profound effect on the dihedral angle between the naphthyl units of the binaphthyl moiety. Given our previous synthetic work with binaphthyl compounds derivatized at the 3,3′ positions,12 and our recent realization of crescent, push–pull, PPV-like chromophores,13 we present here the synthesis of novel enantiopure binaphthyls, in which pyridyl substituents branch out from the 3,3′ positions of the binaphthyl skeleton (Fig. 1, bottom). Here we discuss on the chiroptical properties associated with complexation of the pyridine nitrogen atoms to Pd2+ ions and on the modulation of the optical behavior using acidic vapor as an external trigger.
Results and discussion
Synthesis of the molecular modules
In our design strategy (Fig. 1, bottom), the electron-donating –OR substitutents are in an ortho relationship with respect to the growing π-bridge in the 3,3′ position, achieving efficient conjugation with the π-bridge and charge transfer with the electron-withdrawing unit since resonance delocalization of π-electrons is feasible. The synthetic approach makes use of a convergent approach for the instalment of pyridine endcapping functionalities, and of Horner–Wadsworth–Emmons (HWE) olefination reactions for the efficient and highly stereoselective formation of conjugated double bonds within the π-bridge.13aAmongst the two possible retrosynthetic approaches for the construction of the envisaged molecules, we used a difunctional phosphonate binaphthyl derivative (compound 3 in Scheme 1), in combination with π-extended pyridylaldehydes in HWE olefination reactions. In fact, previous work in our laboratories on similar substrates had revealed that the complementary approach, that is, the use of dialdehyde 4 in combination with a π-extended benzyl pyridyl phosphonate is not viable, since decomposition occurs in the presence of the strong bases required for HWE reactions, when carbon–carbon double bonds are present in the π-extended structures.
 | ||
| Scheme 1 Retrosynthetic approaches to the synthesis push–pull π-extended binaphthyls through HWE olefination. | ||
The synthesis of the key compounds is shown in Scheme 2. Enantiopure starting materials for this work were obtained via multistep syntheses starting from the commercially available, enantiopure (R)-BINOL, and they were chemically transformed under nonracemizing conditions.14 Diphosphonate (R)-3 was obtained from known enantiopure dibromide (R)-5via the Arbuzov reaction, and isolated in pure form by column chromatography. Reaction of (R)-3 with commercially-available 4-pyridylaldehyde 6 gave compound (R)-7 using standard HWE conditions (THF, t-ButOK). The compound was obtained as a mixture of diastereoisomers, with ca. 15% of the cis stereoisomer present, which we were not able to purify further by standard chromatography. The obtainment of a nontrivial quantity of the more sterically congested cis stereoisomer might be rationalized by the fact that the electron-withdrawing pyridine moiety, can stack, in this configuration, over the π-rich binaphthyl unit. Elongation of the π-bridge was carried out making use of the acetal-protected phosphonate 8, which gave the extended pyridyl sinthon 9 by HWE with 4-pyridylaldehyde 6. Small amount of cis stereoisomer could in this case be easily separated by standard chromatography. After deprotection under mild acidic conditions, elongated aldehyde 10 was obtained and used without further purifications. Finally, double HWE olefination between 3 and 10 gave stereopure compound (R)-11 in excellent yields. It was fully characterized by 1H, 13C and 2D NMR spectroscopies and mass spectrometry.
 | ||
| Scheme 2 Synthesis of the molecular modules. | ||
The 1H NMR spectrum of compound (R)-11 is shown in Fig. 2 (top). The NMR analysis demonstrated that the double substitution occurred efficiently and confirmed that, within the limit of detection of the NMR technique, both newly formed carbon–carbon double bonds in compound (R)-11 are in the trans configuration exclusively (with 3JHH coupling constants of ca. 16 Hz).
 | ||
| Fig. 2 Top: Partial 1H NMR spectrum (CDCl3, 300 MHz, 25 °C; aromatic region) of compound (R)-11. Selected peaks are assigned. Bottom: Ball-and-stick rendering of the lowest energy conformation of compound (R)-11 (H atoms omitted for clarity). | ||
The attribution of the signals related to the vinyl protons was made possible especially by evaluation of the HETCOR 2D NMR experiment, allowing us to assign the peaks and calculate the values of the coupling constants, which are essential to discriminate between cis and trans disubstituted carbon–carbon double bonds (Fig. S1†).
The lowest energy conformation (Fig. 2, bottom) for molecule (R)-11, obtained by molecular modelling calculations (see below), display an almost perfectly C2 symmetrical structure, with a dihedral angle between the two naphthyl least-squares planes of 85.6°. Interestingly, the conjugated fragments (pydirine-double bond–phenylene-double bond) extending from the 3,3′ positions are fully coplanar, but such planar system is significantly out of the plane of the naphthyl unit to which it is bonded (dihedral angle equal to 31.0°), owing to the steric effects of the neighbouring OCH3 groups in the 2,2′ positions of the binaphthyl skeleton.
The NMR signal at 8.6 ppm (Fig. 2 top), related to the α-pyridyl protons, and, to a lesser extent, the signal at 7.4 ppm, related to the β-pyridyl protons, are broad, indicating that one or more dynamic processes are in place, becoming slow on the NMR timescale at this temperature and for this instrument operational frequency (300 MHz).15 The processes must exchange the positions of the internal and external α-pyridyl protons, so that they are not anymore equivalent by symmetry. These processes can be in principle due to either: (a) the libration of the pyridine fragments around their own main aryl axis, or (b) the rotation of the entire conjugated fragments (pyridine-double bond–phenylene-double bond) around the neighbouring OCH3 groups, that is, from top to bottom with respect to the naphthyl plane.16,17
Optical and chiroptical characterization of the supramolecular polarization using Pd2+
The coordination of the difunctional ligand (R)-11 with Pd2+ was monitored by means of a full titration of the compound with Pd(MeCN)2Cl2. This Pd2+ salt has been previously reported for the formation of complexes in which two pyridine functionalities displace the labile nitrile ligands to form trans-tetracoordinate, square-planar complexes.18 It is fully soluble in polar organic solvents (MeCN, DMF). The UV-vis titration in MeCN is shown in Fig. 3 (top). The shift of the λmax of the ligand (365 nm), attributed to the π–π* transition, towards longer wavelengths (381 nm), is in agreement with a coordination of the transition metal cation to the pyridine nitrogen atoms, and thus a further polarization of the “push–pull” system. The observed stoichiometry at saturation (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ligand vs. Pd2+) matches with the complete incorporation of the pyridine moieties (two per ligand) into the Pd2+ complexes. The behaviour of the binding profile (inset in Fig. 3, top) is sigmoidal, which is not only indicative of multiple equilibria, but also generally associated with positive cooperativity.19 It is likely that the multiple equilibria in play involve the formation of both oligomeric and cyclic species (see below, NMR titration).
:1 ligand vs. Pd2+) matches with the complete incorporation of the pyridine moieties (two per ligand) into the Pd2+ complexes. The behaviour of the binding profile (inset in Fig. 3, top) is sigmoidal, which is not only indicative of multiple equilibria, but also generally associated with positive cooperativity.19 It is likely that the multiple equilibria in play involve the formation of both oligomeric and cyclic species (see below, NMR titration).
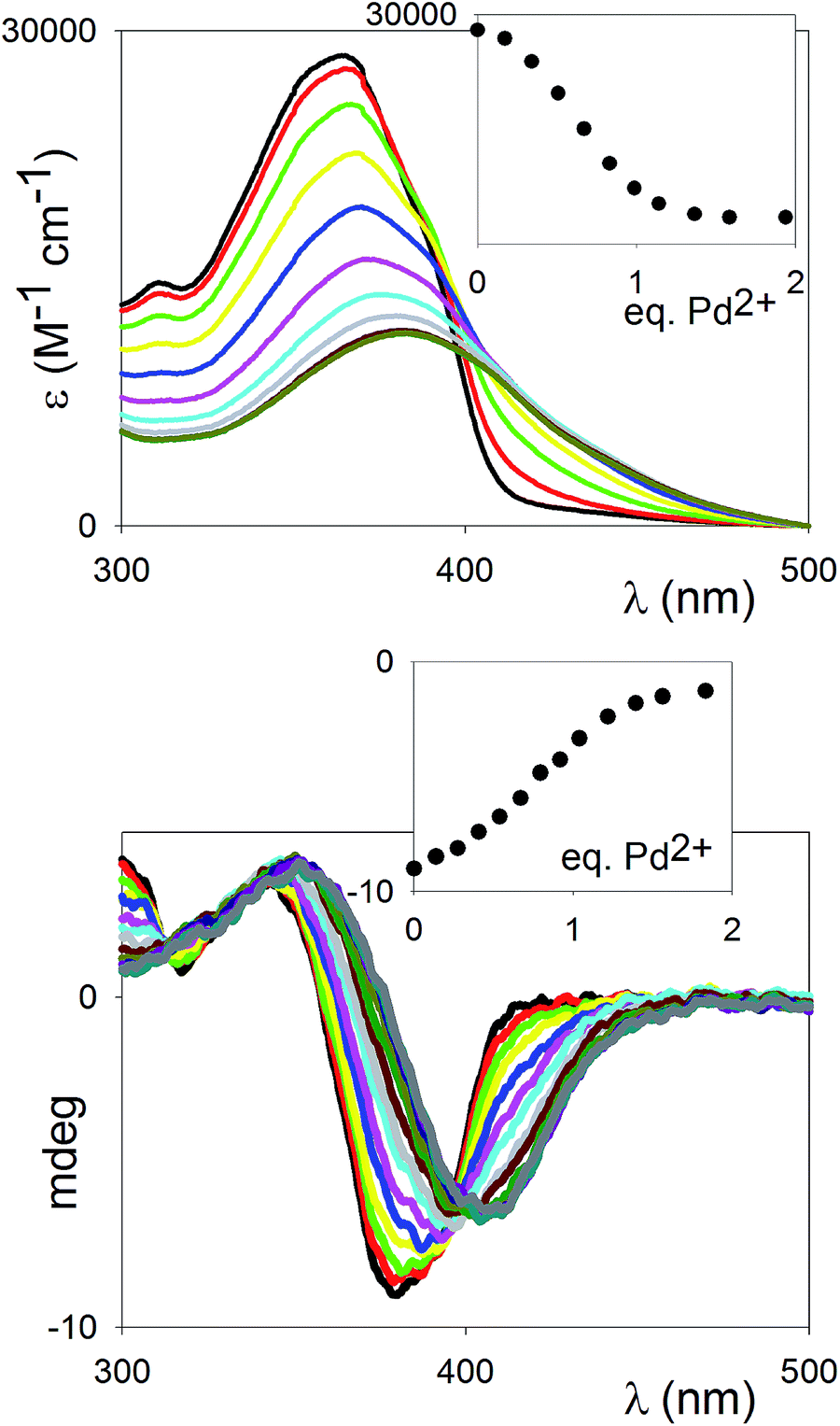 | ||
| Fig. 3 Top: UV titration of ligand (R)-11 (6.65 × 10−5 M) with Pd(MeCN)2Cl2 in MeCN at 25 °C. Inset: titration profile vs. Pd2+ added equivalents at 365 nm. Bottom: CD titration of compound (R)-11 (0.77 × 10−5 M) with Pd(MeCN)2Cl2 in MeCN at 25 °C. Inset: titration profile vs. Pd2+ added equivalents at 380 nm. | ||
A titration experiment in DMF essentially confirmed the behavior observed in MeCN, and differences were found only in the relative intensities of the π–π* band of the free and complexed ligands.
The titration was also monitored by CD spectroscopy (Fig. 3, bottom): an intense exciton couplet signal, classical signature of binaphthyl compounds and due to the two naphthyl chromophores oriented essentially perpendicular to each other, could be observed, with the inversion point corresponding to the λmax of the ligand. The exciton couplet shift towards longer wavelengths upon complexation, and the signal change saturates at ca. 1 eq., in close parallel to the UV-vis titration. The symmetry of the couplet, in terms of intensities of the lowest and highest energy branches, is maintained throughout the titration, pointing to the dominant presence of either uncomplexed or symmetrically dicomplexed species at every stage of the titration, supporting that a highly cooperative binding behaviour is in play.
In order to examine the coordination behavior further, and to confirm the formation of a linear coordination polymer rather than dimeric or oligomeric cyclic species, a 1H NMR titration was performed. The titration could not be made in CD3CN, since using this solvent an insoluble aggregate was formed at the concentrations needed for 1H NMR measurements (at least 10−3 M, two orders of magnitude higher than the UV titrations). Upon addition of Pd2+ (R)-11 in DMF, instead, the aggregate species was completely soluble. The NMR spectra for the titration in d7-DMF are reported in Fig. S2;† general broadening of the signals is observed, with no further changes observed after the addition of 1 eq. of Pd2+, in agreement with the proposed formation of a coordination polymer via pyridine trans coordination to the Pd2+ center. The formation of small quantities of cyclic species, however, cannot be completely ruled out.
Linear and nonlinear optical switching behavior by protonation–deprotonation reaction
The UV-vis absorption spectrum of compound (R)-11 in CHCl3, displays one major band at 367 nm. This band is essentially unshifted in a polar solvent such as CH3CN. By comparison, λmax for 6,6′-disubstituted binaphthyls such as compounds 1 and 2, bearing irreversibly alkylated pyridine nitrogen, is red shifted to a substantial extent (Table 1).The possibility of switching the optical properties of (R)-11via protonation/deprotonation is clearly illustrated in Fig. 4. Upon exposure of the CHCl3 solution of (R)-11 to HCl vapors, the absorption maximum is shifted to 414 nm, a red-shift that can be attributed to protonation of the pyridine moiety with formation of (R)-11·2HCl, in agreement with the red shift observed for the (R)-11·PdCl2 complex. The shift is however considerably larger for the protonated system (47 vs. 16 nm).
 | ||
| Fig. 4 Normalized absorption and emission spectra in CHCl3 of (R)-11, (R)-11·2HCl and (R)-11·2HCl after exposure to NH3 vapors. | ||
The reverse transformation can be induced by treatment of the solution with NH3 vapors, as confirmed by absorption spectroscopy. Interestingly, the protonation–deprotonation process is accompanied by a macroscopic variation in the emissive behaviour. In fact, (R)-11 in CHCl3 displays an intense emission centred at 444 nm which is shifted to 534 nm upon exposure to HCl and restored after treatment with NH3 vapors. A similar interconversion process can be induced by exposure of solid (R)-11 to HCl and ammonia vapors. The initial 560 nm emission of (R)-11 powder is, in fact, shifted to 600 nm after exposure to HCl vapors and restored by treatment with ammonia vapors (Fig. S3†).
The observation of a 50 nm red-shift of the absorption band upon exposure of the CHCl3 solution to HCl vapors prompted us to monitor the second-order NLO properties during the (R)-11/(R)-11·2HCl interconversion. Hyperpolarizabilities of compounds (R)-11 and (R)-11·2HCl in CHCl3 were measured at 1907 nm by electric field-induced second harmonic generation (EFISH)20 and the values are summarized in Table 2.
| Compound | C (M) | μβ in CHCl3 (× 10−48 esu) |
|---|---|---|
| 11 | 10−3 | 170 |
| 11·2HCl | 10−4 | 2000 |
A higher second-order NLO response of (R)-11·2HCl with respect to (R)-11 has been obtained in agreement with the observed absorption red-shift. According to DFT and TDDFT calculations on the doubly protonated form of (R)-11 (see below, Computational studies), the modest variation of the dipole moment from the ground to the excited state of (R)-11·2HCl is not responsible for its observed high second order NLO response compared to (R)-11, which should be instead attributed to both the red shift of the absorption band and the increased oscillator strength. The solid-state interconversion between (R)-11 and (R)-11·2HCl was also monitored by means of the Kurtz–Perry powder method21 at the non-resonant wavelength of 1907 nm. Powdered batches of (R)-11 and (R)-11·2HCl showed SHG efficiency respectively equal and 0.7 times that of standard urea. Low SHG signals for powders, with concomitant high SHG values observed in solution, have been recorded for other binaphthyl residues6 and must be associated with an unfavourable antiparallel or close to antiparallel dipole orientation in the solid state, which is presumably enhanced upon bisprotonation.
Computational studies
DFT and TDDFT calculations on (R)-11 in CHCl3 provided two π → π* excitations at 359 and 357 nm, presenting very high oscillator strengths (f = 2.8 and 1.6, respectively). The former peak is associated principally with HOMO−1 → LUMO and HOMO → LUMO+1 transitions, and the latter to HOMO → LUMO and HOMO−1 → LUMO+1 transitions (see ESI† for a plot of the frontier orbitals). Both pairs of HOMOs and LUMOs are characterized by similar energies but different localization schemes, with HOMO−1 and LUMO+1 more localized on one naphthyl moiety of the molecule, and HOMO and LUMO more localized on the other moiety. As a result, owing to almost symmetrical charge transfers involved in the transitions from one naphthyl moiety to the other one, both absorption peaks are globally characterized by a small charge transfer, as evidenced by a modest increase of the dipole moment from the ground state (μg = 5.33 D) to the excited states (μe = 6.56 and 7.03 D for the first and second peak, respectively).The absorption band of (R)-11·2HCl was well reproduced by DFT and TDDFT calculations on the doubly protonated form of (R)-11 in CHCl3, with two π → π* excitations at 412 and 409 nm (f = 3.0 and 1.4, respectively), very similar in character to those obtained for the neutral compound although characterized by individual transitions with much larger charge transfer (see ESI†). The almost symmetrical charge transfers involved in the transitions from one naphthyl moiety to the other one determine a very small variation of the dipole moment from the ground state (μg = 38.47 D) to the excited states (μe = 34.18 and 34.34 D for the first and second peak, respectively).
CPKS calculations in CHCl3 reproduced well the hyperpolarizability tensor β trend of the neutral and the doubly protonated forms of (R)-11. The vector components along the respective dipole directions were β = 106 and 880 × 10−30 esu, respectively. It is interesting to compare the computed hyperpolarizability of (R)-11 with that of the hypothetical 6,6′-disubstituted compound, analogous to 1 and 2 but functionalized in the same way as (R)-11. CPKS calculations in CHCl3 provided significantly increased hyperpolarizability values (β = 328 and 1540 × 10−30 esu for the neutral and the doubly protonated forms, respectively). Such enhancement should be ascribed to the increased extension of the π-electron delocalized system. In fact, while the dihedral angle between the two naphthyl least-squares planes of this hypothetical compound, 81.5°, was quite similar to that of (R)-11, the conjugated fragment (pydirine-double bond–phenylene-double bond) bonded to the 6,6′ position was essentially coplanar to the naphthyl unit (dihedral angle equal to 5.1°).
Conclusions
We have reported on push–pull binaphthyls of novel conception, in which the π-bridge, bearing at its “pull” end a pyridine unit, extends from the 3,3′-binaphthyl skeleton, bearing alkoxy “push” unit in the 2,2′ positions. These innovative dyes are chiral, securing NLO response, and polarizable through protonation or a coordination event. The HWE olefination methodology has been an efficient tool for the construction of extended, enantiopure binaphthyl derivatives with high isolated yields. The presence of the terminal pyridine units gives the possibility of further polarizing the system, either by weak coordination with a transition metal, or by reversible protonation. In both cases, the observed shift towards lower energies of the absorption major band indicates that the degree of additional polarization achieved is significant. CD spectroscopy helped to elucidate this very highly active CD chromophore, whose characteristic CD couplet shifts in intensity but not in symmetry, as to indicate complete saturation of the coordinating nitrogen atoms of the pyridine ligands. The EFISH values for the system are remarkable, when compared with those obtained for similar systems reported in the literature.Experimental section
General experimental
All available compounds were purchased from commercial sources and used as received. Compounds (R)-5 (ref. 22) and 8 (ref. 23) were prepared as previously described. THF (Na, benzophenone), Et2O (Na, benzophenone) and CH2Cl2 (CaH2) were dried and distilled before use. 1H and 13C NMR spectra were recorded from solutions in CDCl3 on Bruker 200 or AMX300 with the solvent residual proton signal as a standard. Analytical thin layer chromatography was performed on silica gel, chromophore loaded, commercially available plates. Flash chromatography was carried out using silica gel (pore size 60 Å, 230–400 mesh). 1H and 13C NMR spectra were recorded from solutions in CDCl3 on 200–300 MHz or 500 MHz spectrometer with the solvent residual proton signal or tetramethylsilane as a standard. The UV-vis spectroscopic studies were recorded using commercially-available spectrophotometers. Mass spectra were recorded using an electrospray ionization instrument (ESI). Optical rotations were measured on a polarimeter in a 10 cm cell with a sodium lamp (λ = 589 nm) and are reported as follows: [α]rtD (c = mg (mL)−1, solvent). CD spectra were recorded at 25 °C at a scanning speed of 50 nm min−1 and were background corrected. Each spectrum is the instrument average of four consecutive scans. Mass spectra were recorded using an electrospray ionization instrument.Procedure for NMR, UV and CD titrations
The titration experiments were conducted as follows: to a stock solution of chromophore (R)-11 (solution A) were added several aliquots of the Pd2+ salt (solution B). Solution B is formed by the Pd2+ salt (at higher concentration) dissolved in solution A, in order to maintain one species always at the same, constant concentration.Absorption and emission spectroscopy
Electronic absorption spectra were obtained using a Jasco V-530 spectrophotometer. Photoluminescence (PL) measurements were obtained with a SPEX 270 M monochromator equipped with a N2 cooled charge-coupled device exciting either with a monochromated Xe lamp or an Ar+ laser. The spectra were corrected for the instrument response.NLO measurements
EFISH measurements were carried out in CHCl3 and DMF solutions at the concentrations specified in Table 2, at a nonresonant fundamental wavelength of 1907 nm using a Q-switched, mode-locked Nd3+:YAG laser. The 1064 nm initial wavelength was shifted to 1907 nm by a Raman shifter with a high-pressure H2 cell.Computational details
The molecular structures of (R)-11, its bisprotonated form and the analogous 6,6′-disubstituted compounds have been optimized in CHCl3 within the DFT approach, using the PBE0 functional27,28 which has previously been judged well suited for describing the electronic features of a series of organic dyes.29 The 6-311G** basis set was chosen for all atoms. Solvent effects have been taken into account using the conductor-like polarizable continuum model, CPCM.30 Using the PBE0/6-311G** optimized geometries, standard vertical Time Dependent DFT (TDDFT) calculations31–33 have been carried out at the TD-ωB97X/6–311++G** level using the so-called non-equilibrium approach, to determine the absorption wavelengths. The ωB97X34 functional was found to provide the better agreement with the experimental results with respect to other tested functionals (PBE0 and M062X), which gave too high excitation energies in particular for the bisprotonated form. Hyperpolarizabilities have been computed at the same frequency as used in the experiment, using the Coupled Perturbed Kohn Sham (CPKS) approach with the CAM-B3LYP functional,35 which was recently recommended for hyperpolarizability calculations of midsize organic chromophores.36 The reported hyperpolarizability values include the 1/2 factor to be compared with the experimental results. All calculations have been performed with the Gaussian suite of programs.37Acknowledgements
Support from the University of Pavia, MIUR (Programs of National Relevant Interest PRIN grants 2004-033354 and 2009-A5Y3N9), and, from CARIPLO Foundation (2007–2009) and, in part, from INSTM-Regione Lombardia (2010–2012 and 2013–2015) is gratefully acknowledged. D.P. wishes to thank Dr Arvind K. Sharma for experimental assistance, and Prof. Mariella Mella for performing the HETCOR NMR experiment.Notes and references
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS PubMed.
- (a) S. R. Marder, Chem. Commun., 2006, 131–134 RSC; (b) P. A. Sullivan and L. R. Dalton, Acc. Chem. Res., 2010, 43, 10–18 CrossRef CAS PubMed; (c) H. Kang, A. Facchetti, H. Jiang, E. Cariati, S. Righetto, R. Ugo, C. Zuccaccia, A. Macchioni, C. L. Stern, Z. Liu, S. T. Ho, E. C. Brown, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2007, 129, 3267–3286 CrossRef CAS PubMed.
- (a) P. Jonkheijm, P. van der Schoot, A. P. H. J. Schenning and E. W. Meijer, Science, 2006, 313, 80–83 CrossRef CAS PubMed; (b) T. Verbiest, S. Van Elshocht, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, T. Katz and A. Persoons, Science, 1998, 282, 913–915 CrossRef CAS PubMed.
- (a) M. Caricato, A. K. Sharma, C. Coluccini and D. Pasini, Nanoscale, 2014, 7165–7174 RSC; (b) A. Shockravi, A. Javadi and E. Abouzari-Lotf, RSC Adv., 2013, 3, 6717–6746 RSC.
- (a) H.-J. Deussen, E. Hendrickx, C. Boutton, D. Krog, K. Clays, K. Bechgaard, A. Persoons and T. Bjørnholm, J. Am. Chem. Soc., 1996, 118, 6841–6852 CrossRef CAS; (b) G. Koeckelberghs, M. Vangheluwe, I. Picard, L. De Groof, T. Verbiest, A. Persoons and C. Samyn, Macromolecules, 2004, 37, 8530–8537 CrossRef CAS; (c) P. Yan, A. C. Millard, M. Wei and L. M. Loew, J. Am. Chem. Soc., 2006, 128, 11030–11031 CrossRef CAS PubMed; (d) G. Koeckelberghs, T. Verbiest, M. Vangheluwe, L. De Groof, I. Asselberghs, I. Picard, K. Clays, A. Persoons and C. Samyn, Chem. Mater., 2005, 17, 118–121 CrossRef CAS; (e) D. Cornelis, E. Franz, I. Asselberghs, K. Clays, T. Verbiest and G. Koeckelberghs, J. Am. Chem. Soc., 2011, 133, 1317–1327 CrossRef CAS PubMed; (f) G. Yang, Y. Sib and Z. Su, Org. Biomol. Chem., 2012, 10, 8418–8425 RSC.
- H.-J. Deussen, C. Boutton, N. Thorup, T. Geisler, E. Hendrickx, K. Bechgaard, A. Persoons and T. Bjùrnholm, Chem.–Eur. J., 1998, 4, 240–250 CrossRef CAS.
- B. J. Coe, E. C. Harper, K. Clays and E. Franz, Dyes Pigm., 2010, 87, 22–29 CrossRef CAS.
- For supramolecularly polarizable π-conjugated systems: (a) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537–2574 CrossRef CAS PubMed; (b) G. R. Whittell, M. D. Hager, U. S. Schubert and I. Manners, Nat. Mater., 2011, 10, 176–188 CrossRef CAS PubMed; (c) F. Wurthner, J. Schmidt, M. Stolte and R. Wortmann, Angew. Chem., Int. Ed., 2006, 45, 3842–3846 CrossRef PubMed; (d) G. Fabbrini, E. Menna, M. Maggini, A. Canazza, G. Marcolongo and M. Meneghetti, J. Am. Chem. Soc., 2004, 126, 6238–6239 CrossRef CAS PubMed; (e) A. J. Zucchero, P. L. McGrier and U. H. F. Bunz, Acc. Chem. Res., 2010, 43, 397–408 CrossRef CAS PubMed.
- E. Cariati, A. Forni, S. Biella, P. Metrangolo, F. Meyer, G. Resnati, S. Righetto, E. Tordin and R. Ugo, Chem. Commun., 2007, 2590–2592 RSC.
- (a) E. Cariati, C. Botta, S. Danelli, A. Forni, A. Giaretta, U. Giovanella, E. Lucenti, D. Marinotto, S. Righetto and R. Ugo, Chem. Commun., 2014, 14225–14228 RSC; (b) E. Cariati, C. Dragonetti, E. Lucenti, F. Nisic, S. Righetto, D. Roberto and E. Tordin, Chem. Commun., 2014, 1608–1610 RSC.
- (a) C. Gütz, R. Hovorka, C. Stobe, N. Struch, F. Topić, G. Schnakenburg, K. Rissanen and A. Lützen, Eur. J. Org. Chem., 2014, 206–216 CrossRef; (b) C. Gütz, R. Hovorka, C. Stobe, N. Struch, J. Bunzen, G. Meyer-Eppler, Z.-W. Qu, S. Grimme, F. Topić, K. Rissanen, M. Cetina, M. Engeser and A. Lützen, J. Am. Chem. Soc., 2014, 136, 11830–11838 CrossRef PubMed; (c) C.-D. Wu and W. Lin, Inorg. Chem., 2005, 1178–1180 CrossRef CAS PubMed; (d) Y. Cui, H. L. Ngo and W. Lin, Chem. Commun., 2003, 1388–1389 RSC; (e) C.-D. Wua and W. Lin, Dalton Trans., 2006, 4563–4569 RSC.
- (a) A. Bencini, C. Coluccini, A. Garau, C. Giorgi, V. Lippolis, L. Messori, D. Pasini and S. Puccioni, Chem. Commun., 2012, 48, 10428–10430 RSC; (b) C. Coluccini, D. Dondi, M. Caricato, A. Taglietti, M. Boiocchi and D. Pasini, Org. Biomol. Chem., 2010, 8, 1640–1649 RSC; (c) M. Caricato, N. J. Leza, K. Roy, D. Dondi, G. Gattuso, L. S. Shimizu, D. A. Vander Griend and D. Pasini, Eur. J. Org. Chem., 2013, 6078–6083 CrossRef CAS; (d) M. Caricato, A. Olmo, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2012, 68, 7861–7866 CrossRef CAS; (e) M. Caricato, C. Coluccini, D. Dondi, D. A. VanderGriend and D. Pasini, Org. Biomol. Chem., 2010, 8, 3272–3280 RSC; (f) A. Moletti, C. Coluccini, D. Pasini and A. Taglietti, Dalton Trans., 2007, 1588–1592 RSC; (g) C. Coluccini, A. Mazzanti and D. Pasini, Org. Biomol. Chem., 2010, 8, 1807–1815 RSC; (h) S. Colombo, C. Coluccini, M. Caricato, C. Gargiulli, G. Gattuso and D. Pasini, Tetrahedron, 2010, 66, 4206–4211 CrossRef CAS; (i) A. Pacini, M. Caricato, S. Ferrari, D. Capsoni, A. Martínez de Ilarduya, S. Muñoz-Guerra and D. Pasini, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4790–4799 CrossRef CAS; (j) M. Caricato, S. Díez González, I. Arandia Ariño and D. Pasini, Beilstein J. Org. Chem., 2014, 10, 1308–1316 CrossRef PubMed; (k) M. Caricato, N. J. Leza, C. Gargiulli, G. Gattuso, D. Dondi and D. Pasini, Beilstein J. Org. Chem., 2012, 8, 967–976 CrossRef CAS PubMed; (l) M. Agnes, A. Sorrenti, D. Pasini, K. Wurst and D. B. Amabilino, CrystEngComm, 2014, 16, 10131–10138 RSC; (m) M. Crespo Alonso, M. Arca, F. Isaia, R. Lai, V. Lippolis, S. K. Callear, M. Caricato, D. Pasini, S. J. Coles and M. C. Aragoni, CrystEngComm, 2014, 16, 8582–8590 RSC; (n) M. Caricato, A. Deforge, D. Bonifazi, D. Dondi, A. Mazzanti and D. Pasini, Org. Biomol. Chem., 2015 10.1039/c4ob02643h.
- (a) C. Coluccini, A. K. Sharma, M. Caricato, A. Sironi, E. Cariati, S. Righetto, E. Tordin, C. Botta, A. Forni and D. Pasini, Phys. Chem. Chem. Phys., 2013, 15, 1666–1674 RSC; (b) M. Caricato, C. Coluccini, D. A. Vander Griend, A. Forni and D. Pasini, New J. Chem., 2013, 37, 2792–2799 RSC; (c) D. Pasini, P. P. Righetti and V. Rossi, Org. Lett., 2002, 4, 23–26 CrossRef CAS PubMed; (d) L. Garlaschelli, I. Messina, D. Pasini and P. P. Righetti, Eur. J. Org. Chem., 2002, 3385–3392 CrossRef CAS; (e) C. Coluccini, P. Metrangolo, M. Parachini, D. Pasini, G. Resnati and P. Righetti, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5202–5213 CrossRef CAS; (f) C. Coluccini, D. Pasini, P. Righetti and D. A. Vander Griend, Tetrahedron, 2009, 65, 10436–10440 CrossRef CAS.
- H. T. Stock and R. M. Kellogg, J. Org. Chem., 1996, 61, 3093–3105 CrossRef CAS PubMed.
- Other 4-pyridyl-vinylene conjugated systems, similarly to compound (R)-11, show broad α-pyridyl protons at room temperature. See: C. Stangel, A. Bagaki, P. A. Angaridis, G. Charalambidis, G. D. Sharma and A. G. Coutsolelos, Inorg. Chem., 2014, 53, 11871–11881 CrossRef CAS PubMed.
- The low solubility of compound 11 in appropriate solvents for low temperature NMR analysis (e.g., THF, see ref. 12g) prevented us from a more detailed dynamic of the system. Additionally, a thorough investigation of such dynamic behaviour will require the synthesis of a series of model compounds (such as the naphthyl analogue of compound 11, in which the chiral axis is not present) and not, which is outside the purpose of this paper.
- The exchange method allows to calculate values for the rate constant kex of the exchange process from NMR analysis, J. Sandström, Dynamic NMR Spectroscopy, Academic Press, London, 1982, ch. 6, from the approximate expression kex = π(Δν), where Δν is the difference (Hz) between the line width at the temperature Tex, where exchange of sites occurs, and the line width in the absence of exchange (which in our case was assumed the value of the sharp H4-bin signal at 8.3 ppm in Fig. 2). A value of kex = 16 Hz was calculated in our case, which translates, by employing the Eyring equation, into a ΔG≠ = 12 kcal mol−1 value for the energy barrier.
- For examples of pyridine moieties forming trans PdCl2 coordination complexes, see: (a) Z. Qin, M. C. Jennings and R. J. Puddephatt, Inorg. Chem., 2001, 40, 6220–6228 CrossRef CAS PubMed; (b) L. Pirondini, D. Bonifazi, E. Menozzi, E. Wegelius, K. Rissanen, C. Massera and E. Dalcanale, Eur. J. Org. Chem., 2001, 2311–2320 CrossRef CAS; (c) Y. S. Chong, M. D. Smith and K. D. Shimizu, J. Am. Chem. Soc., 2001, 123, 7463–7464 CrossRef CAS PubMed; (d) M. T. Stone and J. S. Moore, J. Am. Chem. Soc., 2005, 127, 5928–5935 CrossRef CAS PubMed; (e) G. Giachi, M. Frediani, W. Oberhauser and E. Passaglia, J. Polym. Sci., Polym. Chem. Ed., 2011, 49, 4708–4713 CrossRef CAS.
- (a) K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, Wiley, New York, NY, 1987. See also: Search PubMed; (b) G. Fernández, L. Sánchez, E. M. Pérez and N. Martín, J. Am. Chem. Soc., 2008, 130, 10674–10683 CrossRef PubMed; (c) L. D. Byers, J. Chem. Educ., 1977, 54, 352–354 CrossRef CAS; (d) G. Ercolani, J. Am. Chem. Soc., 2003, 125, 16097–16103 CrossRef CAS PubMed; (e) J. Hamacek and C. Piguet, J. Phys. Chem. B, 2006, 110, 7783–7792 CrossRef CAS PubMed.
- (a) D. Roberto, R. Ugo, S. Bruni, E. Cariati, F. Cariati, P. Fantucci, I. Invernizzi, S. Quici, I. Ledoux and J. Zyss, Organometallics, 2000, 19, 1775–1788 CrossRef CAS; (b) I. Ledoux and J. Zyss, Chem. Phys., 1982, 73, 203–213 CrossRef CAS; (c) K. D. Singer and A. F. Garito, J. Chem. Phys., 1981, 75, 3572–3580 CrossRef CAS.
- S. K. Kurtz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798 CrossRef CAS.
- C. Coluccini, A. Castelluccio and D. Pasini, J. Org. Chem., 2008, 73, 4237–4240 CrossRef CAS PubMed.
- C. Coluccini, A. K. Sharma, D. Merli, D. Vander Griend, B. Mannucci and D. Pasini, Dalton Trans., 2011, 40, 11719–11725 RSC.
- R. C. Helgeson, G. R. Weisman, J. R. Toner, T. L. Tarnowski, Y. Chao, J. M. Mayer and D. J. Cram, J. Am. Chem. Soc., 1979, 101, 4928–4941 CrossRef CAS.
- Y. Cui, H. L. Ngo and W. Lin, Chem. Commun., 2003, 1388–1389 RSC.
- M. I. Ranasinghe, P. Murphy, Z. Lu, S. D. Huang, R. J. Twieg and T. Goodson III, Chem. Phys. Lett., 2004, 383, 411–417 CrossRef CAS.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- (a) D. Jacquemin, E. A. Perpète, G. E. Scuseria, I. Ciofini and C. Adamo, J. Chem. Theory Comput., 2008, 4, 123–135 CrossRef CAS; (b) D. Jacquemin, V. Wathelet, E. A. Perpète and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420 CrossRef CAS.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- M. E. Casida, J. Mol. Struct.: THEOCHEM, 2009, 914, 3–18 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, J. Chem. Phys., 2008, 128, 084106–084115 CrossRef PubMed.
- T. Yanai, D. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- L. E. Johnson, L. R. Dalton and B. H. Robinson, Acc. Chem. Res., 2014, 47, 3258–3265 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional NMR spectra, absorption and emission spectra, and computational details, copies of NMR and mass spectra of newly synthesized compounds. See DOI: 10.1039/c4ra16876c |
| This journal is © The Royal Society of Chemistry 2015 |