
A novel lithium-ion hybrid capacitor based on an aerogel-like MXene wrapped Fe2O3 nanosphere anode and a 3D nitrogen sulphur dual-doped porous carbon cathode†
Xiao
Tang
a,
Hao
Liu
 a,
Xin
Guo
a,
Shijian
Wang
b,
Wenjian
Wu
b,
Anjon Kumar
Mondal
a,
Chengyin
Wang
c and
Guoxiu
Wang
*a
a,
Xin
Guo
a,
Shijian
Wang
b,
Wenjian
Wu
b,
Anjon Kumar
Mondal
a,
Chengyin
Wang
c and
Guoxiu
Wang
*a
aCentre for Clean Energy Technology, Faculty of Science, University of Technology Sydney, Sydney, NSW 2007, Australia. E-mail: guoxiu.wang@uts.edu.au
bDepartment of Materials Science and Engineering, Dongguan University of Technology, Dongguan, China
cCollege of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, China
First published on 18th July 2018
Abstract
Lithium-ion capacitors (LICs) have emerged as promising energy storage devices with both high energy density and high power density. However, due to the mismatch of charge-storage capacity and electrode kinetics between battery-type anodes and capacitor-type cathodes, the application of lithium-ion capacitors has been limited. In this work, interconnected aerogel-like MXene wrapped Fe2O3 nanospheres have been prepared and investigated as battery-type anode materials for lithium-ion capacitors. In this rationally designed hybrid electrode, the Ti3C2Tx MXene matrix is capable of providing fast transport of electrons and suppressing the volume change of Fe2O3. Simultaneously, Fe2O3 hollow nanospheres offer large specific capacity and prevent restacking of the MXene layers, synergizing to boost the electrochemical performances of such hybrid electrodes. Meanwhile, the three-dimensional (3-D) nitrogen and sulphur dual-doped porous carbon (NS-DPC) derived from biomass has also been fabricated as a capacitor-type cathode material for lithium-ion capacitors. Consequently, the lithium-ion capacitors can demonstrate a high energy density of 216 W h kg−1 at a power density of 400 W kg−1 and a high power density of 20 kW kg−1 at an energy density of 96.5 W h kg−1. This work elucidates that both high energy density and power density can be achieved in hybrid lithium-ion capacitors.
Introduction
With the soaring demand for portable electronic devices, hybrid electric vehicles, and large-scale electricity grid storage, future energy-storage devices are required to possess features of high energy density, high power density and superior cycling stability.1 Lithium-ion batteries (LIBs) can provide a high energy density of ∼150 W h kg−1.2 Nevertheless, intrinsically slow solid-state diffusion and volumetric strain lead to low power density. In contrast, supercapacitors (SCs) are able to deliver a high power density of 2–5 kW kg−1 and an excellent cycling performance. However, as charges are only stored on the surface of the active material, SCs usually suffer from low energy density (3–6 W h kg−1). Hence, the development of new energy-storage devices with features of both LIBs and SCs is highly desirable.3,4Recently, lithium-ion capacitors (LICs) have been proposed to bridge the gap between LIBs and SCs, and to deliver higher power density than LIBs and higher energy density than SCs. LICs have previously been constructed by an LIB-type anode with large capacity, an SC-type cathode with superior rate capability and nonaqueous Li-salt-containing electrolyte that can provide a wide working voltage window.2–4 So far, in order to optimize the electrochemical performance of LICs, various electrode materials have been developed.5 Many carbonaceous materials, including active carbon,3,6 graphene,7–9 dual-doped carbon nanofibers10 and metal–organic-framework derived carbon, have been reported as cathodes for LICs. In addition, for LIB-type anodes, insertion-type materials (TiO2,7,11–13 Nb2O5,14 FeOOH,15 Li4Ti5O12,16–18 and graphite,19–22etc.), conversion-type materials (such as MnO, F-doped Fe2O323 and Fe3O4,8etc.) and alloy-type materials (including Sn-encapsulated nitrogen-rich CNT,24 Si/Cu,25etc.) have been utilized to realize high-performance LICs with both high energy density and power density.
Fe2O3 has attracted tremendous attention due to its high specific capacity, low voltage plateau, low-cost and eco-friendliness.26–29 Nevertheless, the poor ionic/electronic conductivities cause sluggish transport of lithium ions and electrons, therefore leading to inferior rate performance. In addition, huge volume expansion during charge and discharge processes can result in pulverization of the electrode and short cycle life. Particularly, the lack of a continuous conductive matrix and electron transport pathway usually leads to low utilization of active materials.28
Significant efforts have been devoted to preparing nanosized Fe2O3 hollow spheres as electrode materials, which is an effective method to shorten the diffusion pathway during charge/discharge processes.30 However, the low electrical conductivity and inferior cycling stability are still the crucial concerns for Fe2O3-based materials as high-performance electrodes for lithium-ion capacitors.31 Therefore, it is highly desirable to fabricate Fe2O3 composite electrodes with high capacity, good rate performance and long cycling stability.
Several conductive matrices including graphene,28,32 mesoporous carbon33–35 and polymers30,36 were adopted to combine with Fe2O3. Ti3C2Tx MXene (Tx represents the surface functional groups, such as –OH, –F, or ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) with an aerogel-like architecture is emerging as a promising conductive matrix to support Fe2O3. The aerogel-like MXene wrapped Fe2O3 nanosphere (noted as Ti3C2Tx@Fe2O3) nanocomposites have several advantages: (i) Ti3C2Tx MXene, which exhibits excellent conductivity, not only acts as an interconnected network to load Fe2O3 and provide an efficient electron transport pathway, but also imparts some charge storage capacity for the whole composite, since Ti3C2Tx MXene itself is one of the pseudocapacitive anodes for LICs.37–40 (ii) Fe2O3 can be adsorbed onto the aerogel-like Ti3C2Tx MXene with a porous structure and large surface area via ion-exchange and electrostatic interactions, therefore suppressing the detachment of Fe2O3 from the matrix during electrochemical reactions. (iii) MXene layers are capable of confining the volume change of Fe2O3 during charge/discharge processes, avoiding pulverization. Meanwhile, Fe2O3 anchored on the surface of MXene plays a role as a spacer, which can prevent restacking of the MXene layers induced by van der Waals interaction and hydrogen bonds. Thus, the “pillar effect” of Fe2O3 between Ti3C2Tx MXene layers and synergistic effects between the Fe2O3 and the Ti3C2Tx MXene matrix together endow the nanocomposite with excellent electrochemical performances as a promising electrode material for lithium-ion capacitors.
O) with an aerogel-like architecture is emerging as a promising conductive matrix to support Fe2O3. The aerogel-like MXene wrapped Fe2O3 nanosphere (noted as Ti3C2Tx@Fe2O3) nanocomposites have several advantages: (i) Ti3C2Tx MXene, which exhibits excellent conductivity, not only acts as an interconnected network to load Fe2O3 and provide an efficient electron transport pathway, but also imparts some charge storage capacity for the whole composite, since Ti3C2Tx MXene itself is one of the pseudocapacitive anodes for LICs.37–40 (ii) Fe2O3 can be adsorbed onto the aerogel-like Ti3C2Tx MXene with a porous structure and large surface area via ion-exchange and electrostatic interactions, therefore suppressing the detachment of Fe2O3 from the matrix during electrochemical reactions. (iii) MXene layers are capable of confining the volume change of Fe2O3 during charge/discharge processes, avoiding pulverization. Meanwhile, Fe2O3 anchored on the surface of MXene plays a role as a spacer, which can prevent restacking of the MXene layers induced by van der Waals interaction and hydrogen bonds. Thus, the “pillar effect” of Fe2O3 between Ti3C2Tx MXene layers and synergistic effects between the Fe2O3 and the Ti3C2Tx MXene matrix together endow the nanocomposite with excellent electrochemical performances as a promising electrode material for lithium-ion capacitors.
Herein, we report the successful fabrication of an interconnected aerogel-like architecture by distributing porous Fe2O3 hollow nanospheres onto the aerogel-like Ti3C2Tx MXene via a surfactant. The Ti3C2Tx@Fe2O3 hybrid electrodes achieve a large specific capacity and an excellent rate performance. We also explored the capacity contribution from diffusion-controlled and pseudocapacitive currents in the hybrid electrode. Furthermore, three-dimensional (3-D) nitrogen and sulphur dual-doped porous carbon (NS-DPC) has also been prepared as a capacitor-type cathode material for lithium-ion capacitors. Consequently, a 4 V lithium-ion capacitor based on the aerogel-like MXene wrapped Fe2O3 nanosphere anode and the 3-D NS-DPC cathode was assembled, exhibiting a high energy density of 216 W h kg−1 at a power density of 400 W kg−1 and a high power density of 20 kW kg−1 at an energy density of 96.5 W h kg−1.
Experimental section
Synthesis of Fe2O3 hollow nanospheres
The Fe2O3 hollow nanospheres were prepared according to the previously reported method.41 In a typical procedure, 0.42 g of Na4Fe(CN)6, 0.04 g of hexadecyltrimethyl ammonium bromide (CTAB), 0.18 g of (NH4)2S2O8 and 0.18 g of NaH2PO4 were dissolved in 100 mL distilled water. After 30 minutes of magnetic stirring at room temperature, a yellow transparent solution was obtained and then transferred into a Teflon-lined autoclave, followed by a hydrothermal treatment at 180 °C for 8 hours. After cooling down to room temperature, the products were rinsed several times with distilled water and dried at 80 °C overnight. Finally, the pristine Fe2O3 hollow nanospheres can be obtained by annealing the as-prepared products in an Ar atmosphere at 450 °C for 2 hours.Synthesis of aerogel-like Ti3C2Tx MXene
Ti3AlC2 with a particle size of <38 μm was synthesized according to a previous report.42 Then, 3 g of Ti3AlC2 powder was etched in a mixture of lithium fluoride (LiF, 3 g, Alfa Aesar) and 9 M hydrochloric acid (HCl, 30 mL, Fisher Scientific) at ∼35 °C for 24 hours to extract the Al atoms and obtain a multilayered Ti3C2Tx suspension. The obtained suspension was rinsed several times with distilled water and centrifuged (3500 rpm) until the pH of the supernatant was >5. To prepare aerogel-like Ti3C2Tx, distilled water was added into the as-prepared suspension with a weight ratio of Ti3C2Tx to water of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100. Then, the diluted suspension was sonicated under an Ar atmosphere for 1 hour and frozen by liquid nitrogen, followed by a freeze-dry process to obtain aerogel-like Ti3C2Tx.
:100. Then, the diluted suspension was sonicated under an Ar atmosphere for 1 hour and frozen by liquid nitrogen, followed by a freeze-dry process to obtain aerogel-like Ti3C2Tx.
Synthesis of an aerogel-like MXene wrapped Fe2O3 nanosphere nanocomposite
0.7 g Fe2O3 and 0.05 g cetyltrimethylammonium bromide (CTAB) were mixed in 20 mL distilled water. After 30 minutes of sonication, a red solution denoted as solution A can be obtained. Then, 0.3 g aerogel-like Ti3C2Tx was distributed in 10 mL distilled water to prepare solution B. Solution A (CTAB-grafted Fe2O3) was added dropwise into solution B under stirring. The mixture was sonicated for 30 minutes and frozen by liquid nitrogen, followed by a freeze-dry process to obtain an aerogel-like Ti3C2Tx@Fe2O3 nanocomposite. The mass loading ratio of Fe2O3 in the composites was around 70 wt%.Synthesis of 3-D nitrogen and sulphur dual-doped porous carbon (NS-DPC)
As illustrated in Scheme S1 (ESI†), Sterculia lychnophora were first soaked in warm distilled water until a spongy precursor was obtained. Then, the precursor was frozen by liquid nitrogen, followed by a freeze-dry process to obtain cryo-dried Sterculia lychnophora, which were further ground to a powder by using a household blender. The obtained powders (∼4 g) were mixed in a mortar with KHCO3 in a mass ratio of 1:4. The mixed powders were calcined at 800 °C for 5 hours under an argon atmosphere in a tube furnace. After carbonization, the as-prepared products were washed several times with 1 M HCl and distilled water and then dried at 80 °C overnight under vacuum to obtain 3-D nitrogen and sulphur dual-doped porous carbon materials (NS-DPC).
Characterization
The XRD patterns of the as-prepared samples were collected by using a Bruker D8 discover XRD with Cu Kα radiation (40 kV and 40 mA) and a step scan of 0.02°, 5°–80° 2θ ranges and a step time of 0.5 s. Raman spectra were measured using a Renishaw inVia Raman spectrometer system (Gloucestershire, UK) equipped with a Leica DML Bmicroscope (Wetzlar, Germany) and a 17 mW at 633 nm Renishaw helium neon laser source. The specific surface area was determined by the gas sorption technique using a Micromeritics 3Flex analyser based on the Brunauer–Emmett–Teller (BET) method. X-ray photoelectron spectroscopy (XPS) measurements were performed on a Kratos XSAM-800 spectrometer with an Mg Kα radiation source. The morphology of the samples was observed using a scanning electron microscope (SEM) (Zeiss Supra 55VP, Germany) and a transmission electron microscope (TEM) (JEOL JEM-2011, Japan) with an accelerating voltage of 200 kV.Electrochemical tests
The Ti3C2Tx@Fe2O3 hybrid anodes/porous carbon cathodes were prepared by mixing 80% active materials, 10% carbon black, and 10% PVDF used as a binder to form a slurry, which was then coated on copper/aluminium foil, followed by drying at 60 °C in a vacuum oven overnight. For anode tests, half-cells were assembled using Ti3C2Tx@Fe2O3 hybrid anodes and Li foils in 2032 type stainless steel coin cells, where the electrolyte was 1.0 M LiPF6 dissolved in ethylene carbonate and diethyl carbonate (EC/DEC, v/v = 1:1). For cathode tests, half-cells were assembled using a porous carbon cathode and Li foils in 2032 type stainless steel coin cells while other conditions remain the same. Galvanostatic charge–discharge measurements were performed on a LAND battery test system. CV and EIS measurements were conducted on an electrochemical workstation (CHI 660D). All the tests were carried out at room temperature. The mass loadings of the Ti3C2Tx@Fe2O3 hybrid and porous carbon electrodes were about 1.2 and 2 mg cm−2, respectively. Prelithiated Ti3C2Tx@Fe2O3 electrodes were selected for the anodes and porous carbon electrodes were applied as cathodes with a cathode/anode mass ratio of ∼5:1. During the prelithiation process, the anode was directly put in contact with the Li foil in the same electrolyte for 12 hours. Then, the anode was washed with DEC and shifted to the assembly of coin cell-type hybrid capacitors.
Results and discussion
As illustrated in Scheme 1a, Ti3C2Tx MXene was prepared via selectively etching Al atoms, during which functional groups (such as –OH, –F, orO) were attached on the surface of the MXene.38,39 Subsequently, a freeze-drying process was adopted to swell the MXene layers to form an aerogel-like MXene matrix. Meanwhile, the BET surface area was increased from 126 m2 g−1 (etched-Ti3C2Tx) to 367 m2 g−1 (aerogel-like Ti3C2Tx) (shown in Fig. S1, ESI†). However, due to the van der Waals interaction and hydrogen bonding, MXene layers tend to restack again and then lead to reduced surface area.40,43,44 Hence, Fe2O3 hollow nanospheres consisting of numerous subcomponent nanoparticles with a size of 5–10 nm (as shown in Fig. S2, ESI†) were introduced as “pillars” to prevent the restacking of the MXene layers. As illustrated in Scheme 1b, hollow nanospheres tend to collapse, which is caused by volume change during charge/discharge processes without the MXene protection. In contrast, when anchored on the MXene matrix, the volume change of the Fe2O3 hollow nanospheres can be confined by the MXene matrix and the hollow structure of the Fe2O3 nanospheres can buffer the volume change. Thus, the aerogel-like Ti3C2Tx@Fe2O3 nanocomposite is expected to possess satisfactory cycling performance owing to the “pillar effect”.
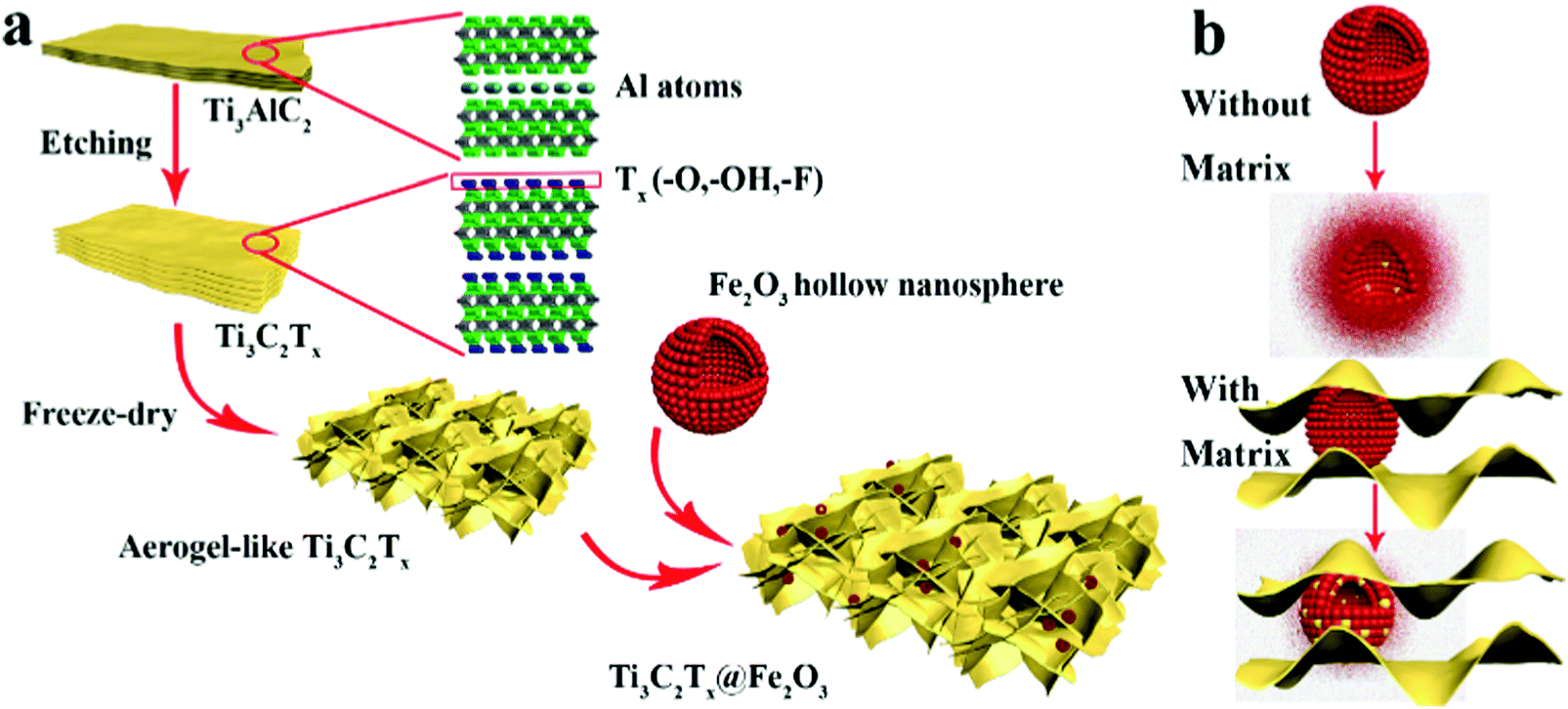 | ||
| Scheme 1 (a) Schematic illustration of the synthesis procedure for the Ti3C2Tx@Fe2O3 nanocomposite. (b) The mechanism of the MXene matrix protection in the Ti3C2Tx@Fe2O3 nanocomposite. | ||
X-ray diffraction (XRD) analysis was performed to investigate the phase of the as-prepared samples. Fig. 1a shows the XRD patterns of the obtained Fe2O3, Ti3C2Tx and Ti3C2Tx@Fe2O3 nanocomposites. In the XRD pattern of the Ti3C2Tx@Fe2O3, the peak at around 7° can be ascribed to the (0001) plane of Ti3C2Tx MXene, suggesting the existence of multilayer sheets in the as-prepared Ti3C2Tx MXene,45–47 whereas other diffraction peaks of the Ti3C2Tx@Fe2O3 are fitted well with hematite (JCPDS card no. 33-0664).27 It is worth noticing that no impurity (such as titanium oxide, etc.) is observed, indicating the successful synthesis of the Ti3C2Tx@Fe2O3 composite without oxidation of Ti3C2Tx. To further confirm the phase and composition of the Ti3C2Tx@Fe2O3 composite, Raman spectra were measured (in Fig. 1b). Since the hematite belongs to the D63d crystal space group, two peaks located at 218 and 282 cm−1 can be indexed to A1g and Eg bands, respectively.48 In addition to two peaks from Fe2O3, the other two peaks at 201 and 380 cm−1 correspond to specific Ti3C2Tx MXene bands.49 No peak shift is observed after combination of Fe2O3 and Ti3C2Tx, indicating the good phase stability of the composite. X-ray photoelectron spectrum (XPS) measurements were also conducted to study the chemical composition and oxidation state of the Ti3C2Tx@Fe2O3 composite. The survey-scan XPS spectrum in Fig. S3 (ESI†) clearly shows the Fe, Ti, C and O signals in the composite sample. As shown in Fig. 1c, the Fe 2p core level XPS spectrum of the Ti3C2Tx@Fe2O3 composite shows two main peaks at 711.4 and 725 eV and two satellite peaks at 719.0 and 732.2 eV, which can be assigned to the characteristic peak of Fe3+ in hematite.28,50,51Fig. 1d shows the high-resolution XPS spectrum of the Ti 2p core level. Two doublets at 455.2 and 458.5 eV correspond to Ti 2p3/2, while another two doublets at 461.2 and 464.1 eV correspond to Ti 2p1/2. For the Ti 2p3/2, there are three deconvoluted peaks at 455.2, 456.9 and 458.5 eV, corresponding to Ti–C bonds, Ti–X bonds and Ti–O bonds on the surface, respectively.52 Similarly, for Ti 2p1/2, three deconvoluted peaks at 461.2, 462.6 and 464.1 eV are observed. Fig. 1e shows the C 1s spectrum, which can be deconvoluted into five peaks at 281.7, 284.8, 286.3, 288.3 and 289.2 eV. The peak at 281.7 eV can be assigned to the C–Ti bond. The peak at 284.8 eV and 286.3 eV can be assigned to C–C and C–O, respectively. The other two peaks situated at 288.3 and 289.2 eV, can be indexed to the signals of CO and O–CO/C–F, respectively.53Fig. 1f shows the O 1s spectrum, which can be deconvoluted into four peaks centred at 529.8, 531.2, 532.8 and 533.1 eV, corresponding to Fe–O, Ti–Ox, Ti–OH and H–O (hydroxide), respectively.54 Thus, the results of XRD, Raman, and XPS spectra convincingly confirm the successful synthesis of Ti3C2Tx@Fe2O3 nanocomposites.
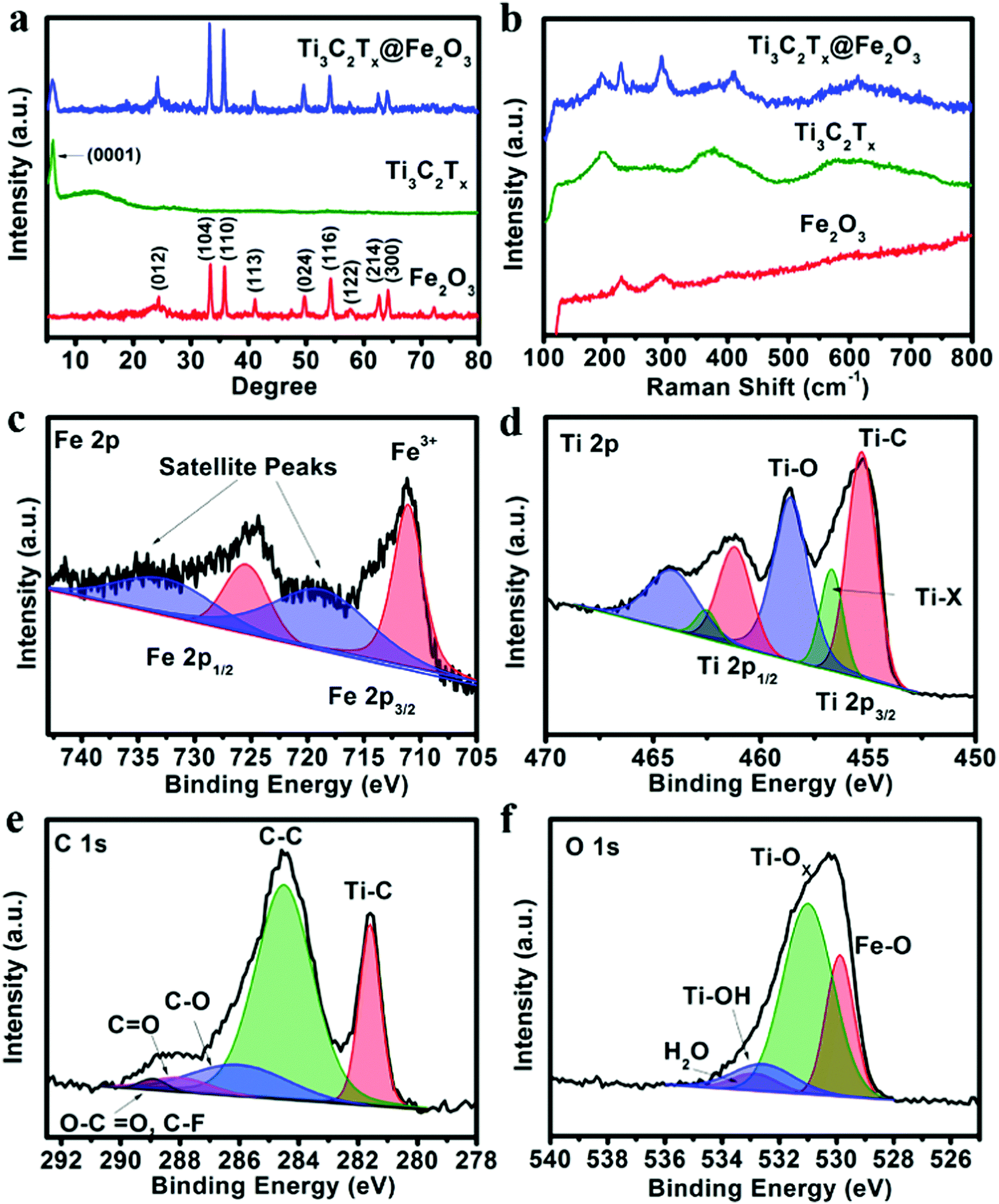 | ||
| Fig. 1 (a) XRD patterns and (b) Raman spectra of the Fe2O3, Ti3C2Tx and Ti3C2Tx@Fe2O3 samples. (c) Fe 2p, (d) Ti 2p, (e) C 1s, and (f) O 1s XPS spectra of the Ti3C2Tx@ Fe2O3 nanocomposite. | ||
The morphologies and crystal structures of Ti3C2Tx@Fe2O3 were further characterized by SEM and TEM. As shown in Fig. 2a, Fig. S2 and S4 (ESI†), the Ti3C2Tx@Fe2O3 composite inherits the morphological characteristics of aerogel-like MXene and porous Fe2O3 hollow nanospheres with a large amount of nanospheres well-distributed in the pores of the aerogel-like MXene, therefore facilitating electron transportation through the interconnected highly-conductive MXene networks. Furthermore, Fig. 2b shows the high-resolution SEM image of the Ti3C2Tx@Fe2O3 composite. The Fe2O3 nanospheres with an average size of 200 nm are observed to be tightly wrapped by few-layer MXene sheets, which is also confirmed by the TEM image (as shown in Fig. 2c). Fig. S5 (ESI†) demonstrates the EDS spectrum of the as-prepared Ti3C2Tx@Fe2O3 composite, confirming the co-existence of Fe, Ti, C, O and F elements. The HRTEM image (Fig. 2d) of the selected area (marked by red circle) in Fig. 2c reveals an interlayer space of about 1 nm, suggesting that the MXene sheets are multilayer. Fig. 2e shows an HRTEM image taken from the selected region (marked by red box) of the Ti3C2Tx@Fe2O3 composite in Fig. 2c. An interplanar lattice fringe of 0.27 nm is detected, which can be attributed to the (104) plane of hematite, again confirming the successful loading of Fe2O3 nanospheres. Moreover, scanning transmission electron microscopy (STEM) analysis is able to provide more information on the detailed morphology and elemental distribution of the as-prepared Ti3C2Tx@Fe2O3 composite. As depicted in Fig. 2f–k, the distributions of the Fe, Ti, O, C and F elements are highly consistent with the contour of the Ti3C2Tx@Fe2O3 composite. Notably, the contour of the Ti element map is observed to cover the contour of the Fe element map, illustrating the evenly distributed Fe2O3 nanospheres on the MXene matrix.
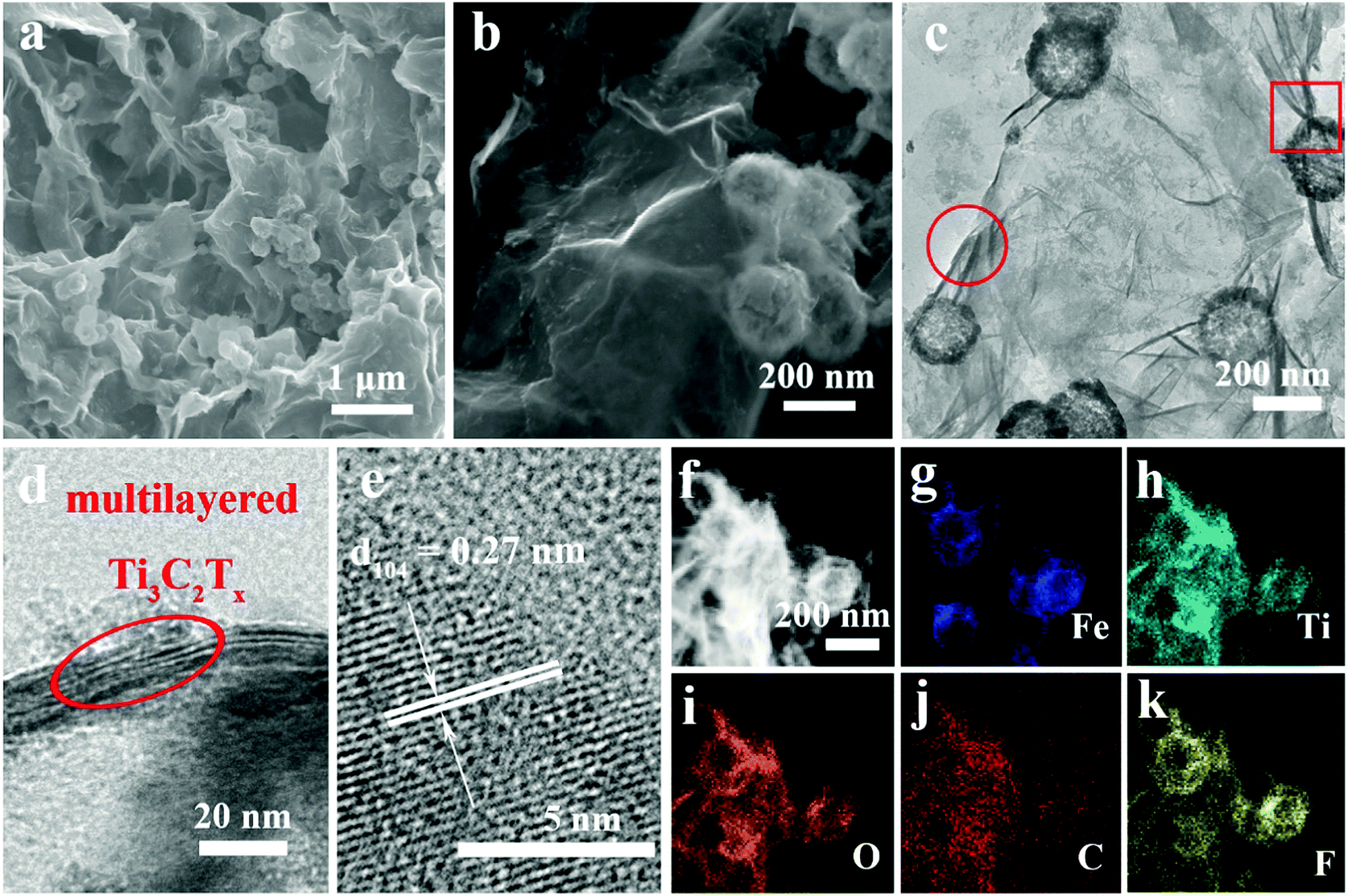 | ||
| Fig. 2 (a and b) SEM images of the aerogel-like Ti3C2Tx@Fe2O3 composite. (c) TEM image of the aerogel-like Ti3C2Tx@Fe2O3 composite. (d and e) HRTEM images of the Ti3C2Tx@Fe2O3 composite. The circle and box in panel (c) correspond to the HRTEM images for the selected area of panel (d) and (e), respectively. (f–k) Elemental mapping of the Ti3C2Tx@Fe2O3 composite. | ||
Owning to the synergistic effect of the highly conductive MXene matrix and porous Fe2O3 hollow nanospheres, the Ti3C2Tx@Fe2O3 anode is expected to exhibit improved rate and cycling performance. As shown in Fig. 3a, a high initial discharge capacity of 1645 mA h g−1 and a charge capacity of 1201 mA h g−1 are achieved at a current density of 0.1 A g−1, corresponding to an initial coulombic efficiency of 73%, which can be ascribed to the formation of a solid electrolyte interphase (SEI) layer. This irreversible capacity loss only occurred in the first few cycles and the coulombic efficiency increased to ∼100% in subsequent cycles. Fig. 3b and Fig. S6 (ESI†) presents the rate performance of the Ti3C2Tx@Fe2O3 anode at different current densities from 0.1 A g−1 to 5 A g−1. It can deliver 1180, 1031, 910, 745 and 625 mA h g−1 at the current densities increasing from 0.1 to 2 A g−1, respectively. Even at a current density of 5 A g−1, a specific capacity of 493 mA h g−1 is retained, demonstrating an enhanced rate performance of the Ti3C2Tx@Fe2O3 anode. This value is markedly higher than those of other reported anode materials, such as 3D-graphene@Fe2O3,28 MXene@SnO237 and Fe2O3@polyaniline.30 We also tested the Ti3C2Tx@Fe2O3 composite synthesized with different mass ratios (Ti3C2Tx:Fe2O3 = 1:1). As shown in Fig. S7 (ESI†), although the rate capability was improved, the capacity of the composite electrode decreased to ∼886 mA h g−1 at a current density of 0.1 A g−1. Meanwhile, as shown in Fig. 3b, although the initial discharge capacity of the pristine Fe2O3 anode is high (∼1960 mA h g−1), it exhibits a poor rate performance (only ∼110 mA h g−1 at the current density of 5 A g−1 with capacity retention of ∼5%). Thus, the combination of Fe2O3 and an MXene matrix has a great advantage compared with the pristine Fe2O3 and MXene. The increase of electroconductivity from the pristine Fe2O3 to Ti3C2Tx@Fe2O3 was further confirmed by Electrochemical Impedance Spectroscopy (EIS) measurement (shown in Fig. 3c). The aerogel-like MXene anode exhibits a small charge transfer resistance (Rct) of ∼40 Ω, indicating its excellent conductivity. After loading Fe2O3 nanospheres on the conductive matrix, the Ti3C2Tx@Fe2O3 anode shows a reduced Rct of ∼60 Ω, which is noticeably lower than that of the purely Fe2O3 anode (∼120 Ω). This proves that the conductive network can effectively increase the electroconductivity, thus leading to an enhanced rate performance. In addition, the long-term cycling stability of a hybrid anode has been observed as shown in Fig. 3d. The Ti3C2Tx@Fe2O3 anode demonstrates 0.042% fading rate per cycle at a current density of 5 A g−1 for up to 500 cycles, which is much lower than that of the bare Fe2O3 anode, and also lower than those of other previously reported Fe2O3-based anodes (such as hierarchical porous Fe2O3 nanosheets,31 polypyrrole-coated Fe2O3@C composites33 and 3D-graphene@Fe2O3,28etc.). In addition, Fig. S8 (ESI†) demonstrates the SEM images of the Ti3C2Tx@Fe2O3 electrode after cycling. It is noted that under the protection of the MXene, the Fe2O3 sphere still preserves the structure after the cycling. The above results convincingly confirm that the MXene matrix can effectively enhance the electrochemical performances of the Ti3C2Tx@Fe2O3 composite anodes.
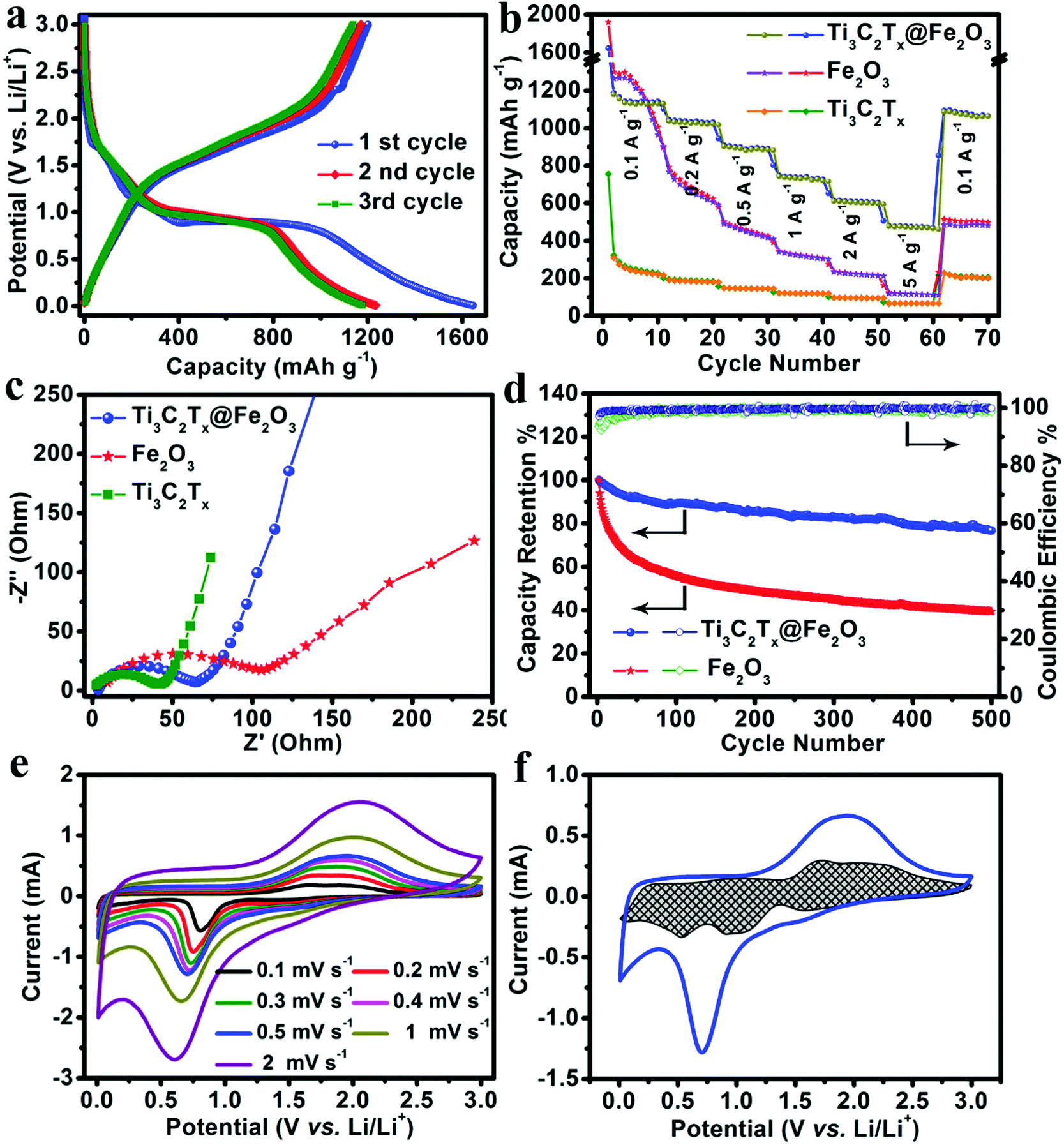 | ||
| Fig. 3 Lithium storage performance of the Fe2O3, Ti3C2Tx and Ti3C2Tx@Fe2O3 electrodes: (a) the first three charge–discharge cycles of the Ti3C2Tx@Fe2O3 hybrid electrode at a current density of 0.1 A g−1. (b) Rate performances and (c) EIS of the Fe2O3, Ti3C2Tx and Ti3C2Tx@Fe2O3 electrodes. (d) Cycling stability of the Fe2O3 and Ti3C2Tx@Fe2O3 electrodes at a current density of 5 A g−1. (e) CV curves of a Ti3C2Tx@Fe2O3 electrode at different scan rates. (f) The capacity separation of the Ti3C2Tx@Fe2O3 hybrid electrode at a scan rate of 0.5 mV s−1. | ||
In order to gain further insight into the electrochemistry of the electrode material, cyclic voltammetry (CV) measurements were performed. Fig. 3e depicts the CV curves of the Ti3C2Tx@Fe2O3 anode collected at different scan rates from 0.1 to 0.2, 0.3, 0.4, 0.5, 1 and 2 mV s−1, which are well-consistent with the charge–discharge profiles. The charge storage behaviours of the Ti3C2Tx@Fe2O3 anode can be studied by separating the capacitive and diffusion-controlled currents viaeqn (1).42
| i(V) = k1ν + k2ν0.5 | (1) |
Ti3C2Tx@Fe2O3 anodes with improved rate and cycling performances have been achieved. Thus, development of matchable lithium-ion capacitor cathodes is highly desired to effectively enhance the electrochemical performances of full lithium-ion capacitors. In this regard, a 3-D nitrogen and sulphur dual-doped porous carbon (NS-DPC) derived from biomass has been synthesized as a cathode material to match Ti3C2Tx@Fe2O3 anodes. As presented in Fig. S11a (ESI†), the XRD pattern of the NS-DPC has no obvious peaks at 23° and 43.2°, which correspond to the (002) and (100) planes, respectively, indicating disordered carbon. The Raman spectrum of the NS-DPC is shown in Fig. S11b (ESI†), where the broad D band suggests a defect-induced structure while the G band indicates the graphitic layers and the tangential vibration of the carbon atoms. The ratio of ID/IG (ID signifies the intensity of D band and IG denotes the intensity of the G band) of the NS-DPC is determined to be 0.85, which indicates more structural distortion and defects in the as-prepared NS-DPC. Fig. S12a and b (ESI†) show the SEM images of the NS-DPC and it can be observed that the as-prepared material shows an interconnected porous architecture with pore sizes ranging from 500 nm to 1 μm. These are beneficial for the rapid diffusion of electrolyte during charge/discharge processes. The porous carbon was further characterized by TEM. Noticeably, large numbers of micro- and mesopores as well as channels have been detected in Fig. S13 (ESI†), suggesting a large surface area of porosity. The material shows a typical type II adsorption–desorption isotherm with a high nitrogen sorption capacity (Fig. S14a, ESI†).55 The Brunauer–Emmett–Teller (BET) surface area and pore volume of NS-DPC is 1807 m2 g−1 and 0.77 cm3 g−1, respectively. The pore size distribution (Fig. S14b, ESI†) is narrow with an average pore size less than 3 nm, again confirming the existence of mesopores. X-ray photoelectron spectra (XPS) were also taken to determine the elemental composition of the obtained NS-DPC (as displayed in Fig. S15, ESI†). The spectrum of N 1s (Fig. S15b, ESI†) can be deconvoluted into two peaks located at 400.2 and 402.2 eV, corresponding to graphitic nitrogen and pyridine nitrogen, respectively. Moreover, in Fig. S15c (ESI†), two fitted peaks can be assigned to carbon-bonded sulphur C–S–C (164.1 eV) and highly oxidized sulphur SOx (167.9 eV), confirming the existence of an S element.56
The electrochemical performance of NS-DPC as the electrode for lithium-ion capacitors was tested in a two-electrode system with lithium foil as a counter and reference electrode and 1.0 M LiPF6 (dissolved in EC/DEC, v/v = 1:1) as an electrolyte. As shown in Fig. 4a, CV measurements were carried out at scan rates from 5 mV s−1 to 50 mV s−1 with a potential window from 2.0 V to 4.5 V. The CV curves of the NS-DPC cathode maintain a good rectangular shape with scan rate increasing from 5 mV s−1 to 50 mV s−1, suggesting an outstanding capacitive behaviour and rate performance. The small humps in the CV curves indicate the existence of a pseudocapacitive process, which can be ascribed to the interaction between the electrolyte and the nitrogen/sulphur atom defects. And this can also be confirmed by Fig. S16 (ESI†), in which 83% of the total capacity is assigned to the capacitive behaviour at a scan rate of 40 mV s−1. Moreover, the CV curves still retain rectangular shapes without distortion as the potential window is increased from 3.5 to 4.5 V, confirming the stable capacitive behaviour of the NS-DPC cathode (Fig. 4b). Linear charge–discharge curves at different current densities from 0.1 A g−1 to 10 A g−1 can be observed for the NS-DPC cathode in Fig. S17 (ESI†), suggesting rapid capacitive behaviour. Fig. 4c presents the rate performance of an NS-DPC cathode, exhibiting ∼140 mA h g−1 at a current density of 0.1 A g−1. This value is much higher than that of porous carbon without any doping (shown in Fig. S18, ESI†), also higher than those of previously reported activated carbon materials in a similar potential window.10 Furthermore, a specific capacity of ∼91 mA h g−1 was achieved even at a high current density of 10 A g−1, corresponding to ∼64% capacity retention with the current density increasing from 0.1 A g−1 to 10 A g−1, which confirms the superior rate capability of the NS-DPC cathodes. Except for excellent rate capability, the NS-DPC cathode material also possesses an outstanding cycling stability, showing a slight increase of ∼3% of the initial capacity for the NS-DPC cathode after 10000 cycles. This may be attributed to the activation of the NS-DPC cathode material, explicitly confirming the great cycling performance of the NS-DPC cathode. It is noted that the excellent capacitive performance can be attributed to the natural features of the biomass, including heteroatoms and natural templates.
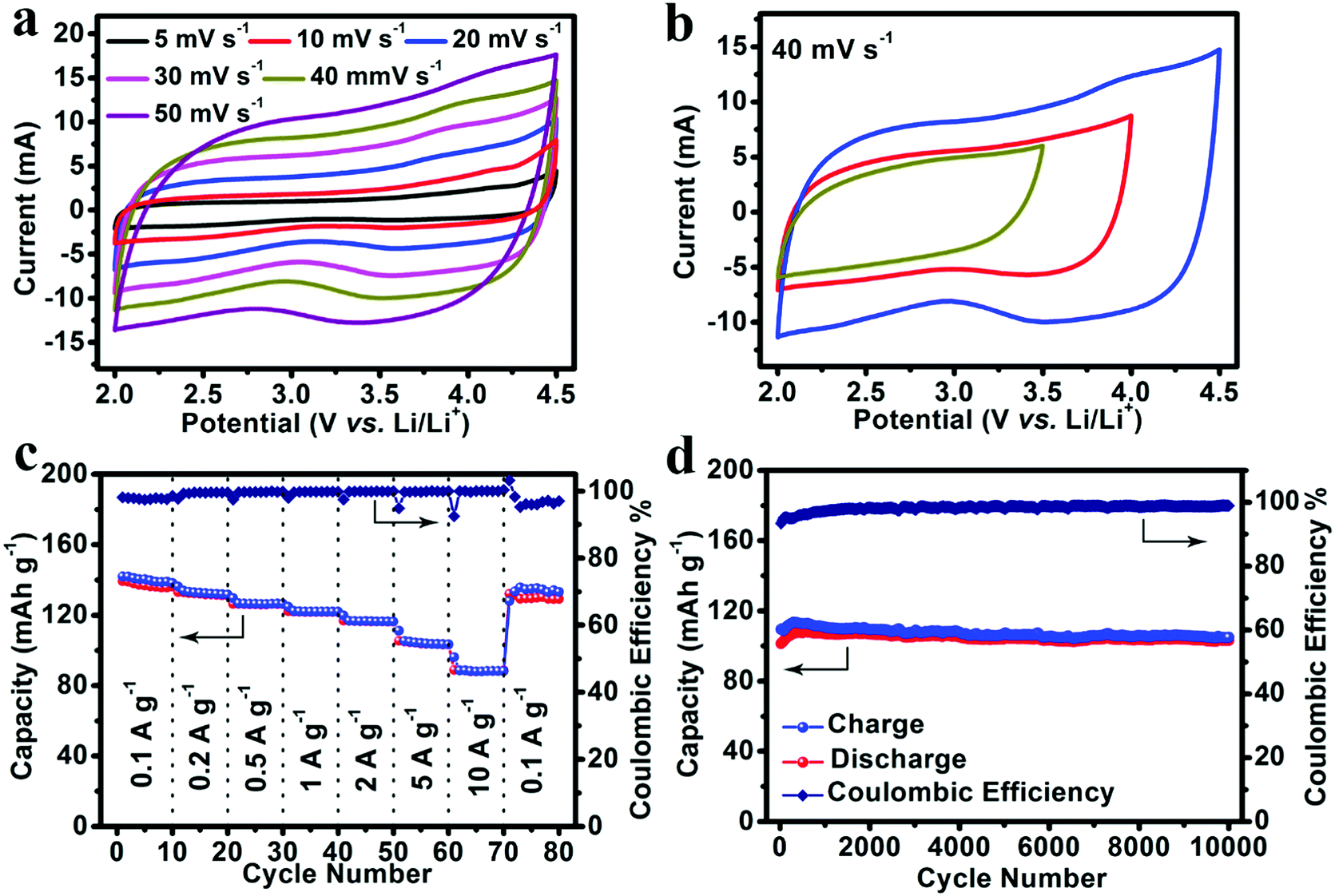 | ||
| Fig. 4 Lithium storage performance of the NS-DPC cathode tested in a LiPF6 electrolyte: (a) CV curves of the NS-DPC cathode at different scan rates. (b) CV curves of the NS-DPC cathode cycled within different potential windows at 40 mV s−1. (c) Rate performance of a NS-DPC cathode. (d) Cycling stability of a NS-DPC cathode at a current density of 5 A g−1. | ||
As an effective method to boost both the energy density and power density, we constructed lithium-ion full capacitors based on the as-prepared NS-DPC cathode and the prelithiated Ti3C2Tx@Fe2O3 anode (Fig. S19, ESI†). The LIC device with 4 V potential window was fabricated by using a 1 M LiPF6 electrolyte. As schematically depicted in Fig. 5a, since LICs combine the characteristics of both LIBs and SCs by utilizing the LIB anode and SC cathode, Li+ ions will intercalate into MXene layers and react with Fe2O3 hollow nanospheres, while the PF6− in the electrolyte is absorbed onto the surface of the NS-DPC cathode during the charge process. The discharge process is the reverse of the charge process. Compared with conventional SCs, LICs have an asymmetric configuration, thus improving the energy density because of the intake of a high-capacity anode with its faradaic based storage mechanism. Meanwhile, unlike LIBs, LICs consume both cations and anions, which can provide faster kinetics than the rocking chair type reaction in LIBs.5,10,57,58Fig. 5b demonstrates the cyclic voltammetry curves of the Ti3C2Tx@Fe2O3//NS-DPC LIC device at scan rates ranging from 5–50 mV s−1 in a voltage window between 0.01 and 4 V. The CV curves display a quasi-rectangular shape even at a high scan rate of 50 mV s−1, suggesting superior capacitive behaviour and fast charge–discharge capability of the as-assembled full lithium-ion capacitor. Furthermore, the prolonged and symmetric charge–discharge curves at different current densities exhibit a near-linear slope with a slight internal resistance (IR) drop (Fig. 5c), again confirming the good capacitive behaviour of the device. A high specific capacity of 109 mA h g−1 (based on the total mass of cathodic and anodic active materials) is achieved at a current density of 0.2 A g−1, which is much higher than other previously reported values for LICs.10,29,59
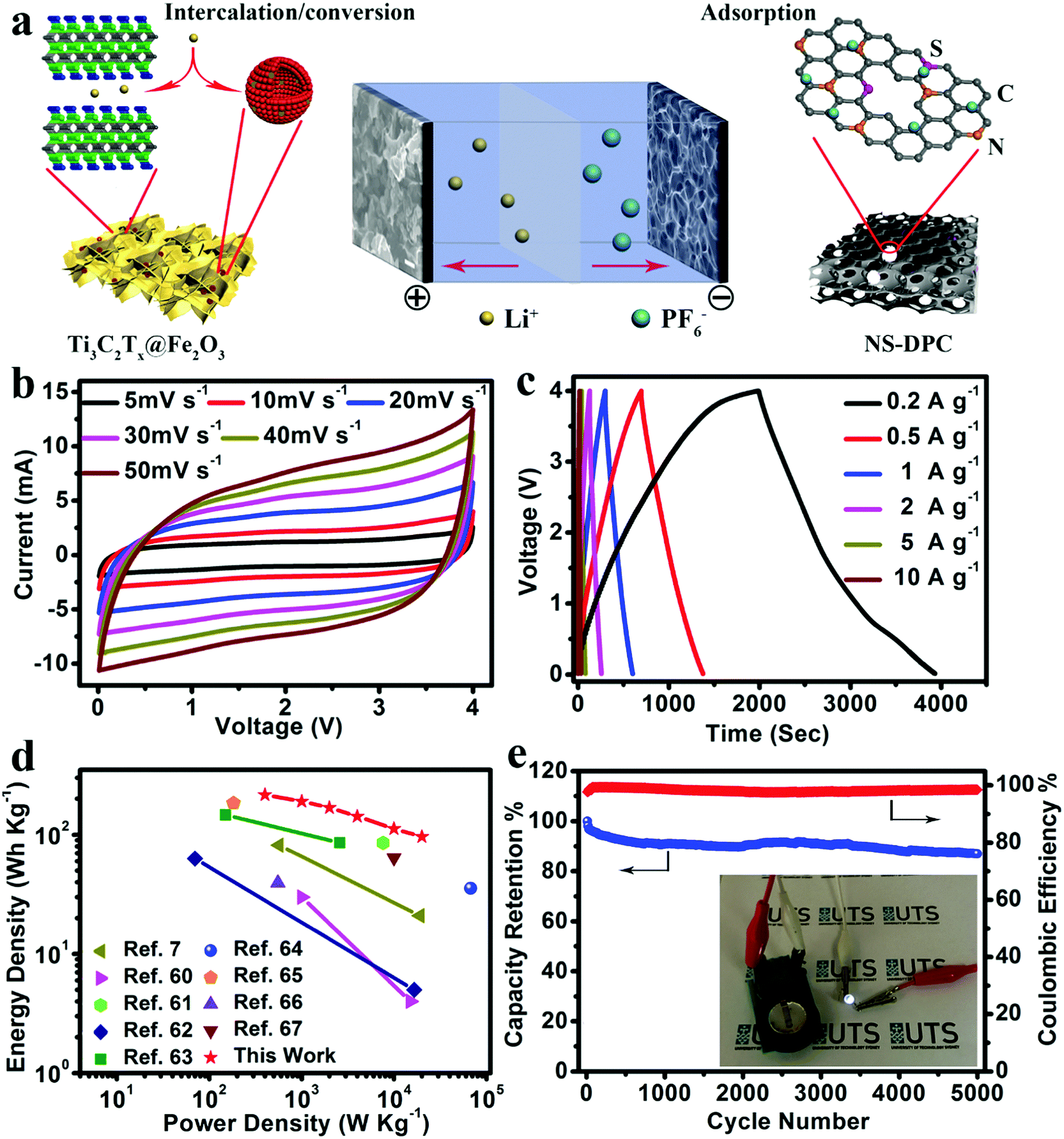 | ||
| Fig. 5 (a) Schematic illustration of the charge-storage mechanisms for the Ti3C2Tx@Fe2O3//NS-DPC LIC device. (b) CV curves and (c) charge–discharge curves of the Ti3C2Tx@Fe2O3//NS-DPC LIC device. (d) Ragone plots of the Ti3C2Tx@Fe2O3//NS-DPC LIC device and other previously reported devices. (e) Cycling stability of the Ti3C2Tx@Fe2O3//NS-DPC device at a current density of 5 A g−1; the inset is a demonstration of the LIC device powering an LED. | ||
Energy density and power density are two important parameters for evaluation of the as-assembled Ti3C2Tx@Fe2O3//NS-DPC LIC device. Thus, the comparisons of energy density and power density of the Ti3C2Tx@Fe2O3//NS-DPC LIC device and other previously reported results are summarized in a Ragone plot (shown in Fig. 5d).60–67 It can be seen that the Ti3C2Tx@Fe2O3//NS-DPC LIC device delivers a high energy density of 216 W h kg−1 at a power density of 400 W kg−1. Even at a high power density of 20 kW kg−1, the device can still achieve 96.5 W h kg−1, which is superior to the previously reported lithium-ion capacitors (see details in Tables S1 and S2 in ESI†). Those include Li4Ti5O12/graphene//AC (4 W h kg−1 at 15 kW kg−1),60 TiO2//graphene (21 W h kg−1 at 19 kW kg−1),7 hard carbon//AC (85.7 W h kg−1 at 7.6 kW kg−1)61 and Nb2O5/C//AC (5 W h kg−1 at 16.5 kW kg−1),62etc. Except for the high energy and power density of the as-assembled device, it also exhibits outstanding cycling stability, retaining 87% of the initial capacity at 5 A g−1 after 5000 cycles, as shown in Fig. 5e. In addition, the as-assembled Ti3C2Tx@Fe2O3//NS-DPC LIC device is able to power a 2.5 V light-emitting diode (LED), suggesting that the Ti3C2Tx@Fe2O3//NS-DPC LIC device possesses promising expectations for practical applications (see inset of Fig. 5e).
Conclusions
In summary, for the first time, we successfully designed and synthesized an interconnected aerogel-like MXene wrapped Fe2O3 nanosphere nanocomposite as an anode material for lithium-ion capacitors. The Ti3C2Tx MXene as a highly conductive matrix affords fast transport of electrons, while the Fe2O3 hollow nanospheres provide large specific capacity, thus synergizing to enhance the electrochemical performance of composite electrodes. Consequently, owing to the nanostructural pillar effect and combined synergistic effects, the Ti3C2Tx@Fe2O3 nanocomposite delivers a high reversible capacity of 1180 mA h g−1 at a current density of 0.1 A g−1 with excellent rate performance and cycling stability. A 4 V lithium-ion capacitor has been assembled based on the MXene wrapped Fe2O3 nanosphere anode and the 3D nitrogen sulphur dual-doped porous carbon cathode, showing a maximum capacity of 109 mA h g−1. The highest energy density of 216 W h kg−1 at a power density of 400 W kg−1 and the highest power density of 20 kW kg−1 at an energy density of 96.5 W h kg−1 have been achieved by Ti3C2Tx@Fe2O3//NS-DPC LIC devices, confirming both high energy density and power density of the fabricated lithium-ion capacitor. These features together endow the Ti3C2Tx@Fe2O3//NS-DPC LIC device with promising energy storage density for practical devices and applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Australian Renewable Energy Agency (ARENA) project (ARENA 2014/RND106) and the Australian Research Council (ARC) through the ARC Discovery Project (DP170100436). X. T. acknowledges the financial support from the China scholarship council (CSC grant number: 201606840117). The authors thank Prof. Yury Gogotsi (Department of Materials Science and Engineering and A. J. Drexel Nanomaterials Institute, Drexel University, Philadelphia, PA19104, USA) for providing MAX phase Ti3AlC2 materials.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef PubMed
.
- D. Dubal, O. Ayyad, V. Ruiz and P. Gomez-Romero, Chem. Soc. Rev., 2015, 44, 1777–1790 RSC
- K. Yuan, T. Hu, Y. Xu, R. Graf, L. Shi, M. Forster, T. Pichler, T. Riedl, Y. Chen and U. Scherf, Mater. Chem. Front., 2017, 1, 278–285 RSC
- M. Cheng, Y. Meng, Q. Meng, L. Mao, M. Zhang, K. Amin, A. Ahmad, S. Wu and Z. Wei, Mater. Chem. Front., 2018, 2, 986–992 RSC
- H. Wang, C. Zhu, D. Chao, Q. Yan and H. J. Fan, Adv. Mater., 2017, 29, 1702093 CrossRef PubMed
- G. G. Amatucci, F. Badway, A. Du Pasquier and T. Zheng, J. Electrochem. Soc., 2001, 148, A930–A939 CrossRef
- H. Wang, C. Guan, X. Wang and H. J. Fan, Small, 2015, 11, 1470–1477 CrossRef PubMed
- F. Zhang, T. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. Chen, Energy Environ. Sci., 2013, 6, 1623–1632 RSC
- Y. Ma, H. Chang, M. Zhang and Y. Chen, Adv. Mater., 2015, 27, 5296–5308 CrossRef PubMed
- Q. Xia, H. Yang, M. Wang, M. Yang, Q. Guo, L. Wan, H. Xia and Y. Yu, Adv. Energy Mater., 2017, 7, 1701336 CrossRef
- Q. Wang, Z. Wen and J. Li, Adv. Funct. Mater., 2006, 16, 2141–2146 CrossRef
- L. Gao, D. Huang, Y. Shen and M. Wang, J. Mater. Chem. A, 2015, 3, 23570–23576 RSC
- H. Kim, M. Y. Cho, M. H. Kim, K. Y. Park, H. Gwon, Y. Lee, K. C. Roh and K. Kang, Adv. Energy Mater., 2013, 3, 1500–1506 CrossRef
- E. Lim, H. Kim, C. Jo, J. Chun, K. Ku, S. Kim, H. I. Lee, I. S. Nam, S. Yoon and K. Kang, ACS Nano, 2014, 8, 8968–8978 CrossRef PubMed
- L. Cheng, H. Q. Li and Y. Y. Xia, J. Solid State Electrochem., 2016, 10, 405–410 CrossRef
- A. Banerjee, K. K. Upadhyay, D. Puthusseri, V. Aravindan, S. Madhavi and S. Ogale, Nanoscale, 2014, 6, 4387–4394 RSC
- V. Aravindan, D. Mhamane, W. C. Ling, S. Ogale and S. Madhavi, ChemSusChem, 2013, 6, 2240–2244 CrossRef PubMed
- K. Leng, F. Zhang, L. Zhang, T. Zhang, Y. Wu, Y. Lu, Y. Huang and Y. Chen, Nano Res., 2013, 6, 581–592 CrossRef
- T. Aida, K. Yamada and M. Morita, Electrochem. Solid-State Lett., 2006, 9, A534–A536 CrossRef
- S. Sivakkumar and A. Pandolfo, Electrochim. Acta, 2012, 65, 280–287 CrossRef
- V. Khomenko, E. Raymundo-Piñero and F. Béguin, J. Power Sources, 2008, 177, 643–651 CrossRef
- J. H. Kim, J. S. Kim, Y. G. Lim, J. G. Lee and Y. J. Kim, J. Power Sources, 2011, 196, 10490–10495 CrossRef
- K. Karthikeyan, S. Amaresh, S. N. Lee, V. Aravindan and Y. S. Lee, Chem. – Asian J., 2014, 9, 852–857 CrossRef PubMed
- J. H. Won, H. M. Jeong and J. K. Kang, Adv. Energy Mater., 2017, 7, 1601355 CrossRef
- X. Liu, H. G. Jung, S. O. Kim, H. S. Choi, S. Lee, J. H. Moon and J. K. Lee, Sci. Rep., 2013, 3, 3183 CrossRef PubMed
- P. Yang, Y. Ding, Z. Lin, Z. Chen, Y. Li, P. Qiang, M. Ebrahimi, W. Mai, C. P. Wong and Z. L. Wang, Nano Lett., 2014, 14, 731–736 CrossRef PubMed
- X. Tang, R. Jia, T. Zhai and H. Xia, ACS Appl. Mater. Interfaces, 2015, 7, 27518–27525 CrossRef PubMed
- T. Jiang, F. Bu, X. Feng, I. Shakir, G. Hao and Y. Xu, ACS Nano, 2017, 11, 5140–5147 CrossRef PubMed
- J. Wang, H. Tang, H. Wang, R. Yu and D. Wang, Mater. Chem. Front., 2017, 1, 414–430 RSC
- J. M. Jeong, B. G. Choi, S. C. Lee, K. G. Lee, S. J. Chang, Y. K. Han, Y. B. Lee, H. U. Lee, S. Kwon and G. Lee, Adv. Mater., 2013, 25, 6250–6255 CrossRef PubMed
- K. Cao, L. Jiao, H. Liu, Y. Liu, Y. Wang, Z. Guo and H. Yuan, Adv. Energy Mater., 2015, 5, 1401421 CrossRef
- Z. Ma, X. Huang, S. Dou, J. Wu and S. Wang, J. Phys. Chem. C, 2014, 118, 17231–17239 CrossRef
- F. Han, D. Li, W. C. Li, C. Lei, Q. Sun and A. H. Lu, Adv. Funct. Mater., 2013, 23, 1692–1700 CrossRef
- N. Zhang, X. Han, Y. Liu, X. Hu, Q. Zhao and J. Chen, Adv. Energy Mater., 2015, 5, 1401123 CrossRef
- G. Gao, L. Yu and H. B. Wu, Small, 2014, 10, 1741–1745 CrossRef PubMed
- Y. Zeng, Y. Han, Y. Zhao, Y. Zeng, M. Yu, Y. Liu, H. Tang, Y. Tong and X. Lu, Adv. Energy Mater., 2015, 5, 1402176 CrossRef
- B. Ahmed, D. H. Anjum, Y. Gogotsi and H. N. Alshareef, Nano Energy, 2017, 34, 249–256 CrossRef
- X. Wang, S. Kajiyama, H. Iinuma, E. Hosono, S. Oro, I. Moriguchi, M. Okubo and A. Yamada, Nat. Commun., 2015, 6, 6544 CrossRef PubMed
- J. Luo, X. Tao, J. Zhang, Y. Xia, H. Huang, L. Zhang, Y. Gan, C. Liang and W. Zhang, ACS Nano, 2016, 10, 2491–2499 CrossRef PubMed
- M. Ghidiu, M. R. Lukatskaya, M. Q. Zhao, Y. Gogotsi and M. W. Barsoum, Nature, 2014, 516, 78–81 Search PubMed
- L. Li, Y. Chu, Y. Liu and L. Dong, J. Phys. Chem. C, 2007, 111, 2123–2127 CrossRef
- X. Xie, M. Q. Zhao, B. Anasori, K. Maleski, C. E. Ren, J. Li, B. W. Byles, E. Pomerantseva, G. Wang and Y. Gogotsi, Nano Energy, 2016, 26, 513–523 CrossRef
- M. Q. Zhao, C. E. Ren, Z. Ling, M. R. Lukatskaya, C. Zhang, K. L. Van Aken, M. W. Barsoum and Y. Gogotsi, Adv. Mater., 2015, 27, 339–345 CrossRef PubMed
- J. Yan, C. E. Ren, K. Maleski, C. B. Hatter, B. Anasori, P. Urbankowski, A. Sarycheva and Y. Gogotsi, Adv. Funct. Mater., 2017, 27, 1701264 CrossRef
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef PubMed
- M. Q. Zhao, X. Xie, C. E. Ren, T. Makaryan, B. Anasori, G. Wang and Y. Gogotsi, Adv. Mater., 2017, 29, 1702410 CrossRef PubMed
- Y. Ma, B. Li and S. Yang, Mater. Chem. Front., 2018, 2, 456–467 RSC
- D. De Faria, S. Venâncio Silva and M. De Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef
- M. Hu, Z. Li, T. Hu, S. Zhu, C. Zhang and X. Wang, ACS Nano, 2016, 10, 11344–11350 CrossRef PubMed
- T. Fujii, F. De Groot, G. Sawatzky, F. Voogt, T. Hibma and K. Okada, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 3195 CrossRef
- S. Xu, C. M. Hessel, H. Ren, R. Yu, Q. Jin, M. Yang, H. Zhao and D. Wang, Energy Environ. Sci., 2014, 7, 632–637 RSC
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef PubMed
- B. Ahmed, D. H. Anjum, M. N. Hedhili, Y. Gogotsi and H. N. Alshareef, Nanoscale, 2016, 8, 7580–7587 RSC
- X. Liang, Y. Rangom, C. Y. Kwok, Q. Pang and L. F. Nazar, Adv. Mater., 2017, 29, 1603040 CrossRef PubMed
- J. Deng, T. Xiong, F. Xu, M. Li, C. Han, Y. Gong, H. Wang and Y. Wang, Green Chem., 2015, 17, 4053–4060 RSC
- Q. Pang, J. Tang, H. Huang, X. Liang, C. Hart, K. C. Tam and L. F. Nazar, Adv. Mater., 2015, 27, 6021–6028 CrossRef PubMed
- D. Xu, D. Chao, H. Wang, Y. Gong, R. Wang, B. He, X. Hu and H. J. Fan, Adv. Energy Mater., 2018, 8, 1702769 CrossRef
- H. Wang, G. Jia, Y. Guo, Y. Zhang, H. Geng, J. Xu, W. Mai, Q. Yan and H. J. Fan, Adv. Mater. Interfaces, 2016, 3, 1600375 CrossRef
- M. Yang, Y. Zhong, J. Ren, X. Zhou, J. Wei and Z. Zhou, Adv. Energy Mater., 2015, 5, 1500550 CrossRef
- N. Xu, X. Sun, X. Zhang, K. Wang and Y. Ma, RSC Adv., 2015, 5, 94361–94368 RSC
- J. Zhang, X. Liu, J. Wang, J. Shi and Z. Shi, Electrochim. Acta, 2016, 187, 134–142 CrossRef
- E. Lim, C. Jo, H. Kim, M. H. Kim, Y. Mun, J. Chun, Y. Ye, J. Hwang, K. S. Ha and K. C. Roh, ACS Nano, 2015, 9, 7497–7505 CrossRef PubMed
- F. Zhang, T. F. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. S. Chen, Energy Environ. Sci., 2013, 6, 1623–1632 RSC
- H. W. Wang, Y. Zhang, H. X. Ang, Y. Q. Zhang, H. T. Tan, Y. F. Zhang, Y. Y. Guo, J. B. Franklin, X. L. Wu, M. Srinivasan, H. J. Fan and Q. Y. Yan, Adv. Funct. Mater., 2016, 26, 3082–3093 CrossRef
- H. L. Wang, Z. W. Xu, Z. Li, K. Cui, J. Ding, A. Kohandehghan, X. Tan, B. Zahiri, B. C. Olsen, C. M. Holt and D. Mitlin, Nano Lett., 2014, 14, 1987–1994 CrossRef PubMed
- R. Ding, L. Qi and H. Y. Wang, RSC Adv., 2013, 3, 12581–12584 RSC
- R. T. Wang, J. W. Lang, P. Zhang, Z. Y. Lin and X. B. Yan, Adv. Funct. Mater., 2015, 25, 2270–2278 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization and electrochemical tests. See DOI: 10.1039/c8qm00232k |
| This journal is © the Partner Organisations 2018 |