
Molecular-level anchoring of polymer cathodes on carbon nanotubes towards rapid-rate and long-cycle sodium-ion storage†
Yuxiang
Hu‡
 ab,
Kai
Zhang‡
b,
Han
Hu
ab,
Songcan
Wang
ab,
Delai
Ye
ab,
Michael J.
Monteiro
b,
Zhongfan
Jia
*b and
Lianzhou
Wang
*ab
ab,
Kai
Zhang‡
b,
Han
Hu
ab,
Songcan
Wang
ab,
Delai
Ye
ab,
Michael J.
Monteiro
b,
Zhongfan
Jia
*b and
Lianzhou
Wang
*ab
aNanomaterials Centre, School of Chemical Engineering, The University of Queensland, QLD 4072, Australia
bAustralian Institute for Bioengineering and Nanotechnology, The University of Queensland, QLD 4072, Australia. E-mail: l.wang@uq.edu.au; z.jia@uq.edu.au
First published on 10th July 2018
Abstract
Nitroxide radical polymers (NRPs) have attracted increasing research interest as promising cathode materials for sodium ion batteries, primarily owing to their high-voltage (ca. 3.47 V vs. Na+/Na), environment compatibility, low-cost, and rich resources. However, such NRP cathodes are subjected to low rate-capacity and poor cycling stability because of their inferior conductivity and dissolubility in the electrolyte. Herein, we report a “molecular glue” strategy to anchor pyrene functionalized NRPs on a highly condutive carbon nanotube (CNT) matrix through π–π interactions between the pyrene functional groups and CNTs. As such, the NRP layer is homogeneously “glued” on the carbon substrates and the non-covalent interaction with CNTs drastically improves the insolubility and electric conductivity of the composite. Benefiting from these structural merits, the new nanocomposite exhibits a record cyclability (92% capacity retention at a high current density of 2.2 A g−1 after 6000 cycles) and remarkable rate capability (78 mA h g−1 at 5.5 A g−1) when evaluated as a high-voltage cathode material for SIBs. This work demonstrates the importance of rational hybridization of radical polymers and carbon substrates for enhanced electrochemical performance and provides insightful guides for designing high-performance polymer-based composites for energy storage applications.
Introduction
To meet the demands of energy storage for large-scale practical applications such as electric vehicles and smart grid systems, efficient and economical energy storage devices are urgently required.1–4 Although lithium-ion batteries (LIBs) have played a dominant role in energy storage in the past few decades, the continuously increasing cost (∼1500 USD kg−1) and the limited reserves of lithium resources (17 ppm of earth's crust) demand the development of alternative battery systems.5–11 Sodium-ion batteries (SIBs) are one of the most promising alternatives because of the low cost and abundance of sodium resources (23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ppm).7,12,13 However, due to the large radius of Na+ (1.02 Å in comparison with 0.75 Å for Li+), SIBs suffer from slow ion diffusion and poor cycling stability, restricting their widespread application14,15 despite tremendous efforts devoted to exploring new electrode materials to overcome the challenges.16,17
000 ppm).7,12,13 However, due to the large radius of Na+ (1.02 Å in comparison with 0.75 Å for Li+), SIBs suffer from slow ion diffusion and poor cycling stability, restricting their widespread application14,15 despite tremendous efforts devoted to exploring new electrode materials to overcome the challenges.16,17
Polymer-based electrodes are promising cathode materials for SIBs due to their inherent advantages, including high renewability, good environment compatibility, low weight, and rich sources.18–21 Previously reported polymer cathode materials for SIBs include carbonyl-based compounds, organosulfonic polymers, and radical polymers.22–27 Amongst them, poly(2,2,6,6-tetramethyl piperidiny-1-oxyl-4-yl methacrylate) (PTMA) represents one type of nitroxide radical polymer (NRP); it has a high operational potential (ca. 3.47 V vs. Na+/Na), high demonstrated capacity, and considerable rate capacity. This is primarily due to the fast and reversible redox reactions of the radical repeating unit (2,2,6,6-tetramethyl piperidine-1-oxyl (TEMPO)), in which the electron exchange rate is on the order of kex = 105–7 M−1 s−1.28–30 Due to the low conductivity of PTMA, conductive carbon materials must be added for improved electrical conductivity of the electrode. However, the poor dispersion of PTMA into carbon materials remains a major hurdle to realize the optimal electrochemical performance, primarily towards high rate-capacity and long-term cycling stability. There are generally two strategies (i.e. covalent or non-covalent functionalization) to enhance the dispersion of the NRPs with carbon additives. Although the covalent functionalization of a carbon substrate with NRPs would produce a composite with good dispersibility, the procedure is destructive toward intact conductivity of the matrix and relatively complex with poor reproducibility.27,28,31–33 Nevertheless, it is desirable to develop a more versatile and effective strategy to improve the adhesiveness of NRPs with the conductive matrix. Recently, through non-covalent π–π interactions, a pyrene functionalized PTMA was reported to bind with reduced graphene sheets (rGO) forming an NRP/rGO composite with molecular level dispersion and consequently improved/intact composite conductivity.34 However, rGO only provided limited electric conductivity due to its structural defects. Therefore, NRP-based composites with non-covalent interactions and a more conductive matrix are promising cathodes towards SIBs, yet no relevant studies have been reported before.
In this work, we report a novel design of cathode materials by molecular-level anchoring of multiple pyrene functionalized PTMAs onto CNTs (Py-PTMA/CNTs) through molecular-level non-covalent π–π interactions (Scheme 1). The new composite can not only produce molecular level connectivity between Py-PTMA and CNTs, but also preserve the excellent electric conductivity of pristine CNTs. As a result, the composite demonstrated a remarkably stable capacity of more than 80 mA h g−1 after 6000 cycles (more than 92% capacity retention) at a high current density of 2.2 A g−1 (equivalent to 20C). Furthermore, the Py-PTMA/CNTs achieved an excellent capacity of 78 mA h g−1 at a very fast charging rate (50C equivalent to current density of 5.5 A g−1), which is so far among the best performances of high-voltage cathodes in SIBs.
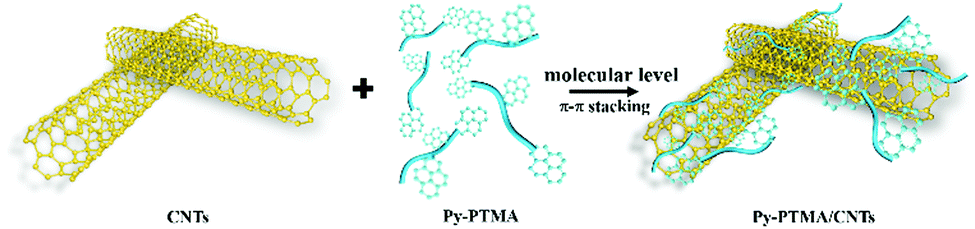 | ||
| Scheme 1 Assembly schematic of the pyrene functionalized Py-PTMA with CNTs via non-covalent π–π stacking interactions. | ||
Experimental
Preparation of the pyrene-functionalized polymer
Poly(2,6,6-tetramethyl piperidine-1-oxyl (TEMPO) methacrylate) (PTMA483) was synthesized using single electron transfer-living radical polymerization followed by oxidation. Briefly, 1.0 g (4.44 × 10−3 mol) of 2,2,6,6-tetramethylpiperidin-4-yl methacrylate (TMPM), 1.0 mg (4.48 × 10−6 mol) of CuBr2, 21.25 μL (7.99 × 10−5 mol) of Me6TREN and 13.15 μL (8.88 × 10−5 mol) of EBiB were dissolved together in isopropanol, which was purged with argon for 20 min. Polymerization was initiated via dropping in a copper wire wrapped stirrer bar at 25 °C. After 4 h polymerization, the polymers were precipitated in petroleum spirit (40–60 °C) twice and oxidized by H2O2 with catalysis of sodium tungstate in methanol for two days. The red PTMA precipitated out was washed with water and methanol, and dried under high-vacuum overnight.A rapid nitroxide radical coupling (NRC) reaction was adopted for the synthesis of P(TMA455-co-PyMA28) (Py-PTMA). In short, 0.10 g (4.16 × 10−4 mol) of PTMA483, 7.6 mg (2.08 × 10−5 mol) of pyren-1-ylmethyl 2-bromopropanoate and 11.1 μL (4.16 × 10−5 mol) of Me6TREN were dissolved in 4.0 mL of toluene/DMSO (v/v, 1/1) solution. After purging the solution with Ar for 20 mins, Cu(I)Br was added in one portion under positive argon pressure at room temperature to start the radical coupling reaction. The resulting mixture was condensed by air blowing until the polymer precipitated out. The polymer was redissolved in DCM and passed through a short basic alumina column to remove residual copper, and the red polymer solution was condensed with rotary evaporation to afford P(TMA455-co-PyMA28).
Formation of polymer CNT composites
The polymer (PTMA or Py-PTMA) and CNTs (6/4 w/w) were dispersed in THF. After stirring and sonicating, the final composite samples PTMA/CNTs and Py-PTMA/CNTs were obtained by evaporation of THF through opening the cap of the vials and then dried under high vacuum at 25 °C for 24 h to remove the solvent.Material characterization
UV-Vis spectra of the polymers were extracted from a Waters 2695 separation module Size Exclusion Chromatograph (SEC) equipped with a Waters 996 photodiode array detector. Electron Paramagnetic Resonance (EPR) spectra were recorded with a Bruker E540 spectrometer operating at X-band (∼9.8 GHz) and using a Bruker ER4122SQE-W1 high sensitivity resonator. Samples were measured in a 1.5 mm internal diameter capillary with the following instrumental parameters, microwave power = 0.635 mW, modulation amplitude = 1 G. The spectra were processed using Bruker Xepr software. X-ray photoelectron spectroscopy (XPS) characterization was performed by Kratos Ultra Axis XPS (Al Kα X-ray). A Renishaw Micro-Raman Spectroscopy System was used to obtain the Raman spectra (laser wavelength of 514 nm). The sample morphology was characterized by field-emission scanning electron microscopy (FE-SEM) (JEOL-7100), transmission electron microscopy (TEM) and high-resolution TEM (HR-TEM) (JEOL, JEM-2100).Electrochemical measurements
The batteries were assembled by utilizing a CR-2032 cell in an argon-filled glove box. A piece of Na metal anode (Sigma) and cathode electrode were stacked with a glass fibre separator (What-man). The electrolyte (50 μL) was made of 1 M NaClO4 and ethylene carbonate (EC)/propylene carbonate (PC) (1:1 v/v). To prepare the cathode, polymer/CNTs, Ketjen Black, and polyvinylidene fluoride (PVdF) were mixed with a weight ratio of 8:1:1 with N-méthyl-2-pyrrolidone (NMP) as a dispersing agent. The mixture slurry was then coated onto aluminium foil and dried at 80 °C under vacuum for 10 h. The areal loading is around 1.5 mg cm−2 with a mass loading of polymer of around 0.7 mg cm−2. The reversible capacity is based on the weight of the active material in the electrode. The charge–discharge electrochemical performance of the batteries was evaluated by a battery tester (LAND-CT2001A). The cyclic voltammograms (CVs) were conducted on an electrochemical station (CHI 600e Shanghai, China) between 2.5 and 3.7 V. Electrochemical impedance spectroscopy (EIS) was conducted using an Autolab (Aut85380) analyzer at an AC voltage of 5 mV in the frequency of 100 kHz and 100 mHz after being fully charged. All the measurements were conducted at 25 °C.
Results and discussion
Synthesis and characterization of PTMA483 (PTMA) and ca. 5 mol% of pyrene functionalized PTMA (Py-PTMA) were carried out according to our previous report.34 Briefly, PTMA483 was synthesized through a single electron transfer-living radical polymerization of 2,2,6,6-tetramethylpiperidin-4-yl methacrylate followed by oxidation.35 The resulting PTMA was further functionalized with pyrene through a copper catalyzed nitroxide radical coupling (NRC) reaction36 between TEMPO radicals and bromine functional pyrene (Fig. S1a, ESI†). TEMPO radicals on PTMA act as both chemical reactive functionalities for the NRC reaction and redox-active groups for energy storage, where, in the latter case, TEMPO radicals undergo a reversible oxidation and form oxoammonium cations (Fig. S1b, ESI†).37 Fig. S2a (ESI†) presents the UV-Vis spectra of PTMA and Py-PTMA, in which both polymers show n–π* absorption of nitroxide radicals at 475 nm, while Py-PTMA demonstrates multiple absorption from 250–350 nm ascribed to the pyrene moieties. The electron spin resonance spectra (ESR) displayed in Fig. S2b (ESI†) were similarly broad for the two polymers due to the spin exchange interaction of dense nitroxide radicals. The slightly lower intensity of Py-PTMA compared to PTMA was due to the loss of nitroxide radicals after trapping by pyrene functionalized compounds.The electrode composites PTMA/CNTs and Py-PTMA/CNTs were prepared via simple dispersing-depositing methods described in the ESI. Field-emission scanning electron microscopy (FE-SEM) was then employed to characterize the morphology of the composites with or without pyrene groups. (Fig. 1). The SEM image of the PTMA/CNT composites (Fig. 1a) exhibited a heterogeneous distribution with large aggregation of the polymer in the mixture. In contrast, the Py-PTMA/CNT composites showed that the structures consisted of CNTs with a coarse surface (inset of Fig. 1b), indicating the conformal coating of Py-PTMA on the CNT walls. The Py-PTMA/CNT composites still keep the one-dimensional structure similar to CNTs, twisting together to form a porous structure (Fig. 1b). Such porous structures will concomitantly allow the rapid diffusion of electrolytes. The TEM image clearly showed that the composite consisted of a homogeneously coated polymer layer on the CNTs, forming a core–sheath morphology (Fig. 1c). Previous molecular dynamic (MD) simulations suggest that stronger adhesion to CNTs was due to the much lower interaction energy of the conjugated vs. non-conjugated aromatic groups.38 Therefore, the pyrene functional groups on our Py-PTMA acting as anchors to provide this excellent binding to the CNTs compared to PTMA without pyrene. Such a tight layer of Py-PTMA on the CNT surface as displayed in HRTEM (Fig. 1d) will facilitate excellent electron transportation between the nitroxide polymer and CNTs.
 | ||
| Fig. 1 FE-SEM images of (a) PTMA/CNTs and (b) Py-PTMA/CNTs composites with enlarged SEM images in the inset, and (c) TEM and (d) HRTEM images of Py-PTMA/CNTs. | ||
Characterization by XPS (Fig. S3, ESI†) also supported the homogeneous attachment of Py-PTMA throughout the CNT substrate. The typical π → π* transition, which is different from the π–π stacking interaction between the polymer and CNTs, of the C 1s spectra around 291 eV was observed in the spectra of the CNTs and PTMA/CNTs, while this typical peak was much weaker for Py-PTMA/CNTs. Since the XPS spectra provide a surface and not bulk signal of the material, the weakened peak of Py-PTMA CNTs can be ascribed to the burial of the pyrene groups in the polymer sheath close to the CNTs. The interaction between the Py-PTMA and CNTs was further investigated by Raman spectroscopy as shown in Fig. 2 with the Raman spectra of Py-PTMA in Fig. S4 (ESI†). Two obvious peaks corresponding to the D and G bands of CNTs were observed for all three samples. The G-band shift in the CNT composites was ascribed to the interaction between pyrene and CNTs.39 The variation from 1587 cm−1 (CNTs) to 1582 cm−1 (Py-PTMA/CNTs) in the Raman spectra supports the close interaction between the pyrene groups and CNTs. The G band (1587 cm−1) for the PTMA/CNTs indicated negligible interaction between the PTMA and CNTs.
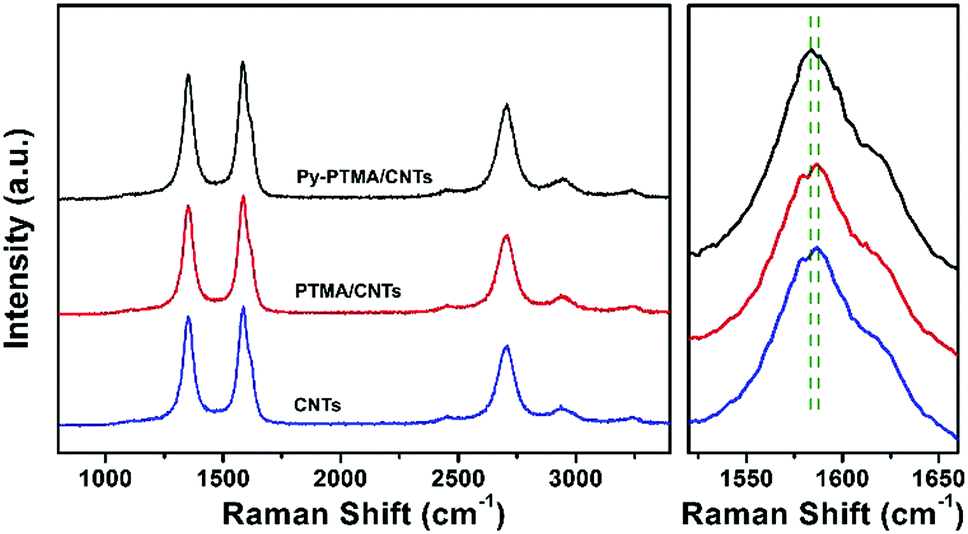 | ||
| Fig. 2 Raman spectra of Py-PTMA/CNTs, PTMA/CNTs and CNTs. | ||
The self-discharge of SIBs caused by the dissolution of organic active materials can drastically reduce both the battery lifetime and columbic efficiency.25 The battery voltage variation over time was measured to verify the stability of both Py-PTMA/CNTs and PTMA/CNTs in the electrolyte, one significant property towards a cathode material in SIBs. Based on the open circuit voltage (OCV) testing after the battery was fully charged (Fig. S5, ESI†), it was found that the Py-PTMA/CNT composite showed a much lower self-discharge rate than the PTMA/CNTs due to the strong π–π interaction of the pyrene functionalized Py-PTMA with the CNTs, in particular in polar electrolytes.38 Conversely, the obvious voltage decline of the PTMA/CNT composite was primarily due to the dissolution of the polymer.35
The electrochemical properties of the Py-PTMA/CNTs and PTMA/CNT electrodes were evaluated and compared by galvanostatic charge–discharge measurements. Fig. 3a shows their charge–discharge curves for the 1st, 50th and 100th cycle at a current density of 110 mA g−1 (equivalent to a rate of 1C). Note, there are generally two reaction plateaus for PTMA around 3.4 V and 2.0 V vs. Na+/Na, but the batteries in this work were tested only between 2.5 V and 3.7 V (vs. Na+/Na) because the lower plateau (∼2.0 V) was too low to be treated as one plateau for the cathode.28 The initial discharge capacity of Py-PTMA/CNTs reached as high as 107 mA h g−1 (theoretical capacity of the Py-PTMA/CNTs is 110 mA h g−1). While having a similar operation potential, the PTMA/CNT composite only delivered 84 mA h g−1 in the initial discharge process (Fig. 3b). Furthermore, the cycling stability of both electrodes was compared in Fig. 3c. It can be clearly observed that the Py-PTMA/CNT electrode maintained a stable capacity after 100 cycles of 105 mA h g−1 with a capacity retention of 98%, while the PTMA/CNT composite suffered from quick and constant capacity decline to around 60 mA h g−1 after 100 cycles. Note that the CNTs contribute negligible capacity towards the overall capacity in the testing voltage range (Fig. S6a, ESI†) with an optimized polymer/CNT weight ratio in the Fig. S6b (ESI†).
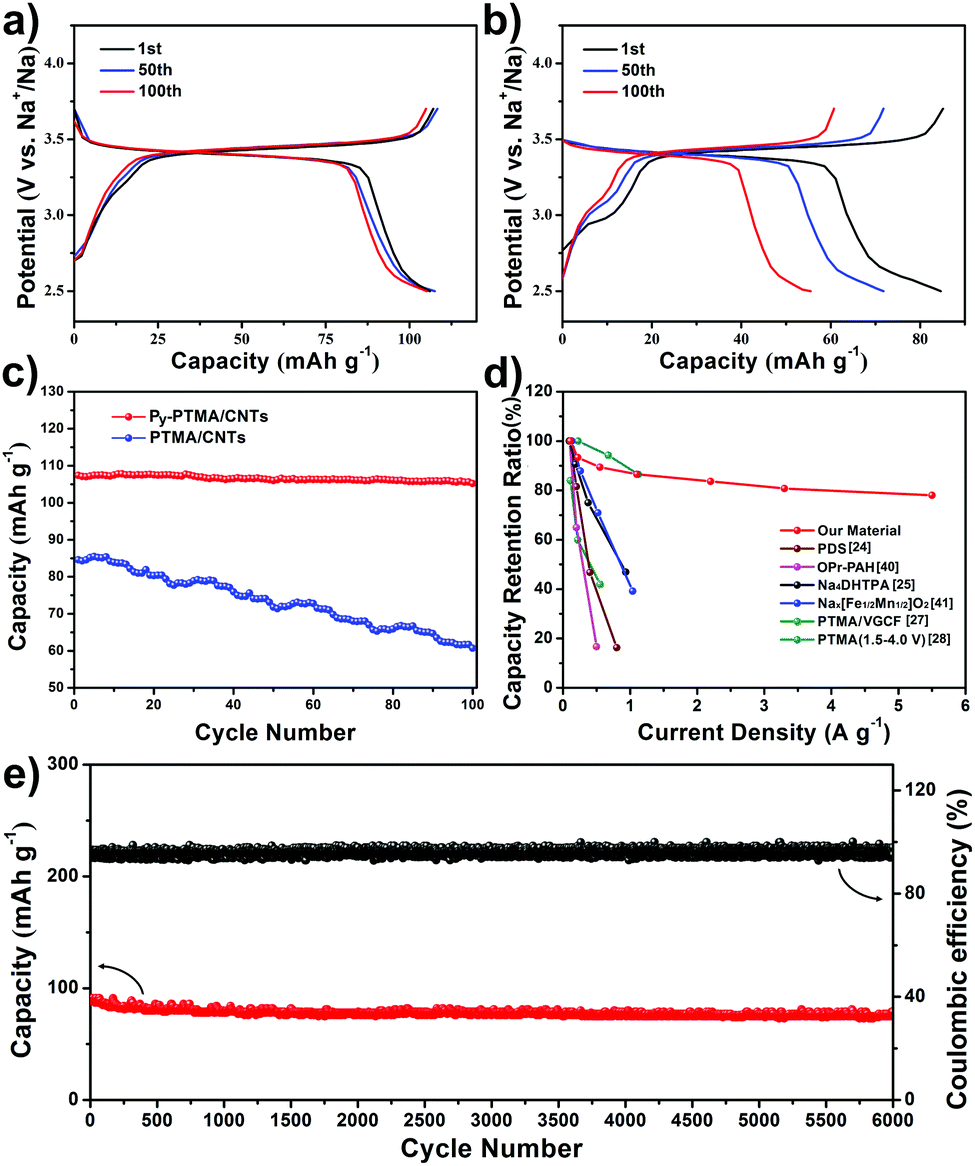 | ||
| Fig. 3 Charge–discharge curves of (a) Py-PTMA/CNTs, and (b) PTMA/CNT batteries at the 1st, 50th, and 100th cycle at 110 mA g−1 (1C-rate). (c) Discharge capacity vs. cycle number for Py-PTMA/CNTs and PTMA/CNTs at 1C. (d) Discharge capacity retention ratio of the Py-PTMA/CNTs and other representative polymer, small-molecular and inorganic cathode materials at different current densities.24,25,27,28,40,41 (e) Long-life cycling performance of the Py-PTMA/CNTs at a current density of 2.2 A g−1 (20C-rate). | ||
Two other critical parameters for the implementation of SIBs are the rate capability and long-cycling stability. Unlike the traditional rocking-chair type SIB system based on large sodium ion insertion/de-insertion, the charge and discharge processes at high current density in the radical-based electrodes corresponded to the fast kinetics reaction of oxidation and reduction between the nitroxide radical and the oxoammonium cation (Fig. S1b, ESI†). In the meantime, the inferior electron conductivity of the radical polymer was substantially increased in the Py-PTMA/CNT cathodes, which leads to the battery high power outputs. The discharge capacity retention performances of the Py-PTMA/CNT electrode under different current densities (rate capability) are compared with those of other representative polymer cathodes, small-molecular cathodes, and inorganic cathodes (in Fig. 3d).24,25,27,28,40,41 Very encouragingly, the capacity retention of the Py-PTMA/CNTs remains very stable with increased current density. Even at a high current density of 5.5 A g−1 (equivalent to 50C), our new Py-PTMA/CNT composite electrode still delivers a high capacity of 78 mA h g−1, indicating ca. 73% capacity retention over a wide range of current densities ranging from 0.11 A g−1 to 5.5 A g−1 (Fig. S6c, ESI†). In strong contrast, other polymer-based cathodes, including the PTMA/CNTs (Fig. S6c, ESI†), small-molecular organics, and transition metal oxide cathodes in SIBs exhibited relatively quick rate performance decay in a smaller current density window (<2 A g−1).24,25,28,40,41 The reaction between the nitroxide radical and oxoammonium has been proved to be highly reversible, favoring a long cycling.42Fig. 3e depicts the coulombic efficiency and cycling behavior of the Py-PTMA/CNT electrode at a high rate of 20C (current density of 2.2 A g−1). Such an electrode initially delivered a discharge capacity of 87 mA h g−1 at 20C. Even after 6000 cycles, it maintained a high capacity of 80 mA h g−1 (92% capacity retention) corresponding to a high coulombic efficiency of 96%. These features further demonstrated the superior stability and reversibility of our newly developed composite cathode materials with a core–sheath electrode microstructure. This represents a significant advance compared to other reported polymer-based cathode materials in SIBs (Table S1, ESI†), including sodium poly(pyrrole-co-(sodium-3-(pyrrol-lyl) propanesulphonate)) (84% capacity retention after 100 cycles at 0.04 A g−1),23 poly-(diphenylaminesulfonic acid sodium) (70% capacity retention after 100 cycles at 0.05 A g−1),24 PTMA with nano-fibers (64% capacity retention after 50 cycles at 0.05 A g−1),27 and oligopyrene (69% capacity retention after 100 cycles at 0.02 A g−1).40
To gain deep insights into the fast charge/discharge process, SEM, electrochemical impedance spectroscopy (EIS), and cyclic voltammograms (CV) were applied to further investigate the electrochemical process of the Py-PTMA/CNT electrode. SEM images of the electrode after 600 and 6000 cycles revealed no obvious changes in morphology (Fig. S7, ESI†). The “molecular glue” interactions of pyrene functionalities allowed the Py-PTMA chains to wrap around the CNTs that formed an independent and strong adhered core–sheath nanostructure. Because of this unique structure, electron transfer was highly favored even under a bulky electrode. Fig. S8 (ESI†) shows the CV curves of the Py-PTMA/CNT and PTMA/CNT electrodes at various sweep rates between 0.1 mV s−1 and 1.0 mV s−1. The oxidation and reduction peaks in the curves were consistent with the charge–discharge plateau in Fig. 3a.
Fig. 4a shows the relationship between the peak current density (ip) and square root of the scan rate (v1/2) of the electrodes, which were derived from the CV curves in Fig. S8 (ESI†). Generally, the electrode ion diffusion coefficient (Dion) could be determined by the dependence of the peak current density on the square root of the scan rate. On the premise of the same material, Dion would be proportional to the slope value of ipvs. v1/2 based on the following equation:43
| ip = 2.69 × 105n3/2C0Polv1/2Dion | (1) |
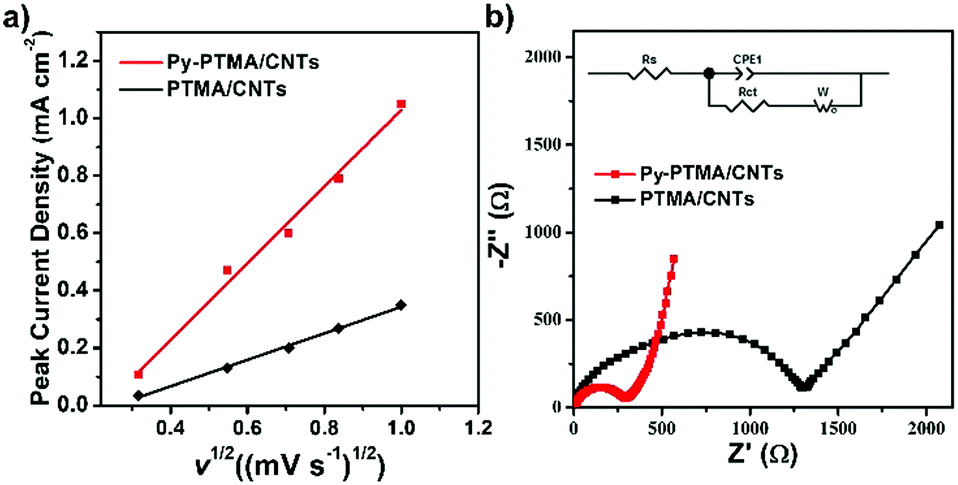 | ||
| Fig. 4 (a) Peak current density (ip) vs. square root of scan rate (v1/2) plots. (b) EIS of the Py-PTMA/CNT (red) and PTMA/CNT (black) electrodes after the 20th cycle; the inset is the equivalent circuit model. | ||
The electron transfer process of nitroxide radicals, for the first time, was also observed through in situ UV-Vis measurement to investigate the electrochemical redox reaction in SIBs. In Fig. 5, a broad adsorption at 470 nm corresponding to the nitroxide radicals was observed in the fresh electrode (grey point in charge–discharge curves), which was consistent with the observation in solution (Fig. S2a, ESI†). This adsorption gradually faded from green to blue lines during the charging process. The complete disappearance of the peak after fully charging (blue point) the battery indicated the depletion of the nitroxide radicals. This was also supported by the disappearance of the ESR signal after full charging of the battery (Fig. S9, ESI†). Whereas after being fully discharged, the UV-Vis adsorption at 470 nm nearly recovered to its original value. The appearance of the ESR signal after being re-discharged also verifies the recovery of nitroxide radicals. Taken together, the results corroborate the highly reversible redox process of nitroxide radicals during the charge/discharge process in our designed Py-PTMA/CNT composites.
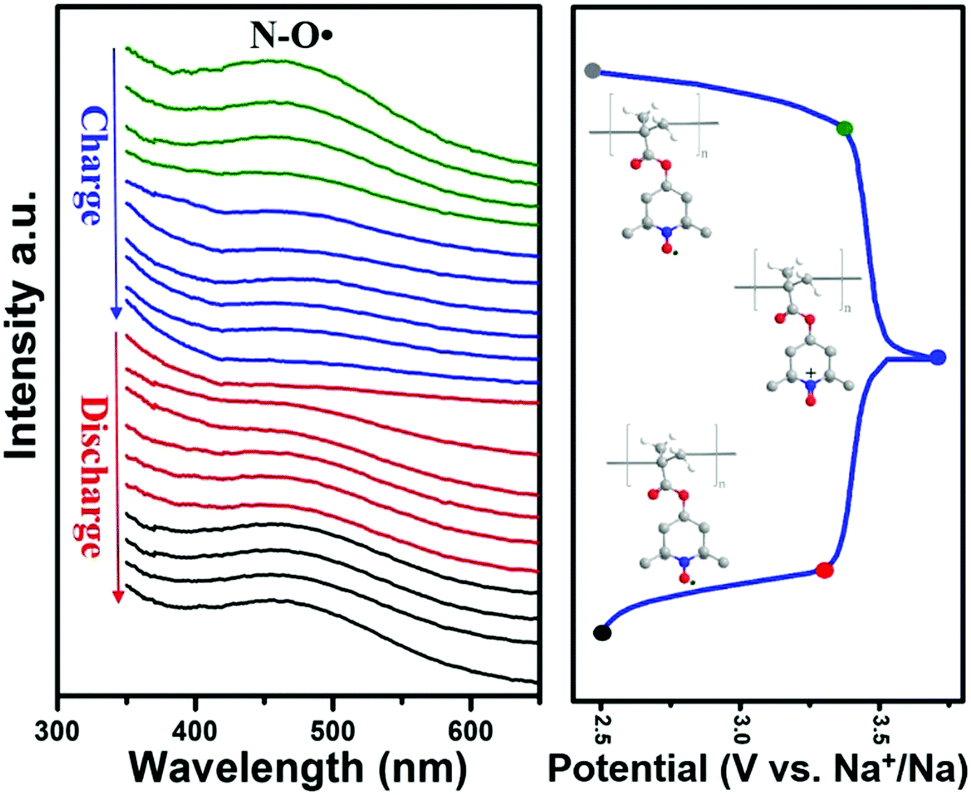 | ||
| Fig. 5 In situ UV-Vis spectra during the galvanostatic charge and discharge process at a current density of 55 mA g−1. | ||
Conclusions
In summary, we have developed a facile method to wrap NRPs onto conductive CNTs using a non-covalent “molecular glue” strategy to form a core–sheath nanostructure. Compared with previous studies on NRPs,21,25,27 this new composite Py-PTMA/CNTs displays remarkably enhanced electrochemical properties. Our Py-PTMA/CNT electrode can deliver a high capacity of 78 mA h g−1 at a very high current density of 5.5 A g−1 and achieve an extraordinary long-term capacity stability with more than 92% capacity retention after 6000 cycles at 2.2 A g−1, which to our knowledge is one of the best reported high-voltage cathode performances in SIBs to date. Detailed electrochemical studies reveal that the electrode structure possesses long-term redox reversibility, high charge and ion transfer kinetics that would be suitable for the next generation of SIBs with long lifetime and high power output. The new findings reported herein should shed light on the future design of new polymer based composite materials for low-cost and high-performance rechargeable battery applications.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Y. Hu and K. Zhang contributes equally to this work. We gratefully acknowledge the financial support from the Australian Research Council through its Discovery and Linkage Programs. Z. J. acknowledges the Australian Research Council for the Future Fellowship (FT130101442) and the award of Foundation Research Excellent Award (FREA) from the University of Queensland. This work was performed in part at the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy and Microanalysis, The University of Queensland (UQ). Y. Hu also acknowledges the support from R. T. P. and a UQ-cent Scholarship.References
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef PubMed.
- C. Liu, F. Li, L.-P. Ma and H.-M. Cheng, Adv. Mater., 2010, 22, E28–E62 CrossRef PubMed.
- J. Wang, H. Tang, L. Zhang, H. Ren, R. Yu, Q. Jin, J. Qi, D. Mao, M. Yang, Y. Wang, P. Liu, Y. Zhang, Y. Wen, L. Gu, G. Ma, Z. Su, Z. Tang, H. Zhao and D. Wang, Nat. Energy, 2016, 1, 16050 CrossRef.
- J. Wang, H. Tang, H. Wang, R. Yu and D. Wang, Mater. Chem. Front., 2017, 1, 414–430 RSC.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nano Lett., 2011, 11, 5421–5425 CrossRef PubMed.
- Y. Hu, T. Zhang, F. Cheng, Q. Zhao, X. Han and J. Chen, Angew. Chem., Int. Ed., 2015, 54, 4338–4343 CrossRef PubMed.
- C. P. Grey and J. M. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef PubMed.
- Y. Hu, B. Luo, D. Ye, X. Zhu, M. Lyu and L. Wang, Adv. Mater., 2017, 29, 1606132 CrossRef PubMed.
- R. Sun, C. Pei, J. Sheng, D. Wang, L. Wu, S. Liu, Q. An and L. Mai, Energy Storage Mater., 2018, 12, 61–68 CrossRef.
- Y. Lai, Y. Jiao, J. Song, K. Zhang, J. Li and Z. Zhang, Mater. Chem. Front., 2018, 2, 376–384 RSC.
- Y. Hu, D. Ye, B. Luo, H. Hu, X. Zhu, S. Wang, L. Li, S. Peng and L. Wang, Adv. Mater., 2018, 30, 1703824 CrossRef PubMed.
- K. Zhang, X. Han, Z. Hu, X. Zhang, Z. Tao and J. Chen, Chem. Soc. Rev., 2015, 44, 699–728 RSC.
- H. Yin, M.-L. Cao, X.-X. Yu, H. Zhao, Y. Shen, C. Li and M.-Q. Zhu, Mater. Chem. Front., 2017, 1, 1615–1621 RSC.
- H. Kim, H. Kim, Z. Ding, M. H. Lee, K. Lim, G. Yoon and K. Kang, Adv. Energy Mater., 2016, 6, 1600943 CrossRef.
- H. Zhou, Energy Environ. Sci., 2013, 6, 2256 RSC.
- Y. Zhu, L. Peng, D. Chen and G. Yu, Nano Lett., 2016, 16, 742–747 CrossRef PubMed.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 742–769 CrossRef.
- S. Muench, A. Wild, C. Friebe, B. Haupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef PubMed.
- J. Qu, R. Morita, M. Satoh, J. Wada, F. Terakura, K. Mizoguchi, N. Ogata and T. Masuda, Chem. – Eur. J., 2008, 14, 3250–3259 CrossRef PubMed.
- J. Yang, Y. Liu, S. Liu, L. Li, C. Zhang and T. Liu, Mater. Chem. Front., 2017, 1, 251–268 RSC.
- Q. Jiang, X. Lan, C. Liu, H. Shi, Z. Zhu, F. Zhao, J. Xu and F. Jiang, Mater. Chem. Front., 2018, 2, 679–685 RSC.
- R. Zhao, L. Zhu, Y. Cao, X. Ai and H. X. Yang, Electrochem. Commun., 2012, 21, 36–38 CrossRef.
- L. Zhu, Y. Shen, M. Sun, J. Qian, Y. Cao, X. Ai and H. Yang, Chem. Commun., 2013, 49, 11370–11372 RSC.
- Y. F. Shen, D. D. Yuan, X. P. Ai, H. X. Yang and M. Zhou, Electrochem. Commun., 2014, 49, 5–8 CrossRef.
- S. Wang, L. Wang, Z. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 5892–5896 CrossRef PubMed.
- T. Liu, K. C. Kim, B. Lee, Z. Chen, S. Noda, S. S. Jang and S. W. Lee, Energy Environ. Sci., 2017, 10, 205–215 RSC.
- Y. Dai, Y. Zhang, L. Gao, G. Xu and J. Xie, Electrochem. Solid-State Lett., 2010, 13, A22 CrossRef.
- J.-K. Kim, Y. Kim, S. Park, H. Ko and Y. Kim, Energy Environ. Sci., 2016, 9, 1264–1269 RSC.
- J. Winsberg, T. Hagemann, T. Janoschka, M. D. Hager and U. S. Schubert, Angew. Chem., Int. Ed., 2017, 56, 686–711 CrossRef PubMed.
- J. L. Hodgson, M. Namazian, S. E. Bottle and M. L. Coote, J. Phys. Chem. A, 2007, 111, 13595–13605 CrossRef PubMed.
- V. Georgakilas, M. Otyepka, A. B. Bourlinos, V. Chandra, N. Kim, K. C. Kemp, P. Hobza, R. Zboril and K. S. Kim, Chem. Rev., 2012, 112, 6156–6214 CrossRef PubMed.
- M. Baibarac, M. Lira-Cantú, J. Oró-Solé, N. Casañ-Pastor and P. Gomez-Romero, Small, 2006, 2, 1075–1082 CrossRef PubMed.
- W. Guo, Y.-X. Yin, S. Xin, Y.-G. Guo and L.-J. Wan, Energy Environ. Sci., 2012, 5, 5221–5225 RSC.
- K. Zhang, Y. Hu, L. Wang, M. J. Monteiro and Z. Jia, ACS Appl. Mater. Interfaces, 2017, 9, 34900–34908 CrossRef PubMed.
- K. Zhang, Y. Hu, L. Wang, J. Fan, M. J. Monteiro and Z. Jia, Polym. Chem., 2017, 8, 1815–1823 RSC.
- Z. Jia, C. A. Bell and M. J. Monteiro, Macromolecules, 2011, 44, 1747–1751 CrossRef.
- H. Nishide and T. Suga, Electrochem. Soc. Interface, 2006, 4, 32–36 Search PubMed.
- M. Yang, V. Koutsos and M. Zaiser, J. Phys. Chem. B, 2005, 109, 10009–10014 CrossRef PubMed.
- Y. Zhang, S. Yuan, W. Zhou, J. Xu and Y. Li, J. Nanosci. Nanotechnol., 2007, 7, 2366–2375 CrossRef PubMed.
- S. C. Han, E. G. Bae, H. Lim and M. Pyo, J. Power Sources, 2014, 254, 73–79 CrossRef.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512–517 CrossRef PubMed.
- A. Vlad, J. Rolland, G. Hauffman, B. Ernould and J.-F. Gohy, ChemSusChem, 2015, 8, 1692–1696 CrossRef PubMed.
- J. Yang, X. Han, X. Zhang, F. Cheng and J. Chen, Nano Res., 2013, 6, 679–687 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8qm00163d |
| ‡ Y. Hu and K. Zhang contributed equally to this work. |
| This journal is © the Partner Organisations 2018 |