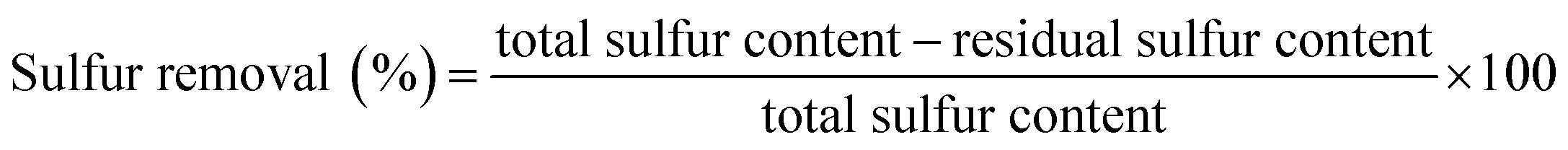DOI:
10.1039/C5RA23272D
(Paper)
RSC Adv., 2016,
6, 140-147
Heterogenization of homogenous oxidative desulfurization reaction on graphene-like boron nitride with a peroxomolybdate ionic liquid†
Received
5th November 2015
, Accepted 7th December 2015
First published on 8th December 2015
Abstract
A peroxomolybdate ionic liquid (PMo IL) was synthesized and further immobilized on graphene-like hexagonal boron nitride (PMo IL/G-h-BN). Compared with the homogenous PMo IL, the heterogeneous catalyst presented an enhanced catalytic and recyclable performance in oxidative desulfurization. The influence of different desulfurization systems, different carriers and reaction conditions indicated that the PMo IL/G-h-BN exhibited a good catalytic activity with hydrogen peroxide as the oxidant in oxidation of dibenzothiophene (DBT). The catalytic activity of PMo IL/G-h-BN shows a superior performance compared to those of other carriers supported with PMo IL catalysts, reaching 98.5% sulfur removal on DBT. This G-h-BN heterogenization of the IL strategy not only reduced the usage amount of IL active species greatly, but also enhanced the recycling performance of the catalyst. The mechanism of adsorption and catalytic oxidative desulfurization was investigated by gas chromatography-mass spectrometry (GC-MS).
1. Introduction
With the rapid expansion of the automobile industry, the consumption of fuel oil has increased greatly. Sulfur compounds in fuel oil, which can be burned into SOx, is the main cause of acid rain, PM 2.5, and some other environmental problems.1,2 Thus, removal of sulfur compounds from fuel oil has become an important issue around the world. Conventionally, sulfur compounds were removed by hydrodesulfurization (HDS),3,4 which is effective for most sulfur compounds. However, for aromatic sulfur compounds, its harsh reaction conditions,5,6 including high reaction temperature, high pressure and hydrogen are needed, limiting its further application. Under this circumstance, non-hydrodesulfurization methods such as oxidative desulfurization (ODS),7–14 adsorptive desulfurization (ADS),15–19 extractive desulfurization (EDS)20–23 and biodesulfurization (BDS) were researched widely.24 Among all these methods, ODS has been proved to be a promising one due to its mild reaction condition and high activity to aromatic sulfur compounds. In ODS process, sulfur compounds can be oxidized to their corresponding sulfones or sulfoxides and then was separated by extractants or adsorbents.25–28
Task-specific ionic liquids (TSILs), owing to their negligible vapor pressure, thermal chemical stability and good extracting ability, etc. are regarded as remarkable desulfurization catalysts.29–34 However, the large amount consumption of ILs and their poor recycling are inevitable.35 To solve these problems, heterogenization of homogeneous TSILs onto supports has been employed.36–38 The most common supports were mesoporous silica,39–44 mesoporous polymers,45 alumina,46,47 activate carbon,48 etc. Though supported ILs catalysts can reduce the usage amount of IL active species greatly and enhanced recycling performance, seldom supported IL catalysts exhibited much better superiority than homogeneous catalysts of ILs themselves.45 More recently, graphene-like hexagonal boron nitride (G-h-BN) has attracted increasing interests due to its layer-structure and high specific surface area.49–51 When G-h-BN was applied as a support for immobilization of TSILs, its large specific surface area can provide high-dispersion of ILs. Moreover, the excellent adsorption ability of G-h-BN to aromatic sulfur compounds can provide a micro-environment with high-concentration of substrates, which can enhance desulfurization efficiency.
In this work, a peroxomolybdate ionic liquid [C14H29P(C6H13)3]2Mo2O11 (PMo IL) was prepared and immobilized on G-h-BN to obtain a heterogeneous catalyst (PMo IL/G-h-BN). The two components show excellent synergetic effects on adsorption and catalytic oxidative desulfurization.
2. Experimental
2.1. Materials
Boric acid (H3BO3, A.R. grade), urea (CO(NH2)2, A.R. grade), hydrogen peroxide (H2O2, 30 wt%), n-octane (A.R. grade), molybdic acid sodium (Na2MoO4·2H2O, A.R. grade) were purchased from Shanghai Sinopharm Chemical Reagent Co., Ltd. [C14H29P(C6H13)3]Cl, dibenzothiophene (DBT, 98%), benzothianaphthene (BT, 99%), 4,6-dimethyldibenzothiophene (4,6-DMDBT, 97%), tetradecane (99%) were purchased from Sigma-Aldrich.
2.2. Catalyst preparation
2.2.1. Synthesis of G-h-BN. A mixed solution was prepared by dissolving 0.01 mol of H3BO3 and 0.24 mol of urea in a mixture of 30 mL ultra-pure water and 30 mL ethanol. Then, the homogenous solution was heated under stirring to volatilize solvents at 40 °C. Subsequently, the obtained solid mixtures were heated from 30 °C to 900 °C at a heating rate of 5 °C min−1 in a tube furnace and maintained at 900 °C for 2 h.52
2.2.2. Synthesis of peroxomolybdate ionic liquid [C14H29P(C6H13)3]2Mo2O11 (PMo IL). An aqueous solution (A) by dissolving 5 mmol of Na2MoO4·2H2O in 10 mL of ultra-pure water followed by adding 6 mL of 30% hydrogen peroxide (H2O2) dropwise in an ice bath with stirring continuously. Then diluted hydrochloric acid was added slowly until the pH of the solution adjusted to 4.3. Simultaneously, 10 mmol of [C14H29P(C6H13)3]Cl was dissolved in 20 mL of ethanol which was added into the above solution A. After stirring for 24 h, the stationary solution presents two phases. Then the upper phase was removed and washed by CH2Cl2, and finally dried in vacuum at 60 °C overnight.53
2.2.3. Synthesis of PMo IL/G-h-BN catalysts. A certain amount of PMo IL was dissolved in 5 mL of ethanol followed by adding G-h-BN, and then kept stirring for 5 h. After stirring, the mixture was dried at 50 °C to volatilize the ethanol and finally dry at 100 °C. Based on different mass percentages, the as-prepared catalysts were marked as 5% PMo IL/G-h-BN, 10% PMo IL/G-h-BN, 20% PMo IL/G-h-BN and 50% PMo IL/G-h-BN, respectively. Here, 5% PMo IL/G-h-BN (as an example) means the mass percentage of PMo IL and G-h-BN is 5% and 95% in this catalyst, respectively.
2.3. Characterization
Fourier transform infrared (FT-IR) spectra of the catalysts were recorded on a Nicolet Nexus 470 Fourier transform infrared pectrometer, using KBr pellets at room temperature. The different loading amount of PMo IL/G-h-BN was measured by a Thermo Scientific DXR Smart Raman spectrometer with a excitation light of 532 nm. G-h-BN and PMo IL/G-h-BN were analyzed by X-ray diffraction (XRD) using D8 Advance X-ray diffraction with Cu Kα radiation (λ = 1.54 Å) in the range of 2θ = 10–80°. The structure and morphology of the catalysts were examined by Leo Supra 35VP field emission scanning electron microscope (SEM) at 2–15 keV accelerating voltage. Transmission electron microscopy (TEM) experiments were performed on a Hitachi H-700 Transmission electron microscope. Atomic force microscopy (AFM) studies were used to investigate the number of layers of G-h-BN. Brunauer–Emmett–Teller (BET) surfaces were obtained from a nitrogen adsorption–desorption isotherm. X-ray photoelectron spectroscopy (XPS) spectra were recorded on a ESCALAB250 (Thermo VG, UK), in which standard monochromatic Al Ka excitation was 1486.6 eV. Gas chromatography-mass spectrometry (GC-MS) was taken to research the mechanism of adsorption coupled with oxidative desulfurization.
2.4. Oxidation of sulfur compounds in model oil
2.4.1. Preparation of model oil. DBT was dissolved in 1000 mL of n-octane. The sulfur content of DBT in model oil was 500 ppm. With the same method, other model oils were prepared by dissolving 4,6-DMDBT and BT in n-octane, with the corresponding S-content of 500 ppm and 250 ppm, respectively.
2.4.2. Oxidative desulfurization process. The oxidative desulfurization experiments were performed in a 40 mL home-made two-necked flask in water bath. In a typical process, the water bath was heated to 60 °C. After catalyst was added to the flask, 5 mL model oil and a certain amount of 30 wt% H2O2 were injected to the flask, and then the mixture was stirred with magnetic agitation in the thermostatic water bath. After reaction, the upper phase (model oil) was periodically withdrawn and analyzed by a gas chromatography (Agilent 7890A, HP-5 column, 30 m long × 0.32 mm inner diameter (id) 0.25 μm film thickness), coupled with a flame ionization detector (GC-FID). Sulfur removal was used to indicate the conversion of sulfur-compounds, which was calculated as following.
3. Results and discussion
3.1. Catalyst characterizations
FT-IR spectra of PMo IL, G-h-BN and different PMo IL/G-h-BN samples were shown in Fig. 1. For PMo IL, the peak around 2926 cm−1 can be indexed to C–H stretching vibration and 980 cm−1 band is the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration modes. 884 and 915 cm−1 bands are peroxy O–O stretching vibration modes. 641 cm−1 band is [Mo(O)2] symmetrical stretching vibration mode, while the 585 cm−1 band is [Mo(O)2] asymmetrical stretching vibration mode.53 For G-h-BN, the absorption band centered around 1381 cm−1 can be attributed to the in-plane B–N transverse optional modes of hexagonal boron nitride,52 while the last peak around 804 cm−1 can be attributed to N–B stretching vibration mode.54 A broad peak can be seen at 3405 cm−1 belonging to –OH vibrations in G-h-BN. After immobilizing PMo IL, the characteristic bands of G-h-BN are easily observed and similar to the pure G-h-BN in spite of the different contents of PMo IL. The C–H stretching vibration peak of PMo IL can be found in the IR spectrum of PMo IL/G-h-BN with the mass ratio of the PMo IL increased. These results indicated that PMo IL has uniformly dispersed on the surface of G-h-BN. Moreover, G-h-BN was intact after supported IL.
O stretching vibration modes. 884 and 915 cm−1 bands are peroxy O–O stretching vibration modes. 641 cm−1 band is [Mo(O)2] symmetrical stretching vibration mode, while the 585 cm−1 band is [Mo(O)2] asymmetrical stretching vibration mode.53 For G-h-BN, the absorption band centered around 1381 cm−1 can be attributed to the in-plane B–N transverse optional modes of hexagonal boron nitride,52 while the last peak around 804 cm−1 can be attributed to N–B stretching vibration mode.54 A broad peak can be seen at 3405 cm−1 belonging to –OH vibrations in G-h-BN. After immobilizing PMo IL, the characteristic bands of G-h-BN are easily observed and similar to the pure G-h-BN in spite of the different contents of PMo IL. The C–H stretching vibration peak of PMo IL can be found in the IR spectrum of PMo IL/G-h-BN with the mass ratio of the PMo IL increased. These results indicated that PMo IL has uniformly dispersed on the surface of G-h-BN. Moreover, G-h-BN was intact after supported IL.
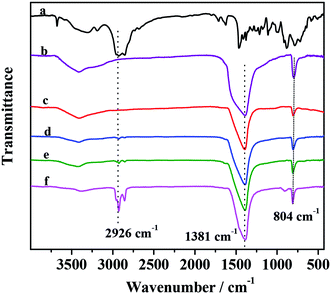 |
| | Fig. 1 FT-IR spectra of catalysts (a) PMo IL; (b) G-h-BN; (c) 5% PMo IL/G-h-BN; (d) 10% PMo IL/G-h-BN; (e) 20% PMo IL/G-h-BN; (f) 50% PMo IL/G-h-BN. | |
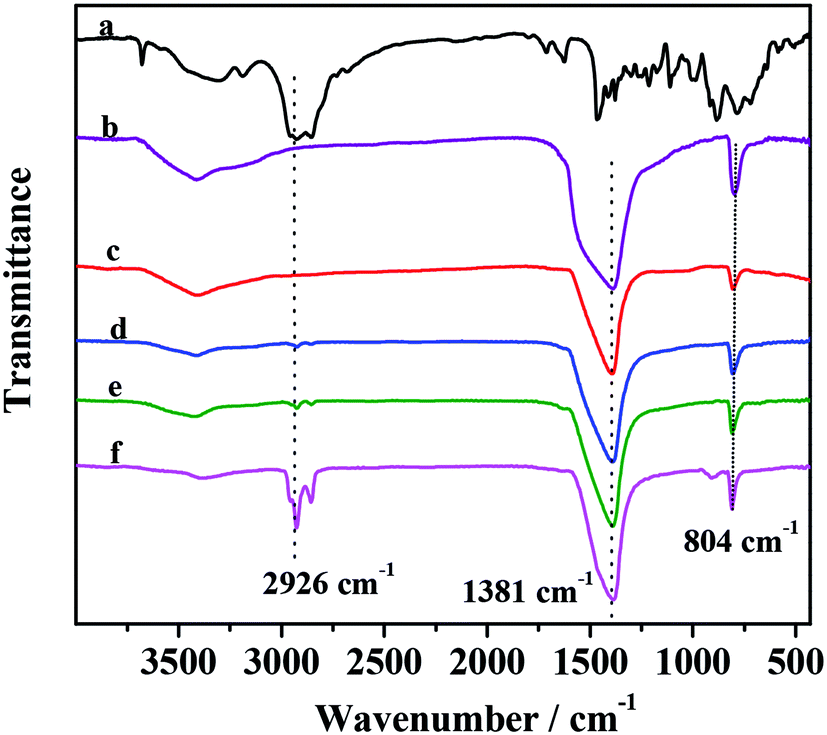
Additional structural information concerning the presence of PMo IL and G-h-BN unit in the catalysts was obtained from Raman spectra (Fig. 2). It was observed obviously that there was a peak in Raman spectra of supported catalysts around 1372 cm−1, which was assigned to the E2g tangential mode of G-h-BN.55 In the sample with different PMo IL content, the E2g value was the same to G-h-BN, proving that the structure of G-h-BN was not changed after fixing with PMo IL. The peak at 2926 cm−1 was the C–H stretching vibration, which became more intense and defined with the increase of PMo IL content in the catalysts indicating that the PMo IL had been immobilized onto the G-h-BN.
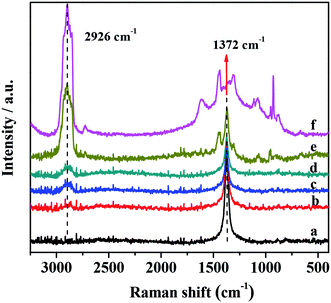 |
| | Fig. 2 Raman spectra of catalysts (a) G-h-BN; (b) 5% PMo IL/G-h-BN; (c) 10% PMo IL/G-h-BN; (d) 20% PMo IL/G-h-BN; (e) 50% PMo IL/G-h-BN; (f) PMo IL. | |
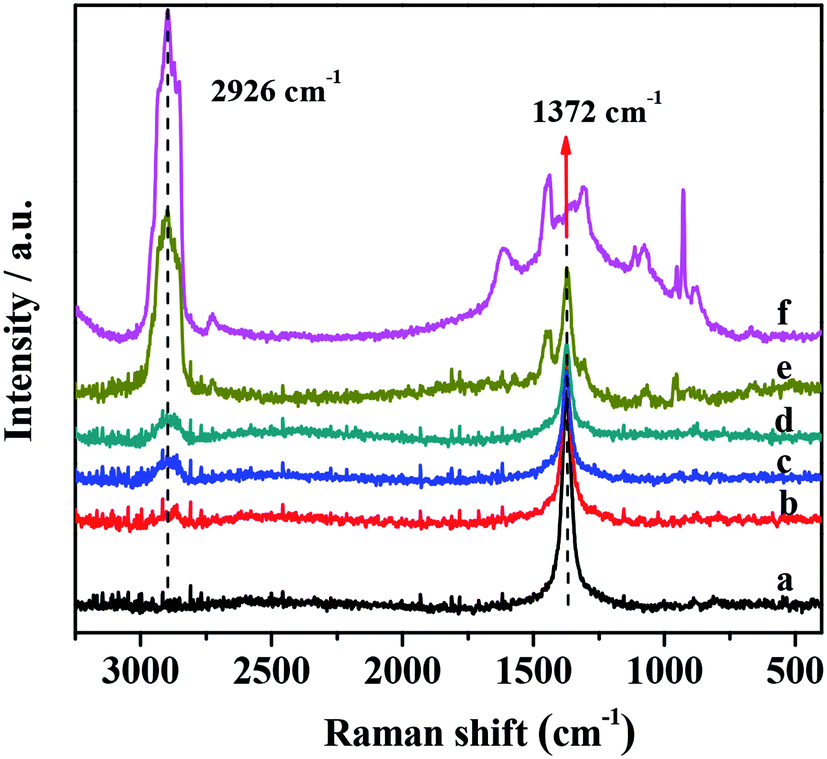
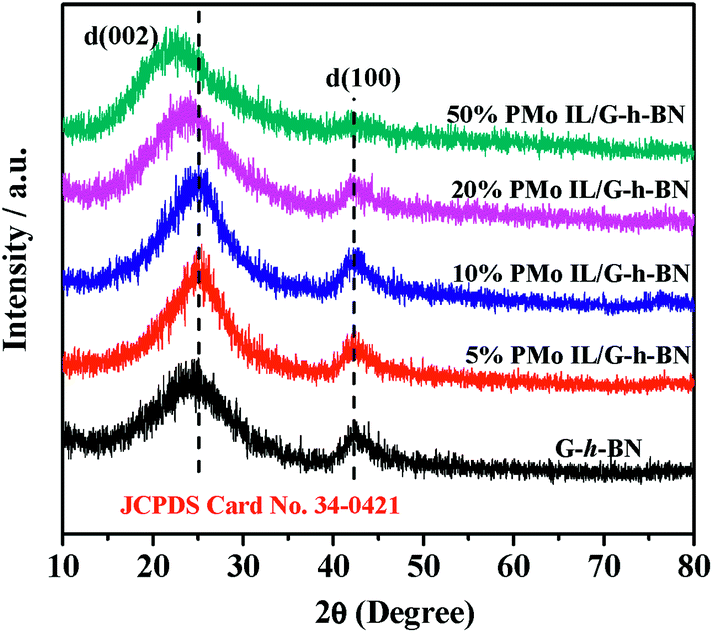
The XRD patterns of G-h-BN and PMo IL/G-h-BN were shown in Fig. 3, the peaks (002) and (100) of G-h-BN were readily indexed to the standard hexagonal phase of h-BN (JCPDS card no. 34-0421), proving the as-prepared BN is a hexagonal structure. When 5%, 10%, 20% and 50% PMo IL were loaded onto G-h-BN, a small gradual shift from 2θ = 24° to low angles could be observed. This indicates that the interdistance between the (002) basal planes can be increased through intercalation of the IL in the inter-layer space.56 With the increment of loading amount of PMo IL, the amount of IL in the inter-layer space of G-h-BN increased as well. The electrostatic attraction between PMo IL and the surface G-h-BN become stronger and interlayer distance may increase.
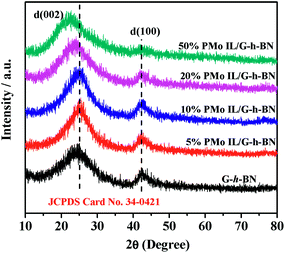 |
| | Fig. 3 XRD of catalysts. | |
3.2. Influence of the loading dose of PMo IL on sulfur removal
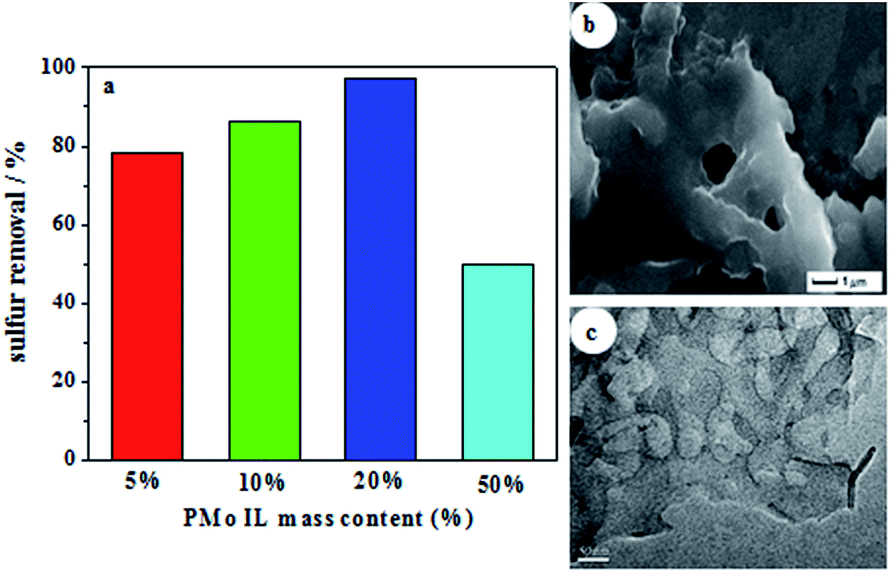
Sulfur removals of different PMo IL mass content on G-h-BN were depicted in Fig. 4a. Sulfur removal improved gradually with the increase of the PMo IL content. Among the results, the sulfur removal with 20% PMo IL/G-h-BN could reach the highest (97.2%), which was attribute to its abundant exposed active site. But when the content of PMo IL was 50%, sulfur removal decreased to 46.7%, which may be caused by the reason that the excess PMo IL made G-h-BN aggregate, leading to the decrease of sulfur removal. In order to prove this hypothesis, SEM and TEM images of 50% PMo IL/G-h-BN were performed (Fig. 4b and c) and the results indicated that 50% PMo IL/G-h-BN aggregated seriously. In order to achieve deep desulfurization, the mass content of 20% PMo IL turned out to be an appropriate amount in the following study.
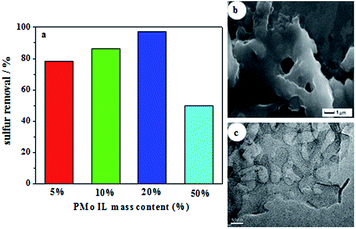 |
| | Fig. 4 Investigation of IL content on sulfur removal (a) sulfur removal of different catalysts (b) SEM image of 50% PMo IL/G-h-BN; (c) TEM image of 50% PMo IL/G-h-BN. Reaction conditions: T = 60 °C; n(O)/n(S) = 4.0; V(oil) = 5 mL; m(catalyst) = 0.05 g; t = 80 min. | |
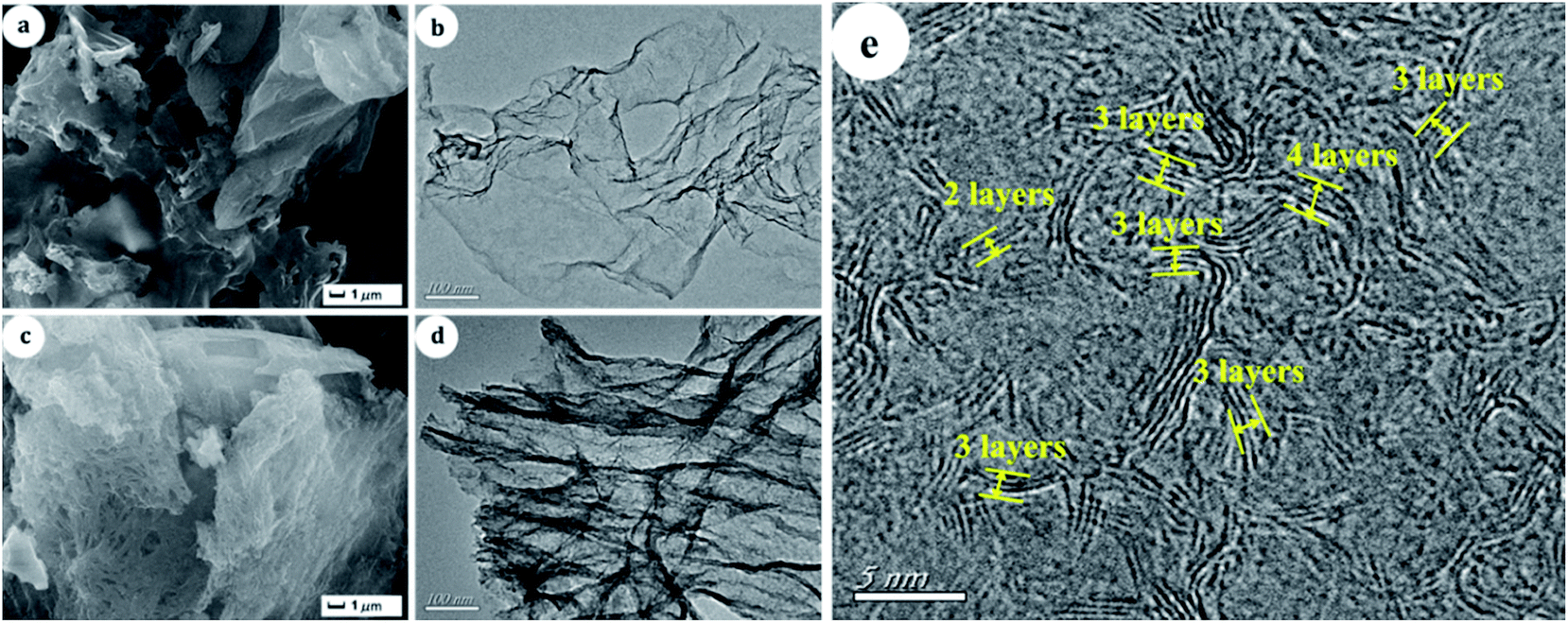
For comparison, the morphology of G-h-BN and 20% PMo IL/G-h-BN were displayed in Fig. 5. SEM and TEM images of G-h-BN (Fig. 5a and b) presented a lamellar crystal without stacking faults existing on the surface. Furthermore, from the HRTEM image of G-h-BN (Fig. 5e), it can be seen that G-h-BN was a few-layer material with the layer number of 2–4, which further proved by the AFM (Fig. S1†). The results of AFM indicated that the height of G-h-BN flake is about 0.8–1.5 nm, corresponding to the number of 2–4 layers.57 This special layered structure presents large BET surface. Compared with the commercial bulk BN (10 m2 g−1), the specific surface of G-h-BN was 456 m2 g−1. In addition, after immobilizing 20% PMo IL on G-h-BN, the structure of G-h-BN was not destroyed. Only a little crispation of G-h-BN could be observed (Fig. 5c and d). The layered structure and high dispersion of PMo IL on G-h-BN was found, which led to the highest catalytic activity.
 |
| | Fig. 5 SEM and TEM images of samples (a) and (c): SEM of G-h-BN and 20% PMo IL/G-h-BN; (b) and (d): TEM images of G-h-BN and 20% PMo IL/G-h-BN; (e): HRTEM image of G-h-BN. | |
To obtain detailed information about the chemical properties of surface elements, XPS spectra of as-prepared catalysts have been taken (Fig. S2†). All elements such as B, N, C, O, P, Mo could be detected in the spectrum, and the binding energies for B 1s and N 1s peaks were 190.3 eV and 398.2 eV, which matched with previous report for a hexagonal phase BN.57
Through the surface wettability of G-h-BN and 20% PMo IL/G-h-BN determination (Fig. 6), we found another reason can be attribute to the high catalytic activity in ODS. When a water droplet contacted the sheet of G-h-BN, a contact angle of 97° was appeared, implying its moderate hydrophilicity. While a lowered contact angle of 75° was detected with a water droplet contacting the sheet of 20% PMo IL/G-h-BN, indicating better hydrophilicity than G-h-BN. With n-octane as the testing droplet, the contacted angles of G-h-BN and 20% PMo IL/G-h-BN were almost zero, demonstrating superwettability for n-octane. Therefore, after immobilization of PMo IL on G-h-BN, this catalyst showed better wettability for both water and n-octane, similar to amphiphilic catalyst, which played a vital role of enhancing contact of both H2O2 and sulfur compounds and presented high catalytic activity. As we all know, oxidative desulfurization of fuels with aqueous H2O2 as oxidant is a typical water/oil biphase reaction. In the absence of phase-transfer co-catalyst, numerous catalysts exhibit low activity because of phase partitioning effect. So the as-prepared 20% PMo IL/G-h-BN acts as an amphiphilic catalyst.
 |
| | Fig. 6 Contact angle for a water (a and b) and n-octane (c and d) droplet on the surface of samples (a, c): G-h-BN and (b, d): 20% PMo IL/G-h-BN. | |
3.3. Influence of different carriers on sulfur removal of DBT
In order to highlight the superiority of G-h-BN carrier, the homogeneous catalysts of the ILs themselves and the catalysts of PMo IL supported on different carriers were introduced to the oxidative desulfurization experiments (Table 1). Sulfur removal was 5.1% without any catalyst. Compared with 97.2% (TOF = 7.71 h−1) sulfur removal of PMo IL/G-h-BN, sulfur removal of ILs themselves was only 45.1% (TOF = 3.58 h−1) under the same reaction conditions, indicating that heterogeneous catalysts of ILs functionalized on G-h-BN could exhibit much higher activities in oxidative desulfurization than homogeneous catalysts of the ILs themselves. Moreover, the usage amount of IL was very low with G-h-BN as support. When commercial BN active carbon and SBA-15 were used to support PMo IL, the sulfur removals were only 43.6%, 45.4% and 42.2% with the same reaction condition of PMo IL/G-h-BN, corresponding to TOF 3.46, 3.6 and 3.35 h−1, respectively. After optimizing reaction conditions, although the catalytic activity of PMo IL/activate carbon and PMo IL/SBA-15 could be enhanced, reaching 66.4% and 94.6% sulfur removals, the TOF decreased to 1.18 and 1.68 h−1. These results demonstrated that G-h-BN was a better carrier with relative high TOF based on PMo IL owning to its unique graphene-like hexagonal structure and high adsorption capacity of DBT.58 Simultaneously, excellent synergetic effects between G-h-BN and PMo IL also enhanced the catalytic activity of PMo IL/G-h-BN in ODS system.
Table 1 Effect of different supports on sulfur removala
| Entry |
Catalytic system |
mcatalyst (g) |
Sulfur removal (%) |
TOF (h−1) b |
| Reaction condition: T = 60 °C; n(O)/n(S) = 4.0; V(oil) = 5 mL; t = 80 min; the mass ratio of PMo IL in PMo IL/G-h-BN is 20%. Based on PMo IL. t = 3 h. |
| 1 |
Without catalyst |
— |
5.1 |
— |
| 2 |
PMo IL |
0.01 |
45.1 |
3.58 |
| 3 |
PMo IL/G-h-BN |
0.05 |
97.2 |
7.71 |
| 4 |
PMo IL/commercial BN |
0.05 |
43.6 |
3.46 |
| 5 |
PMo IL/activate carbon |
0.05 |
45.4 |
3.6 |
| 6 |
PMo IL/SBA-15 |
0.05 |
42.2 |
3.35 |
| 7 |
PMo IL/activate carbon |
0.1 |
64.7, 66.4c |
2.57, 1.18c |
| 8 |
PMo IL/SBA-15 |
0.1 |
69.6, 94.6c |
2.76, 1.68c |
3.4. Optimization of oxidative desulfurization parameters
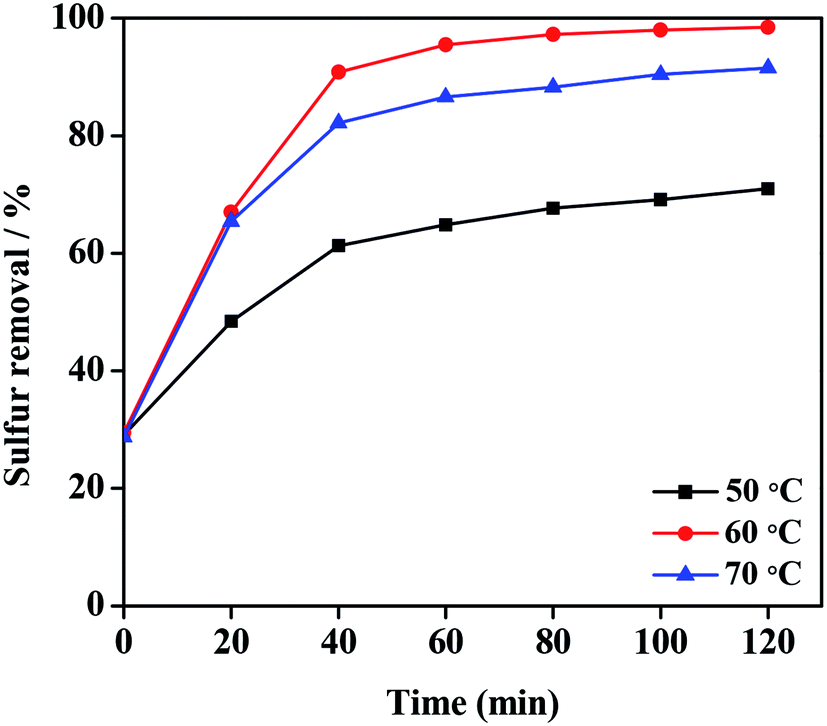
The sulfur removal of DBT was tested at different temperature to investigate the performance of 20% PMo IL/G-h-BN in the desulfurization system (Fig. 7). Temperature was a main factor of reaction conditions in the desulfurization system, and the sulfur removal was improved by prolonging the reaction time at all temperatures. The value at time zero reflected the absorption capability of PMo IL/G-h-BN to sulfur compounds from model oil at room temperature in 15 min. At 50 °C, the catalytic activity could not reach good effect, which may result from low efficiency under 50 °C. Similarly, the sulfur removal at 70 °C was not so good as that at 60 °C, which may be attribute to that the higher temperature caused the faster self-decomposition of H2O2. Thus 60 °C was chosen as the proper temperature and 80 min as the proper reaction time. In order to find the most appropriate amount of oxidant, the desulfurization effect was studied under different n(H2O2)/n(DBT) molar rate (O/S) at 60 °C with 20% PMo IL/G-h-BN as the catalyst, and the results were shown in Fig. S3.† The desulfurization performance became better with the increase of molar rate to O/S = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The optimal value of O/S = 4 was higher than the stoichiometric value, it was because that there were two competing reactions existing forever between the oxidation of DBT by H2O2 and the nonproductive self-decomposition of H2O2.59
:1. The optimal value of O/S = 4 was higher than the stoichiometric value, it was because that there were two competing reactions existing forever between the oxidation of DBT by H2O2 and the nonproductive self-decomposition of H2O2.59
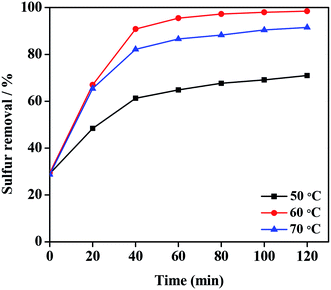 |
| | Fig. 7 Effects of temperature and reaction time on sulfur removal. Reaction condition: Voil = 5 mL, m(catalyst) = 0.05 g, n(O)/n(S) = 4.0. | |
3.5. Influence of different sulfur-containing compounds on sulfur removal
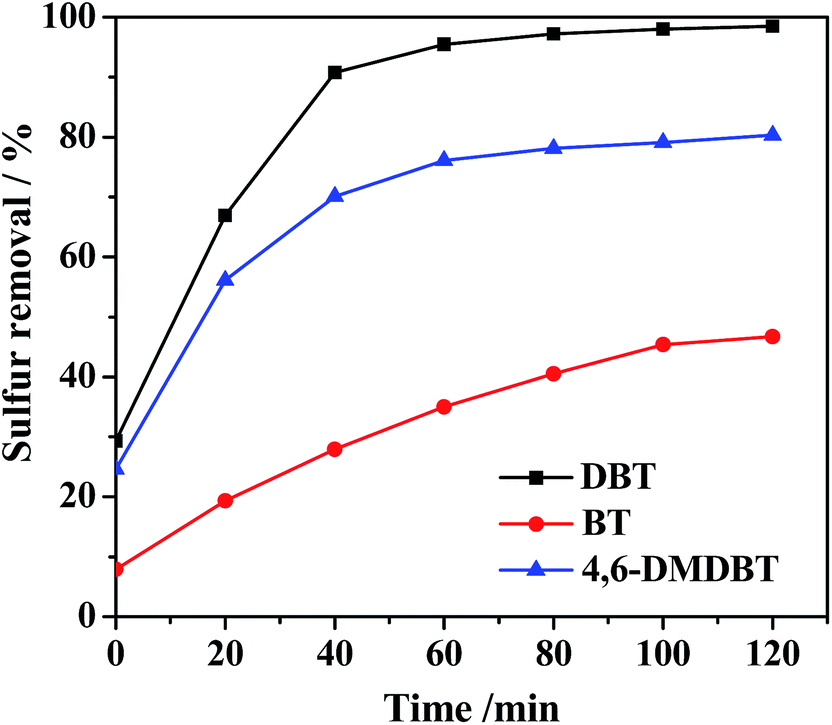
To study the effect of the 20% PMo IL/G-h-BN on the various sulfur compounds, the oxidation of three refractory sulfur compounds such as DBT, 4,6-DMDBT and BT were evaluated. As shown in Fig. 8, the value at time zero reflected the capability of PMo IL/G-h-BN to adsorb sulfur compounds from model oil at room temperature in 15 min. Sulfur removals of DBT, 4,6-DMDBT and BT were 29.3%, 24.6% and 7.9%, respectively. After addition of H2O2, a combination of adsorption and catalytic oxidation system was established and increased sulfur removal sharply, achieving 98.5%, 80.3% and 46.7% sulfur removal in 120 min. The reactivity of sulfur removal decreased in the order: DBT > 4,6-DMDBT > BT. The reasons may be attributed to the following. The adsorption capacity of 20% PMo IL/G-h-BN of DBT and 4,6-DMDBT was better than BT, and the electron density of DBT, 4,6-DMDBT and BT was 5.758, 5.760 and 5.739.60 Hence the beginning reaction rate of DBT and 4,6-DMDBT was faster than the reaction rate of BT, and the final sulfur removal was also more effective. Meanwhile the smaller space steric hindrance facilitated the best effect of sulfur removal of DBT.
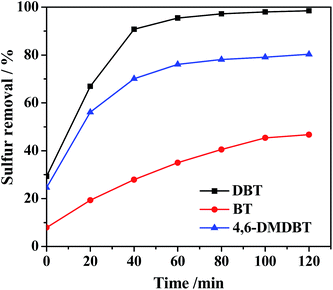 |
| | Fig. 8 Effects of different sulfur-containing compounds on sulfur removal. Reaction condition: T = 60 °C; n(O)/n(S) = 4.0; V(oil) = 5 mL; m(catalyst) = 0.05 g. | |
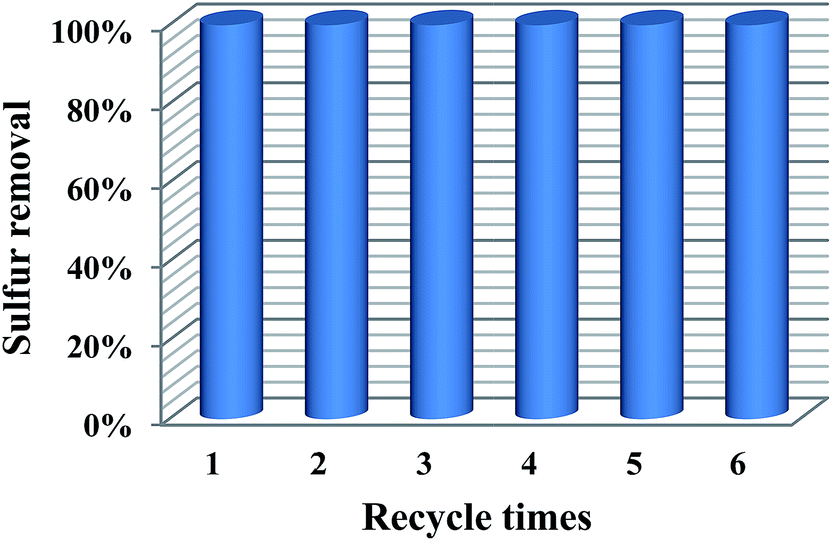
3.6. The recycling performance of the catalyst
Recycling performance is an important criterion of an excellent catalyst. Thus, it is necessary to investigate the recycling performance of 20% PMo IL/G-h-BN. After the first run, the catalyst was separated by centrifugalization. And then the catalyst was placed in a 100 °C oven to make the regenerated catalyst fully dried before the next run. Subsequently, the regenerated catalyst, fresh model oil, and H2O2 were added into the reaction vessel for the next circle. The results in Fig. 9 indicated that the 20% PMo IL/G-h-BN could be recycled six times without significant decline of reactivity.
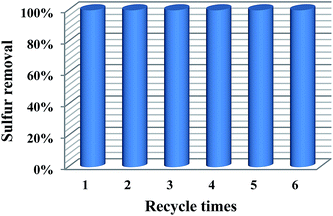 |
| | Fig. 9 Recycling capability of the catalyst. | |
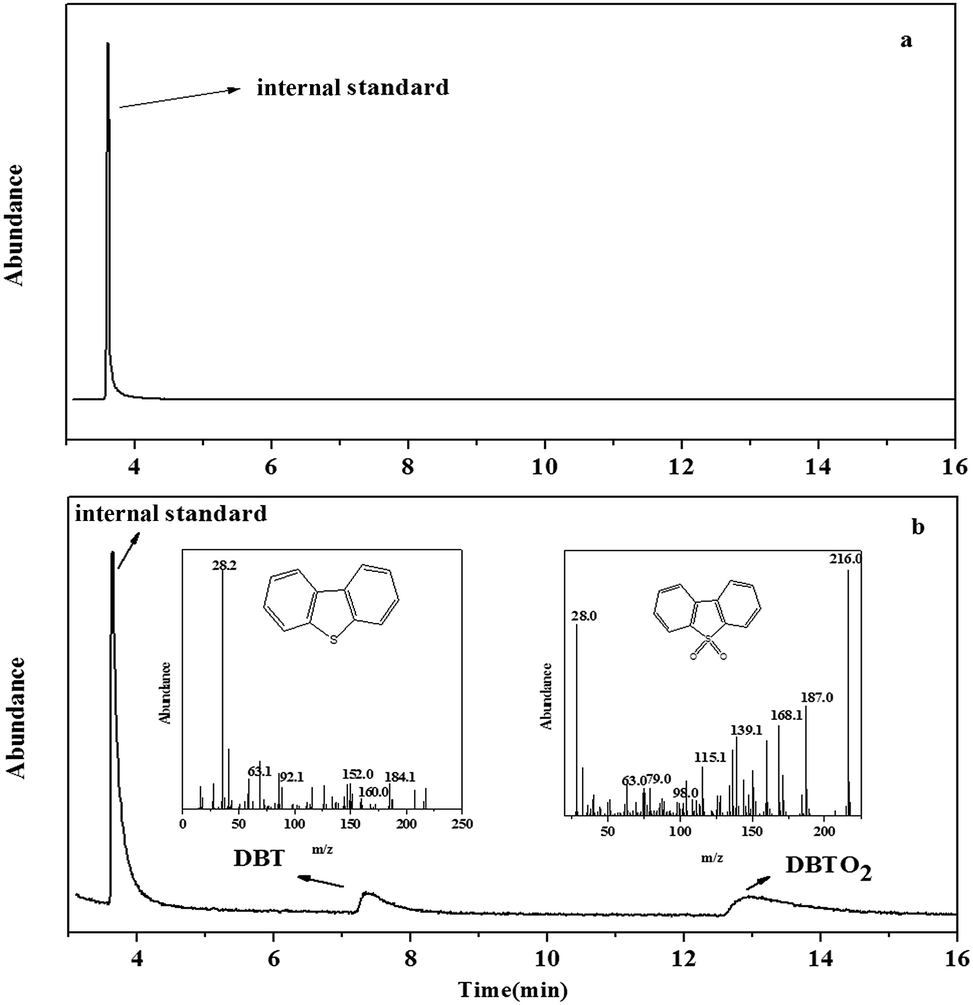
3.7. Investigation of reaction mechanism
In order to research the mechanism of adsorption and catalytic oxidative desulfurization, the components in model oil and catalyst were analyzed by GC-MS. After the reaction, the upper layer oil was analyzed directly, while the catalyst phase was centrifugalized and re-extracted by tetrachloromethane (CCl4). And then the CCl4 was analyzed by GC-MS. There was no sulfur compound in the oil, which was proved by GC-MS (Fig. 10a). The results in Fig. 10b showed that DBT and DBTO2 were existed in the catalyst. So we proposed that DBT was firstly adsorbed on the surface of PMo IL/G-h-BN and then oxidized to corresponding sulfone by H2O2.
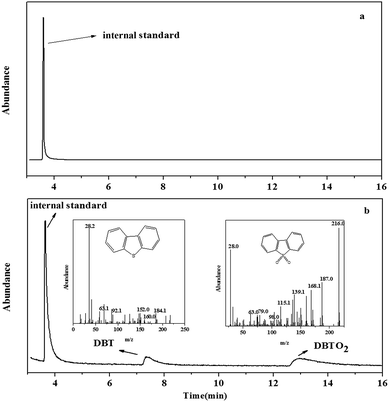 |
| | Fig. 10 GC-MS analysis of oil (a) and catalyst phase (b). | |
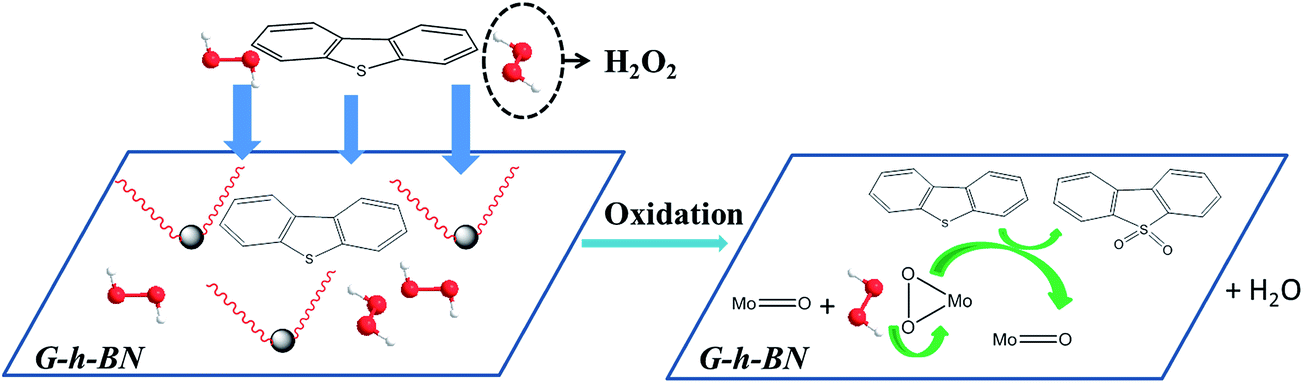
On the basis of abovementioned results, the mechanism of this reaction was speculated in Scheme 1. The excellent activity of 20% PMo IL/G-h-BN was not only assigned to the large surface of G-h-BN and high dispersion of PMo IL, but also attribute to the special two-dimensional structure of G-h-BN. DBT and H2O2 can be adsorbed to the surface of catalyst. Meanwhile, the lone pair electrons in S atom or the occupied π-electrons of planer-structure DBT molecules may donate to the virtual orbitals of B, enhancing the absorbability of the catalyst to substrates.9 Similarly, H2O2 was adsorbed on the surface of the catalyst and combined with the PMo IL to oxidize DBT. The large surface area of G-h-BN led to high dispersion of PMo IL and exposed more active sites for adsorption of DBT and H2O2. Partial high concentration of DBT on the surface of catalyst can be formed, which can enhance the contact and reaction of DBT with peroxo species. Synchronously, once DBT was oxidized on the surface of catalyst, the unoxidized substrates in the oil phase might be transferred onto the surface of catalyst to keep the relative high concentration of DBT and H2O2. On the basis of these results, the overall mechanism of the adsorption and catalytic oxidation desulfurization can be supposed as following. DBT and H2O2 were adsorbed onto the surface of catalyst to form a partial high concentration; H2O2 reacted with the Mo2O112− to form corresponding peroxo species (abbreviated as Mo(O)2), then Mo(O)2 further reacted with adjacent adsorbed DBT on the surface of catalyst and produced DBTO2.
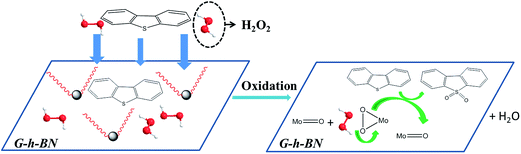 |
| | Scheme 1 The proposed mechanism of oxidation of DBT on 20% PMo IL/G-h-BN. | |
4. Conclusions
In summary, immobilization of PMo IL on G-h-BN was designed and characterized by a series of analysis methods. And 20% PMo IL/G-h-BN was found the better reaction activity in the experiment. PMo IL/G-h-BN catalyst exhibited much higher catalytic activity than PMo IL themselves and other carrier catalysts. The sulfur removal of DBT-containing model oil could achieve to 98.5% at 60 °C. The reaction mechanism was discussed in detail, and GC-MS was also used to further research the reaction mechanism and products. The superiorities of this desulfurization system are as following. (1) Large specific surface area of G-h-BN can make the usage of PMo IL decrease greatly and the active sites highly dispersed on the surface of G-h-BN. (2) G-h-BN owns prominent adsorption ability of DBT and H2O2 with special layered structure, which can form a reaction microenvironment of local high concentrations on the surface of catalysts. (3) The recycling of PMo IL/G-h-BN is easy and the regenerated catalyst could be recycled six times with no significant decrease in its reaction activity. This work indicated that immobilization IL catalysts would continue to be widely used in catalytic oxidative desulfurization system.
Acknowledgements
We thank the National Nature Science Foundation of China (Nos. 21376111, 21576122, 21506083), Six Big Talent Peak in Jiangsu province (JNHB-004), the Natural Science Foundation of Jiangsu Province (BK20131207), the State Key Laboratory of Heavy Oil Processing (SKLOP201402004).
References
- W. S. Zhu, C. Wang, H. P. Li, P. W. Wu, S. H. Xun, W. Jiang, Z. G. Chen, Z. Zhao and H. M. Li, Green Chem., 2015, 17, 2464–2472 RSC
 .
. - F. T. Li, R. H. Liu, J. H. Wen, D. S. Zhao, Z. M. Sun and Y. Liu, Green Chem., 2009, 11, 883–888 RSC .
- Y. S. Chi, C. P. Li, Q. Z. Jiao, Q. S. Liu, P. F. Yan, X. M. Liu and U. Welz-Biermann, Green Chem., 2011, 13, 1224–1229 RSC .
- L. Wang, Y. N. Zhang, Y. L. Zhang, Z. X. Jiang and C. Li, Chem.–Eur. J., 2009, 15, 12571–12575 CrossRef CAS .
- M. Lewandowski, Appl. Catal., B, 2014, 160–161, 10–21 CrossRef CAS .
- Z. G. Wang, S. L. Chen, J. N. Pei, A. C. Chen, J. H. Zhang, Z. M. Xu and J. B. Benziger, AIChE J., 2014, 60, 3267–3275 CrossRef CAS .
- C. Y. Jiang, J. J. Wang, S. T. Wang, H. y. Guan, X. H. Wang and M. X. Huo, Appl. Catal., B, 2011, 106, 343–349 CrossRef CAS .
- H. Y. Lü, Y. N. Zhang, Z. X. Jiang and C. Li, Green Chem., 2010, 12, 1954–1958 RSC .
- W. S. Zhu, B. L. Dai, P. W. Wu, Y. H. Chao, J. Xiong, S. H. Xun, H. P. Li and H. M. Li, ACS Sustainable Chem. Eng., 2015, 3, 186–194 CrossRef CAS .
- P. W. Wu, W. S. Zhu, A. M. Wei, B. L. Dai, Y. H. Chao, C. F. Li, H. M. Li and S. Dai, Chem.–Eur. J., 2015, 21, 15421–15427 CrossRef CAS .
- X. D. Tang, T. Hu, J. J. Li, F. Wang and D. Y. Qing, RSC Adv., 2015, 5, 53455–53461 RSC .
- J. T. Zhang, W. S. Zhu, H. M. Li, W. Jiang, Y. Q. Jiang, W. L. Huang and Y. S. Yan, Green Chem., 2009, 11, 1801–1807 RSC .
- F. T. Li, C. G. Kou, Z. M. Sun, Y. J. Hao, R. H. Liu and D. S. Zhao, J. Hazard. Mater., 2012, 205–206, 164–170 CrossRef CAS PubMed .
- W. H. Lo, H. Y. Yang and G. T. Wei, Green Chem., 2003, 5, 639–642 RSC .
- J. J. Gao, X. Y. Chen, N. N. Ren, W. J. Wu, C. X. Li, H. Meng and Y. Z. Lu, AIChE J., 2013, 59, 2966–2976 CrossRef CAS .
- K. S. Triantafyllidis and E. A. Deliyanni, Chem. Eng. J., 2014, 236, 406–414 CrossRef CAS .
- J. Xiao, X. X. Wang, M. Fujii, Q. J. Yang and C. S. Song, AIChE J., 2013, 59, 1441–1445 CrossRef CAS .
- G. Miao, F. Y. Ye, L. M. Wu, X. L. Ren, J. Xiao, Z. Li and H. H. Wang, J. Hazard. Mater., 2015, 300, 426–432 CrossRef CAS .
- W. L. Li, Q. F. Liu, J. M. Xing, H. S. Gao, X. C. Xiong, Y. G. Li, X. Li and H. Z. Liu, AIChE J., 2007, 53, 3263–3268 CrossRef CAS .
- A. Bösmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz and P. Wasserscheid, Chem. Commun., 2001, 2494–2495 RSC .
- Y. Nie, C. X. Li, A. J. Sun, H. Meng and Z. H. Wang, Energy Fuels, 2006, 20, 2083–2087 CrossRef CAS .
- C. Asumana, G. R. Yu, Y. W. Guan, S. D. Yang, S. Z. Zhou and X. C. Chen, Green Chem., 2011, 13, 3300–3305 RSC .
- C. P. Li, D. Li, S. S. Zou, Z. Li, J. M. Yin, A. L. Wang, Y. N. Cui, Z. L. Yao and Q. Zhao, Green Chem., 2013, 15, 2793–2798 RSC .
- M. Soleimani, A. Bassi and A. Margaritis, Biotechnol. Adv., 2007, 25, 570–596 CrossRef CAS .
- X. C. Chen, D. D. Song, C. Asumana and G. R. Yu, J. Mol. Catal. A: Chem., 2012, 359, 8–13 CrossRef CAS .
- R. Sundararaman and C. Song, Ind. Eng. Chem. Res., 2014, 53, 1890–1899 CrossRef CAS .
- D. S. Zhao, Z. M. Sun, F. T. Li, R. Liu and H. D. Shan, Energy Fuels, 2008, 22, 3065–3069 CrossRef CAS .
- C. H. Ma, B. Dai, C. X. Xu, P. Liu, L. L. Qi and L. L. Ban, Catal. Today, 2013, 211, 84–89 CrossRef CAS .
- H. S. Gao, C. Guo, J. M. Xing, J. M. Zhao and H. Z. Liu, Green Chem., 2010, 12, 1220–1224 RSC .
- H. Y. Lü, S. N. Wang, C. L. Deng, W. Z. Ren and B. C. Guo, J. Hazard. Mater., 2014, 279, 220–225 CrossRef .
- B. Y. Zhang, Z. X. Jiang, J. Li, Y. N. Zhang, F. Lin, Y. Liu and C. Li, J. Catal., 2012, 287, 5–12 CrossRef CAS .
- J. M. Yin, J. P. Wang, Z. Li, D. Li, G. Yang, Y. N. Cui, A. L. Wang and C. P. Li, Green Chem., 2015, 17, 4552–4559 RSC .
- Y. Zhao and G. X. Hu, RSC Adv., 2013, 3, 2234–2240 RSC .
- Y. Nie, Y. X. Dong, L. Bai, H. F. Dong and X. P. Zhang, Fuel, 2013, 103, 997–1002 CrossRef CAS .
- A. Riisager, R. Fehrmann, S. Flicker, R. van Hal, M. Haumann and P. Wasserscheid, Angew. Chem., Int. Ed., 2005, 44, 815–819 CrossRef CAS .
- Y. Xie, Z. F. Zhang, S. Q. Hu, J. L. Song, W. J. Li and B. X. Han, Green Chem., 2008, 10, 278–282 RSC .
- S. H. Xun, W. S. Zhu, D. Zheng, L. Zhang, H. Liu, S. Yin, M. Zhang and H. M. Li, Fuel, 2014, 136, 358–365 CrossRef CAS .
- J. P. Mikkola, P. Virtanen, H. Karhu, T. Salmi and D. Y. Murzin, Green Chem., 2006, 8, 197–205 RSC .
- I. S. Amiinu, X. M. Liang, Z. K. Tu, H. N. Zhang, J. W. Feng, Z. M. Wan and M. Pan, ACS Appl. Mater. Interfaces, 2013, 5, 11535–11543 CAS .
- J. Zhang, A. J. Wang, X. Li and X. H. Ma, J. Catal., 2011, 279, 269–275 CrossRef CAS .
- Y. Chen and Y. F. Song, ChemPlusChem, 2014, 79, 304–309 CrossRef CAS .
- M. Zhang, M. Li, Q. Chen, W. S. Zhu, H. P. Li, S. Yin, Y. N. Li and H. M. Li, RSC Adv., 2015, 5, 76048–76056 RSC .
- J. Zhang, A. J. Wang, Y. J. Wang, H. Y. Wang and J. Z. Gui, Chem. Eng. J., 2014, 245, 65–70 CrossRef CAS .
- X. T. Zhang, G. Q. Luo, M. Y. Zhu, L. H. Kang, F. Yu and B. Dai, RSC Adv., 2015, 5, 76182–76189 RSC .
- F. J. Liu, L. Wang, Q. Sun, L. F. Zhu, X. J. Meng and F. S. Xiao, J. Am. Chem. Soc., 2012, 134, 16948–16950 CrossRef CAS .
- P. T. M. Do, A. J. Foster, J. G. Chen and R. F. Lobo, Green Chem., 2012, 14, 1388–1397 RSC .
- Z. Liu and C. Lv, RSC Adv., 2014, 4, 10221–10227 RSC .
- J. Gamage McEvoy and Z. S. Zhang, Appl. Catal., B, 2014, 160–161, 267–278 CrossRef CAS .
- A. Lopez-Bezanilla, J. Huang, H. Terrones and B. G. Sumpter, Nano Lett., 2011, 11, 3267–3273 CrossRef CAS .
- K. Uosaki, G. Elumalai, H. Noguchi, T. Masuda, A. Lyalin, A. Nakayama and T. Taketsugu, J. Am. Chem. Soc., 2014, 136, 6542–6545 CrossRef CAS .
- C. J. Huang, W. Q. Ye, Q. W. Liu and X. Q. Qiu, ACS Appl. Mater. Interfaces, 2014, 6, 14469–14476 CAS .
- A. Nag, K. Raidongia, K. P. S. S. Hembram, R. J. Datta, U. V. Waghmare and C. N. R. Rao, ACS Nano, 2010, 4, 1539–1544 CrossRef CAS .
- W. S. Zhu, P. W. Wu, Y. H. Chao, H. M. Li, F. Zou, S. H. Xun, F. X. Zhu and Z. Zhao, Ind. Eng. Chem. Res., 2013, 52, 17399–17406 CrossRef CAS .
- X. B. Wang, C. Y. Zhi, L. Li, H. B. Zeng, C. Li, M. Mitome, D. Golberg and Y. Bando, Adv. Mater., 2011, 23, 4072–4076 CrossRef CAS PubMed .
- L. Song, L. J. Ci, H. Lu, P. B. Sorokin, C. H. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Nano Lett., 2010, 10, 3209–3215 CrossRef CAS PubMed .
- W. Lei, D. Portehault, D. Liu, S. Qin and Y. Chen, Nat. Commun., 2013, 4, 1777–1783 CrossRef PubMed .
- A. Ismach, H. Chou, D. A. Ferrer, Y. Wu, S. McDonnell, H. C. Floresca, A. Covacevich, C. Pope, R. Piner, M. J. Kim, R. M. Wallace, L. Colombo and R. S. Ruoff, ACS Nano, 2012, 6, 6378–6385 CrossRef CAS PubMed .
- J. Xiong, W. S. Zhu, H. P. Li, W. J. Ding, Y. H. Chao, P. W. Wu, S. H. Xun, M. Zhang and H. M. Li, Green Chem., 2015, 17, 1647–1656 RSC .
- M. Zhang, W. S. Zhu, S. H. Xun, H. M. Li, Q. Q. Gu, Z. Zhao and Q. Wang, Chem. Eng. J., 2013, 220, 328–336 CrossRef CAS .
- S. Otsuki, T. Nonaka, N. Takashima, W. Qian, A. Ishihara, T. Imai and T. Kabe, Energy Fuels, 2000, 14, 1232–1239 CrossRef CAS .
Footnote |
| † Electronic supplementary information (ESI) available: X-ray photoelectron spectroscopy (XPS), Atomic Force Microscope (AFM) and effects of the amount of H2O2 on sulfur removal. See DOI: 10.1039/c5ra23272d |
|
| This journal is © The Royal Society of Chemistry 2016 |