
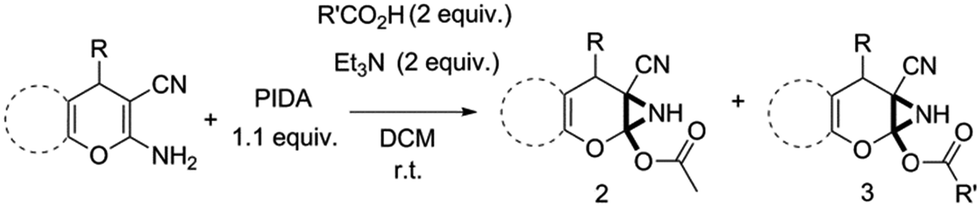
Facile synthesis of functionalized 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetates: a catalyst free approach to access the pyran fused 2-acetoxy-NH-aziridines†
Prasun Mukherjee and
Asish R. Das*
Department of Chemistry, University of Calcutta, Kolkata-700009, India. E-mail: ardchem@caluniv.ac.in; ardas66@rediffmail.com; Fax: +91 3323519754; Tel: +91 3323501014 Tel: +91 9433120265
First published on 16th December 2015
Abstract
Novel 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetate scaffolds were synthesized directly from 2-amino-3-cyano-4-H-pyrans as well as 2-amino-3-cyano-spiropyrans using iodobenzene diacetate (PIDA) as oxidant at room temperature in the absence of any catalyst. Essentially, the enamine fragment of the reactants reacts with PIDA, which makes this reaction a new way to synthesize pyran fused 2-acetoxy-NH-aziridines. These remarkably stable pyran fused 2-acetoxy-NH-aziridines can be used in SAR studies in pharmaceutical and medicinal chemistry. Ready availability of the starting materials, operational simplicity, absence of metal catalyst, mild reaction conditions, and simple workup procedure are the other significant features of this reaction.
Introduction
Aziridines, inherently strained aza-heterocycles, are fundamental precursors to access diverse types of heterocycles due to their strong affinity towards numerous ring opening reactions.1 Despite their high reactivity due to ring strain, the aziridine structural motif can be found in many natural products and synthetic compounds with widespread biological activity.2 For instance mitomycins, ficellomycins, azinomycins, FR and FK compounds (Fig. 1) which contain a fused aziridine fragment as their key structural motif, are well known for their potent antitumor and antibiotic activities.3 In addition, aziridines are also frequently used as chiral auxiliaries and ligands.4 Regardless of their occurrence as biologically relevant compounds, fused aziridine heterocycles are important from a synthetic viewpoint as they can be transformed into numerous other key heterocycles.5 In addition many fused aziridine heterocycles exhibit photochromic properties.6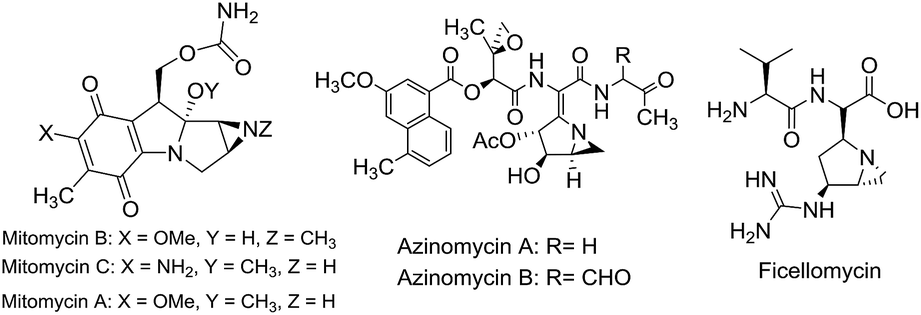 | ||
| Fig. 1 Some bioactive natural products possessing aziridine ring as the crucial structural motif. | ||
Consequently several methods have been developed over the past decades in order to synthesize aziridines and its fused analogues.7 The main synthetic routes of those rely on transfer of suitable nitrogen sources to olefins, transfer of suitable carbon sources to imines, reduction of azirines and intramolecular cyclization of amine derivatives. Notably direct synthesis of NH-aziridines is preferred as N-protected aziridines require additional deprotection steps in order to access the NH-form. However, there are hardly any methods to synthesize pyran-fused NH-aziridines,8 although the resultant scaffold will not only be surplus of biological activity due to the fusion of pyran ring with aziridine but also possess great synthetic utility.
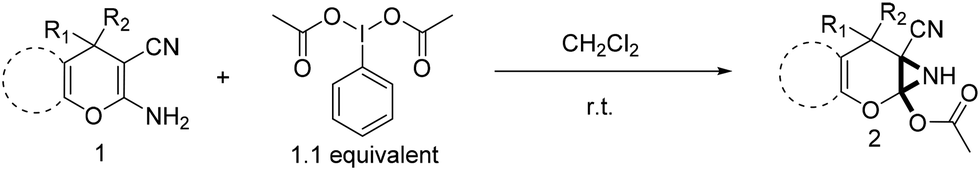
Recently synthesis of azirines from enamine derivatives using hypervalent iodine reagents was reported in literature.9 2-Amino-4-H-pyrans and 2-amino-spiropyrans are privileged compounds, well known for their widespread biological activity.10 Interestingly, they possess an inbuilt enamine structural feature which can be utilized in construction of aziridine molecular scaffold and the consequence will be the formation of a pyran fused aziridine motif. Continuing our efforts on the development of new and efficient methodologies to synthesize bioactive compounds,11 herein we wish to report an operationally very simple and robust synthesis of novel pyran-fused 2-acetoxy-NH-aziridines by reacting the enamine fragment of 2-amino-3-cyano-4-H-pyrans and 2-amino-3-cyano-spiropyrans with iodobenzene diacetate (PIDA) at room temperature in short reaction time (Scheme 1) without the involvement of any catalyst. To the best of our knowledge this is the first report for one step synthesis of pyran-fused 2-acetoxy-NH-aziridines from 2-amino-3-cyano-4-H-pyrans and 2-amino-3-cyano-spiropyrans. This protocol is essentially metal free and uses PIDA as the sole oxidant which is readily available and less toxic.
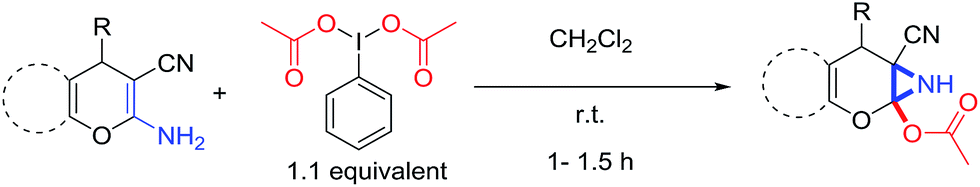 | ||
| Scheme 1 PIDA promoted synthesis of 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetates. | ||
Result and discussion
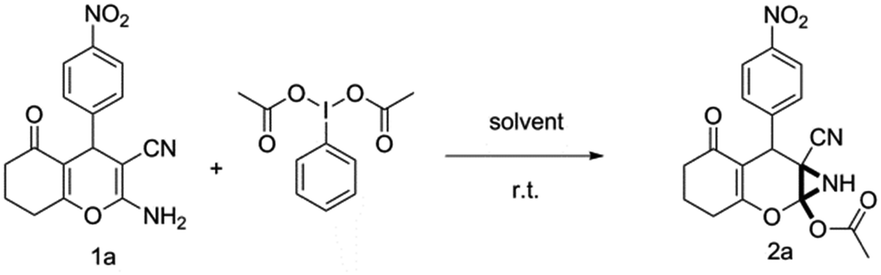
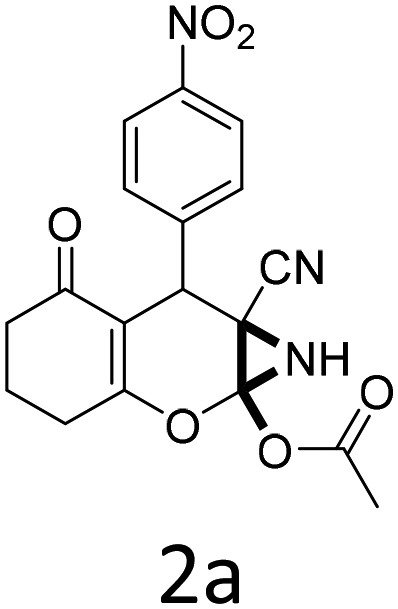
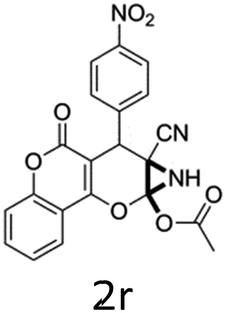
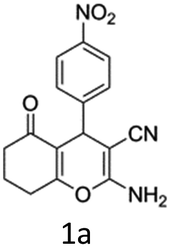
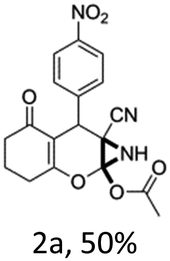
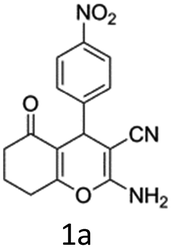
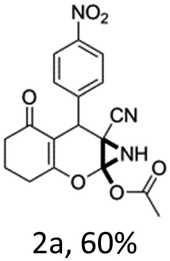
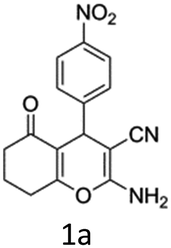
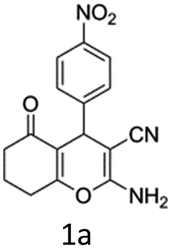
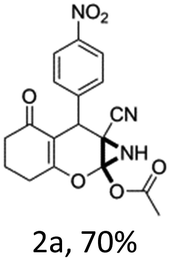
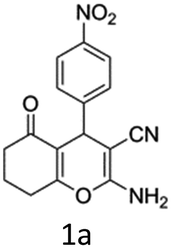
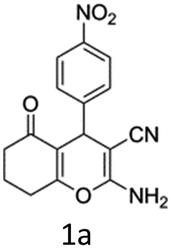
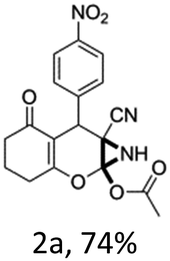
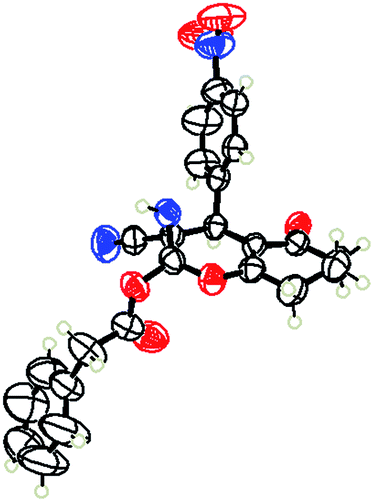
To begin with, a wide variety of functionalized 4-H-pyrans and spiropyrans were easily synthesized using known procedures.12 Taking 2-amino-4-(4-nitrophenyl)-5-oxo-5,6,7,8-tetrahydro-4H-chromene-3-carbonitrile (1a) as the model substrate we have then investigated the feasibility of the proposed transformation by treating 1.00 mmol of it with PIDA (1 mmol) in 3 ml of dichloromethane (DCM) at room temperature. The complete consumption of the starting material 1a, monitored by TLC, took only 1 h. Immediate column chromatography of the reaction mixture gave an off white compound in 80% yield (Table 1, entry 1). The 1H NMR spectrum of the compound showed the presence of signals resembling to methyl H's of acetate group at δ 2.257 ppm and a singlet at δ 2.450 ppm with the disappearance of the signal for the amino group of the starting material. In our delight, the structure of this compound was finally identified and confirmed by X-ray crystallographic analysis as a 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetate derivative (Fig. 2a). The X-ray crystallographic analysis also enables us to understand the relative stereochemistry of the compound 2a and permit us to assign the configuration of the C4 centre of compound 2a as R. It can be seen from the ortep diagram of compound 2a that the p-nitrophenyl group and the aziridine ring bear a syn relationship with each other.
|
|||||
|---|---|---|---|---|---|
| Entry | PIDA (mmol) | Solvent | T (°C) | Time (h) | Yieldb (%) |
| a 1 mmol of 1a was taken in 3 ml of solvent in all the cases.b Yield of the isolated product. | |||||
| 1 | 1.0 | DCM | r.t. | 1 | 80 |
| 2 | 1.1 | DCM | r.t. | 1 | 92 |
| 3 | 1.2 | DCM | r.t. | 1 | 90 |
| 4 | 1.5 | DCM | r.t. | 1 | 72 |
| 5 | 1.1 | DCE | r.t. | 2 | 70 |
| 6 | 1.1 | Toluene | r.t. | 2.5 | 68 |
| 7 | 1.1 | Dioxane | r.t. | 4 | 60 |
| 8 | 1.1 | THF | r.t. | 4 | 64 |
| 9 | 1.1 | Acetonitrile | r.t. | 2 | 55 |
| 10 | 1.1 | DCM | 0 | 10 | 72 |
 | ||
| Fig. 2 Ortep diagram of single crystal of compounds 2a, 2h and 2j. (a) ORTEP diagram of single crystal of compound 2a (CCDC 1404567), (b) ORTEP diagram of single crystal of compound 2j (CCDC 1404581), (c) ORTEP diagram of single crystal of compound 2h (CCDC 1404569). | ||
With this promising result in hand, optimization of the reaction condition was then initiated for the synthesis of 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetate by changing various reaction parameters (Table 1). Use of 1.1 mmol of PIDA increased the product yield to 92% (Table 1, entry 2). Further increase in yield was not obtained when 1.2 mmol PIDA was used (Table 1, entry 3). Additional increase in the amount of PIDA resulted in the low yield of the product (Table 1, entry 4). When the reaction was performed in other solvents, e.g., dichloroethane (DCE), toluene, dioxane, tetrahydrofuran (THF), acetonitrile, the reaction time as well as yield of the product was found to be greatly affected and in all cases a decrease in the product yield was observed (Table 1, entry 5–9). Further investigation showed that decreasing the temperature to 0 °C reduced the rate of the reaction and very less amount of product was obtained after a prolonged time (Table 1, entry 10).
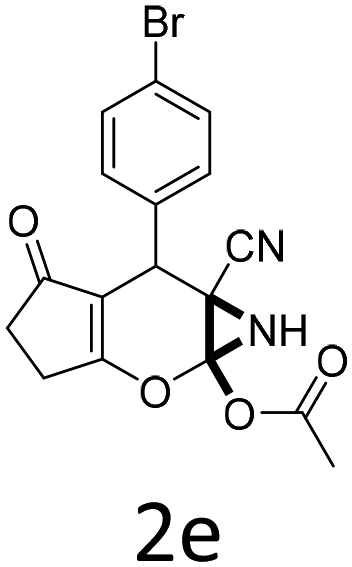
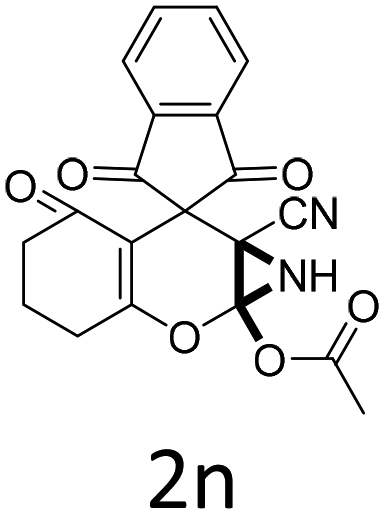
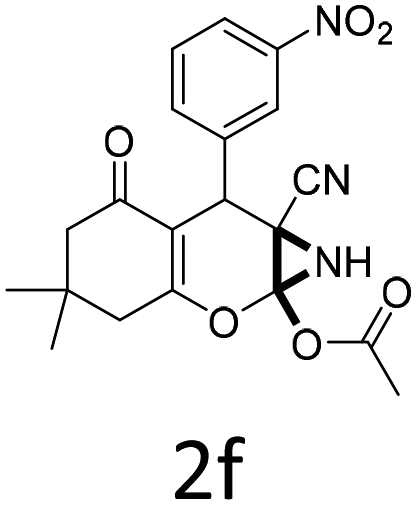
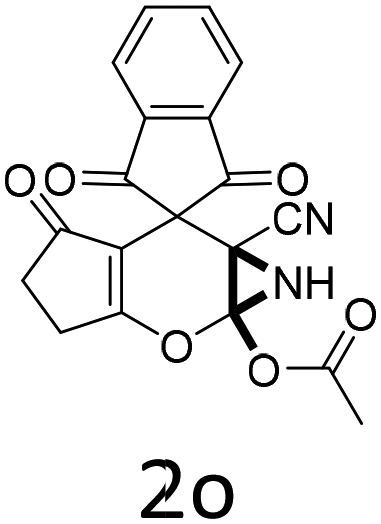
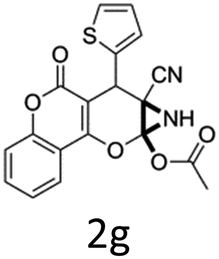
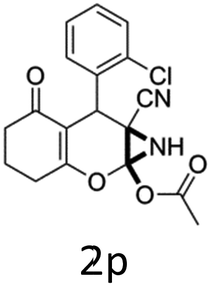
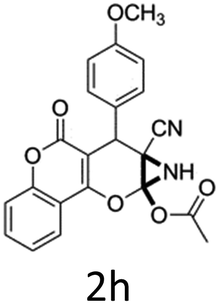
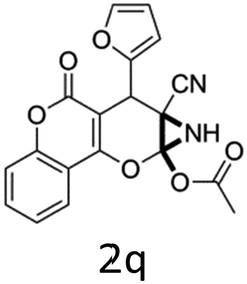
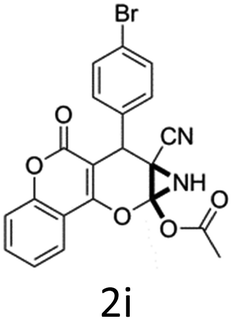
Having identified the optimized reaction conditions (Table 1, entry 2), we then investigated the prospect and restriction of this methodology as exemplified in Table 2. A wide range of diversified 4-H-pyrans (1 mmol) were reacted with 1.1 mmol of iodobenzene diacetate in 3 ml of DCM at room temperature to synthesize functionalized 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetate. Various 4-H-pyrans, synthesized from different aromatic aldehydes having strong electron donating as well as withdrawing functional groups, hetero aromatic aldehydes and aliphatic aldehyde as well were subjected to this reaction. This method has found to possess well tolerance towards different substituents in the 4-position of the pyran ring. However a small decrease in the product yield was observed when thiophene ring was present as the substituent (Table 2, 2g). When the nitrile group of the reactants was replaced by –CO2Et group, complete conversion of the starting material observed at the same condition but we failed to isolate and identify any compounds due to the presence of multiple intimate components in the reaction mixture. Different active methylene compounds, e.g., cyclohexane-1,3-dione, dimedone, cyclopentane-1,3-dione, 4-hydroxycoumarin, 4-hydroxy-6-methyl-2-pyrone, were employed to derive various substituted 4-H-pyrans. The reaction proceeded well reliably, affording good to excellent product yield in all the cases (Table 2, 2a–2p). Notably the coumarin and pyrone moiety have found to possess well tolerance towards PIDA. To further explore substrate scope, spiropyrans were subjected to this reaction under the same optimized conditions. The reaction was found to be equally efficient affording excellent yields (Table 2, 2n & 2o).
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Reactant | Product | Time (h) | Yield (%) | Entry | Reactant | Product | Time (h) | Yield (%) |
| a Reaction conditions: 1a (1 mmol), PIDA (1.1 mmol), DCM (3 ml), r.t. | |||||||||
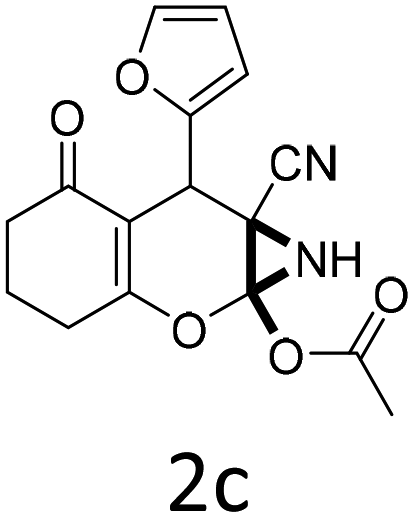
| 1 | 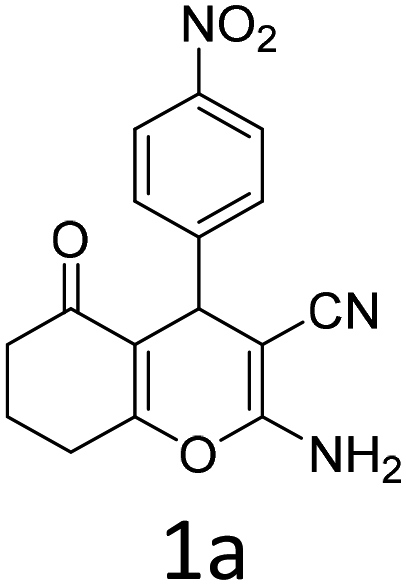 |
 |
1 | 92 | 10 |  |
 |
1.5 | 85 |
| 2 |  |
 |
1 | 89 | 11 | 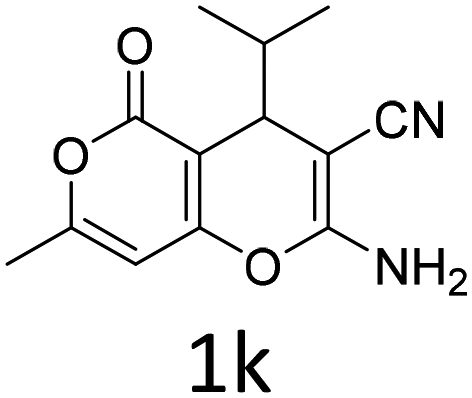 |
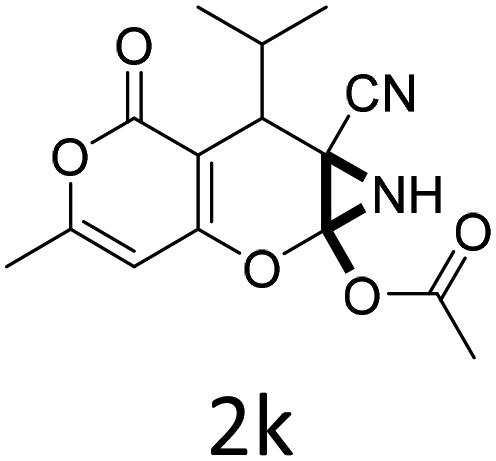 |
1.5 | 84 |
| 3 | 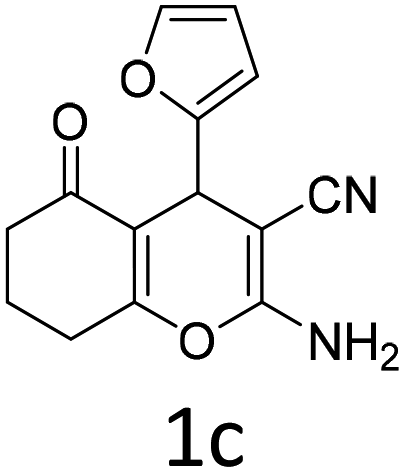 |
 |
1 | 84 | 12 | 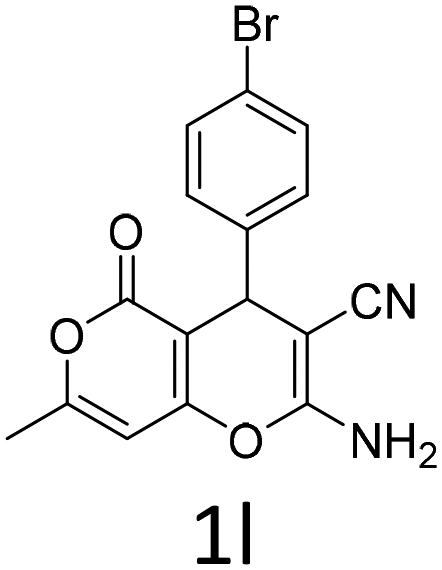 |
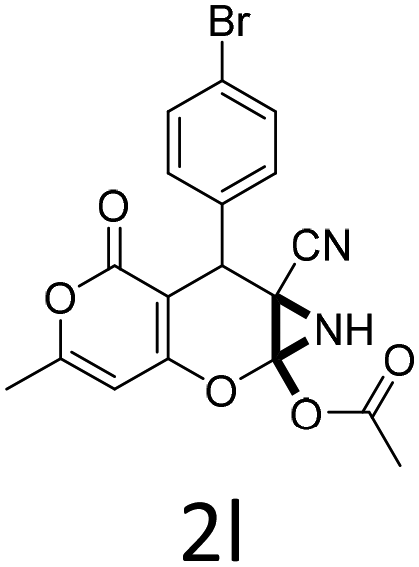 |
1.5 | 86 |
| 4 | 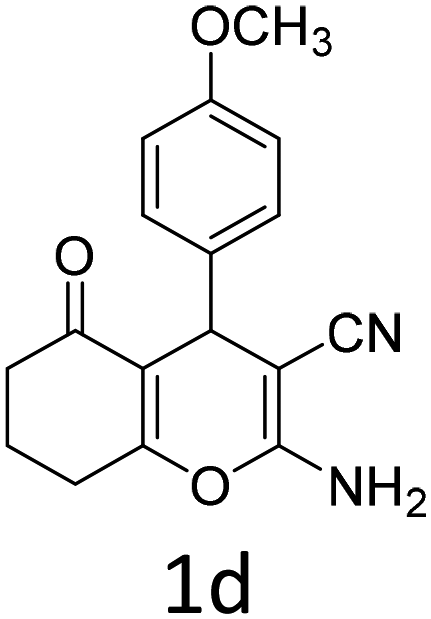 |
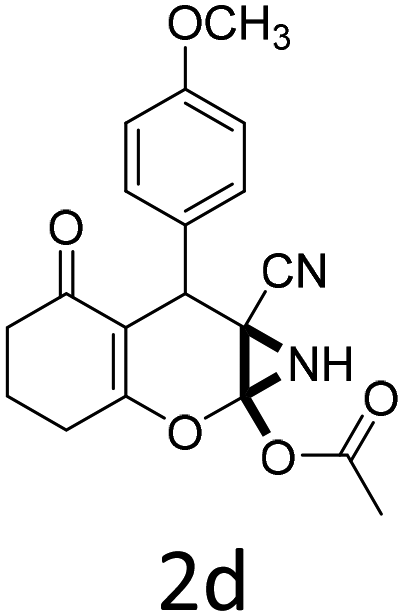 |
1 | 90 | 13 | 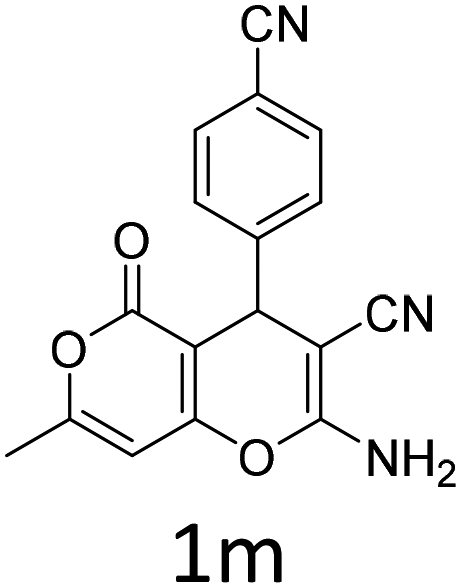 |
 |
1.5 | 88 |
| 5 | 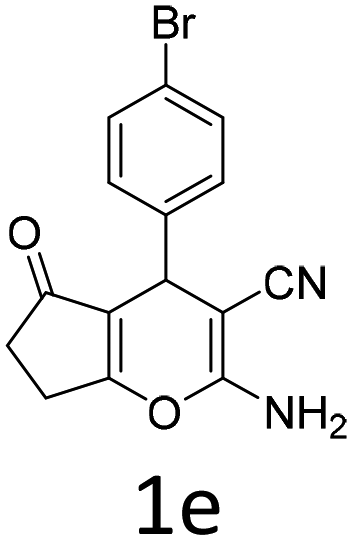 |
 |
1 | 90 | 14 | 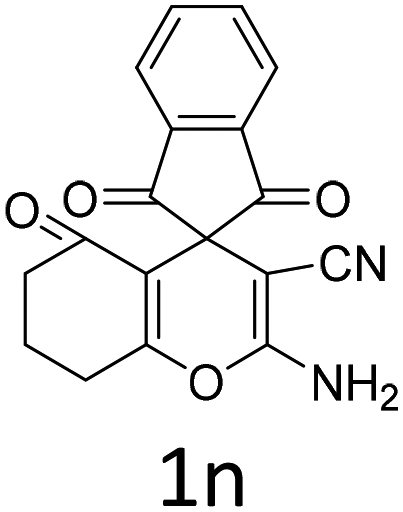 |
 |
1.5 | 88 |
| 6 | 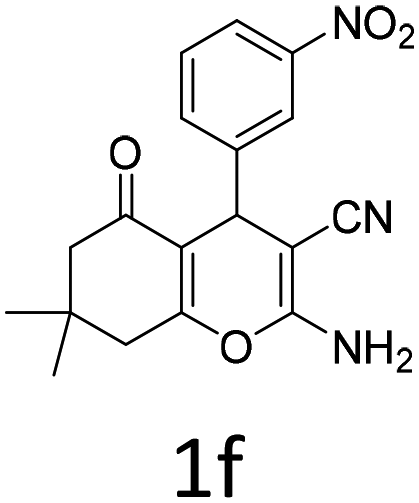 |
 |
1 | 89 | 15 | 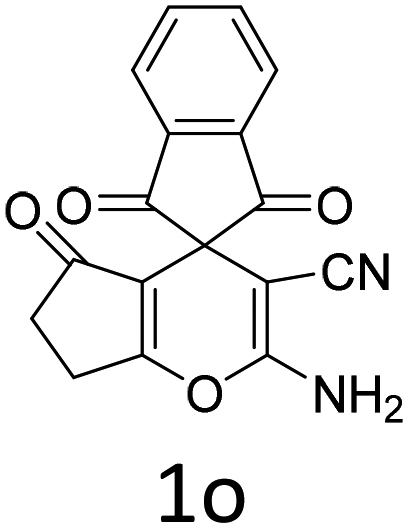 |
 |
1.5 | 86 |
| 7 | 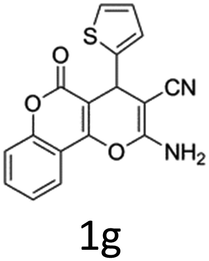 |
 |
1.5 | 76 | 16 | 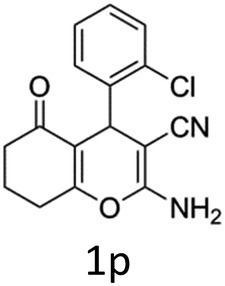 |
 |
1 | 94 |
| 8 | 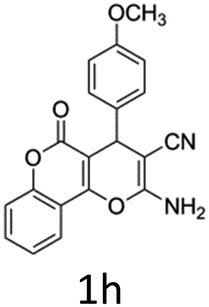 |
 |
1.5 | 90 | 17 | 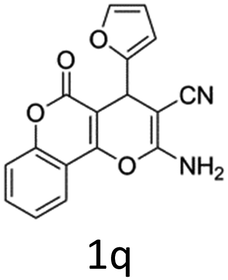 |
 |
1.5 | 86 |
| 9 | 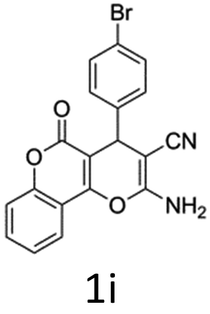 |
 |
1.5 | 88 | 18 | 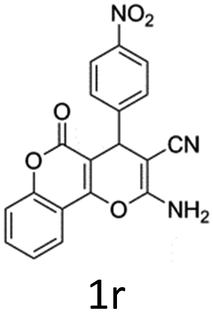 |
 |
1.5 | 90 |
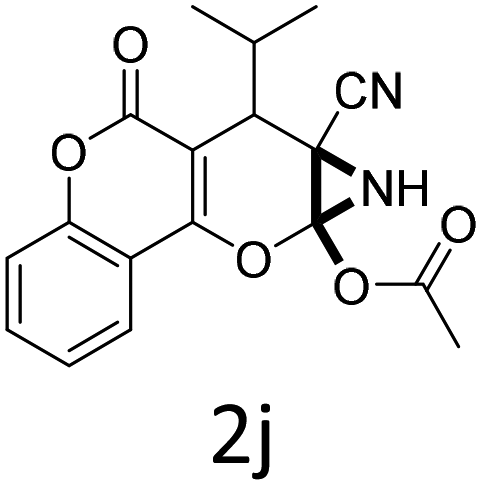
It is noteworthy to mention here that the product 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetate was stable enough and unaffected by silica gel (mesh size 100–200) during column chromatography. The structure of the products was well characterized by spectral (1H, 13C NMR, IR, HRMS) analysis. The structural motif was fully confirmed by X-ray crystallographic analysis of compound 2a, 2h and 2j (Fig. 2). The configuration of C4 centre of 2h and 2j was found to be S and R respectively.
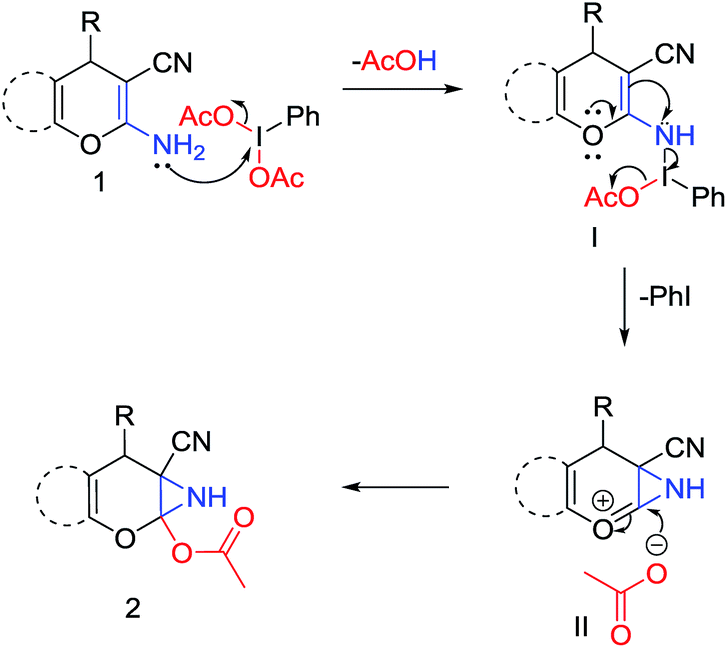
A plausible mechanism13 for this reaction is depicted in Scheme 2. Initial reaction between the amino group of the starting material (1) and PIDA results in the formation of N-iodo intermediate I. The electron donating resonance effect of oxygen atom of the pyran ring then assists the attack of the double bond to the electrophilic nitrogen center and consequently intermediate I converted to II by releasing iodobenzene and an acetate anion. In the next step nucleophilic attack of that acetate anion at the C-2 position of the pyran ring results in the formation of the desired product 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetate (2). The attachment of both –CN and pyran oxygen to the enamine double bond of the molecular scaffold is probably the key reason for the specific formation of 2-acetoxy-NH-aziridines. During the azirine synthesis, as reported by Zhao et al.,9a,b the reaction essentially stops after azirine ring formation, but in this case electron releasing ability of the pyran oxygen atom strongly disfavours the formation of azirine ring and eventually the reaction takes the course of formation of 2-acetoxy-NH-aziridines. The role of strong electron withdrawing –CN group is to preserve the enamine form in the starting materials.9b
 | ||
| Scheme 2 A plausible mechanism of the PIDA promoted synthesis of 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetate. | ||
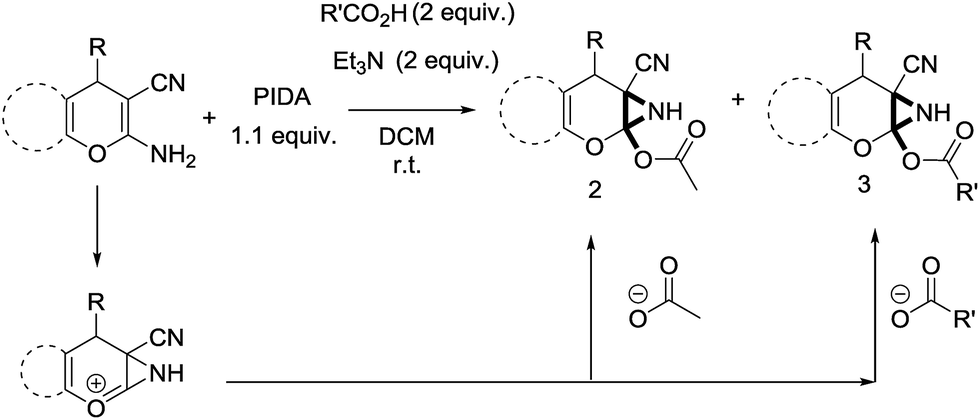
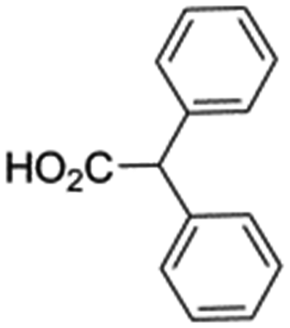
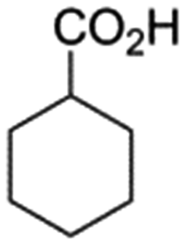
In support of the proposed reaction mechanism we have attempted to trap the intermediate II by carrying out control experiments. The reaction was performed in presence of other carboxylate anion by employing 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of triethylamine (2 equiv.) and a carboxylic acid (2 equiv.) in the reaction mixture (Scheme 3). The outcome of this experiment is shown in Table 3. All reactions were carried out in optimized reaction condition in presence of aliphatic as well as aromatic carboxylic acids. When cyclohexanecarboxylic acid, phenylacetic acid and diphenylacetic acid were employed products 3a–c, r were obtained as the minor product in low but reasonable yields along with product 2 (Table 3, entry 1–4).
:1 mixture of triethylamine (2 equiv.) and a carboxylic acid (2 equiv.) in the reaction mixture (Scheme 3). The outcome of this experiment is shown in Table 3. All reactions were carried out in optimized reaction condition in presence of aliphatic as well as aromatic carboxylic acids. When cyclohexanecarboxylic acid, phenylacetic acid and diphenylacetic acid were employed products 3a–c, r were obtained as the minor product in low but reasonable yields along with product 2 (Table 3, entry 1–4).
 | ||
| Scheme 3 Control experiments in support of the proposed mechanism. | ||
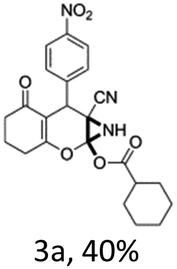
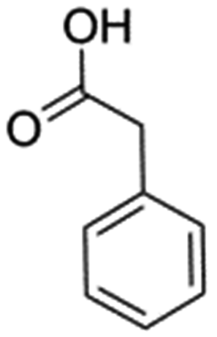


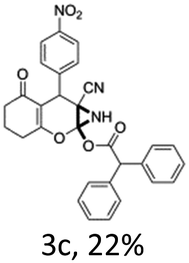
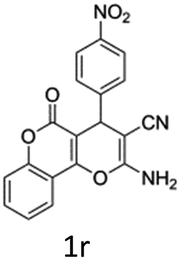
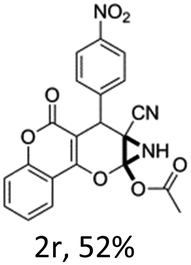
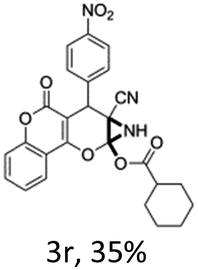


However when the reaction was carried out in presence of aromatic acids, 3 was not obtained and the yield of 2 was also affected. The structural motif of compound 3a–c, r were well characterized through spectral analysis (1H, 13C NMR, IR, HRMS) and fully confirmed by X-ray crystallographic analysis of a single crystal of compound 3b (Fig. 3). The S configuration of the C4 centre of 3b was established from X-ray crystallographic analysis. These experimental observations evidently suggest the formation of the intermediate II during the reaction. However employing aromatic acids, the reaction did not ends up with the formation of product 3 is not very clear and further exploration is under process.
 | ||
| Fig. 3 Ortep diagram of single crystal of compounds 3b (CCDC 1434523). | ||
From the X-ray crystallographic analysis of 2a, 2h, 2j and 3b it was established that all the compounds, synthesized using this protocol, possess a syn relationship between aziridine ring and the aliphatic or aromatic substituents of the C4 centre. This information further clarified the sterochemical course of the attack of acetate or a carboxylate to the intermediate II (Scheme 4). The aromatic or aliphatic substituent of the C4 chiral center controls the regioselective nucleophilic attack of the carboxylate. The β-attack of the carboxylate to the intermediate-II at the α-position of positively charged oxygen center i.e. from the same side of R group is sterically inhibited and so did not proceed at all to realize the compound 4 However, attack by the carboxylate ion to the intermediate II from the opposite side of R group i.e. from the α-face is sterically favourable and eventually took place at ease to the α-position of positively charged oxygen center regioselectively and ultimately afforded compound 2 and 3 in a diastereoselective manner.
 | ||
| Scheme 4 Stereospecific nucleophilic attack of carboxylate. | ||
Experimental section
General procedure for the synthesis of 4-H-pyrans
Triethylamine (1 drop) was added to a solution of aromatic aldehydes (2 mmol), malononitrile (2 mmol), and 1,3-diketones (2 mmol) in EtOH (5 ml) and the reaction mixture was refluxed for 15 min. The precipitate that formed was filtered off, washed with water (5 × 10 ml) and EtOH (3 × 5 ml), and finally recrystallized from EtOH. The crystallized pure compounds were then subjected for the synthesis of 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetates.General procedure for the synthesis of spiropyrans
Triethylamine (1 drop) was added to a solution of ninhydrin (2 mmol), malononitrile (2 mmol), and 1,3-diketones (2 mmol) in EtOH (5 ml) and the reaction mixture was refluxed for 15 min. The precipitate that formed was filtered off, washed with water (5 × 10 ml) and EtOH (3 × 5 ml), and finally recrystallized from EtOH. The crystallized pure compounds were then subjected for the synthesis of 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetates.General procedure for the synthesis of 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetate 2
To a stirring suspension of 2-amino-3-cyano-4-H-pyrans (1 mmol) in 3 ml of DCM, 1.1 mmol of PIDA was added and stirring was continued at r.t. After the total consumption of the starting material, indicated by TLC (20–40% ethyl acetate in petroleum ether), the reaction mixture was directly subjected for column chromatography (15–30% ethyl acetate in petroleum ether) to afford the pure product. Same procedure was applied in case of 2-amino-3-cyano-spiropyrans. The isolated products were characterized by spectral (1H NMR, 13C NMR, IR, HRMS), and X-ray crystallographic analysis.General procedure for the synthesis of 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl carboxylate 3
To a stirring mixture of 2-amino-3-cyano-4H-pyrans (1 mmol), triethylamine (2 mmol) and carboxylic acids (2 mmol) in 3 ml of DCM, 1.1 mmol of PIDA was added and stirring was continued at r.t. After the total consumption of the starting material, indicated by TLC (20–40% ethyl acetate in petroleum ether), the reaction mixture was directly subjected for column chromatography (15–30% ethyl acetate in petroleum ether) to afford the pure product. The isolated products were characterized by spectral (1H NMR, 13C NMR, IR, HRMS), and X-ray crystallographic analysis.Conclusion
In conclusion, 4-H-pyrans have been successfully hybridized into novel 6-cyano-2-oxa-7-azabicyclo[4.1.0]hept-3-en-1-yl acetate molecular scaffold. The methodology appeared to be the first example of one step synthesis of pyran fused 2-acetoxy-NH-aziridines from 4-H-pyrans and spiropyrans using iodobenzene diacetate as the sole oxidant and involving no other reagent or catalyst. The diastereoselective nature of this reaction makes this protocol highly interesting from synthetic chemistry aspect. The product of this reaction is well fascinating as it is a pyran fused 2-acetoxy-NH-aziridine molecular scaffold possessing high functionalization, and we are sure that this methodology as well as this molecular architecture will grab the attention of the chemists.Acknowledgements
We gratefully acknowledge the financial support from U.G.C. and University of Calcutta. P. M. thanks U.G.C. New Delhi, India for the grant of his Senior Research Fellowship. Crystallography was performed at the DST-FIST, India-funded Single Crystal Diffractometer Facility at the Department of Chemistry, University of Calcutta.References
- (a) J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247 RSC; (b) X. E. Hu, Tetrahedron, 2004, 60, 2701 CrossRef CAS; (c) W. McCoull and F. A. Davis, Synthesis, 2000, 1347 CrossRef CAS; (d) C. Schneider, Angew. Chem., Int. Ed., 2009, 48, 2082 CrossRef CAS PubMed; (e) G. S. Singh, M. D'hooghe and N. de Kimpe, Chem. Rev., 2007, 107, 2080 CrossRef CAS PubMed.
- (a) K. Nagaoka, M. Matsumoto, J. Oono, K. Yokoi, S. Ishizeki and T. Nakashima, J. Antibiot., 1986, 39, 1527 CrossRef CAS PubMed; (b) F. Reusser, Biochemistry, 1977, 16, 3406 CrossRef CAS PubMed; (c) K. Masuda, A. Suzuki, T. Nakamura, S. Takagaki, K. Noda, K. Shimomura, H. Noguchi and F. Shibayama, Jpn. J. Pharmacol., 1989, 51, 219 CrossRef CAS PubMed; (d) T. R. Witty and W. A. Remers, J. Med. Chem., 1973, 16, 1281 CrossRef.
- (a) K. Shimomura, T. Manda, S. Mukumoto, K. Masuda, T. Nakamura, T. Mizota, S. Matsumoto, F. Nishigaki, T. Oku, J. Mori and F. Shibayama, Cancer Res., l988, 48, 1166 Search PubMed; (b) F. M. D. Ismail, D. O. Levitsky and V. M. Dembitsky, Eur. J. Med. Chem., 2009, 44, 3373 CrossRef CAS PubMed.
- D. Tanner, Angew. Chem., Int. Ed. Engl., 1994, 33, 599 CrossRef.
- (a) S. Bonham, L. O'Donovan, M. P. Carty and F. Aldabbagh, Org. Biomol. Chem., 2011, 9, 6700 RSC; (b) S. C. Bergmeier and S. J. Katz, J. Comb. Chem., 2002, 4, 162–166 CrossRef CAS PubMed; (c) H. W. Heine and R. P. Henzel, J. Org. Chem., 1969, 34, 171 CrossRef CAS; (d) A. Padwa and E. Glazer, J. Org. Chem., 1973, 38, 284 CrossRef CAS; (e) S. C. Bergmeier and D. M. Stanchina, J. Org. Chem., 1997, 62, 4449–4456 CrossRef CAS PubMed.
- (a) R. B. King and I. Haiduc, J. Am. Chem. Soc., 1972, 94, 4046 CrossRef; (b) A. Padwa and E. Vega, J. Org. Chem., 1975, 40, 175 CrossRef CAS.
- (a) S. J. Brois, J. Org. Chem., 1962, 27, 3532 CrossRef CAS; (b) H. E. Baumgarten, R. L. Zey and U. Krolls, J. Am. Chem. Soc., 1961, 83, 4469 CrossRef CAS; (c) I. D. G. Watson, L. Yu and A. K. Yudin, Acc. Chem. Res., 2006, 39, 194 CrossRef CAS PubMed; (d) W. Lwowsky, Angew. Chem., Int. Ed., 1967, 6, 897 CrossRef; (e) J. U. Jeong, B. Tao, I. Sagasser, H. Henniges and K. B. Sharpless, J. Am. Chem. Soc., 1998, 120, 6844 CrossRef CAS; (f) Z. Li, K. R. Conser and E. N. Jacobsen, J. Am. Chem. Soc., 1993, 115, 5326 CrossRef CAS; (g) L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881 CrossRef CAS PubMed; (h) L. Casarrubios, J. A. Pérez, M. Brookhart and J. L. Templeton, J. Org. Chem., 1996, 61, 8358 CrossRef CAS; (i) D.-K. Wang, L.-X. Dai and X.-L. Hou, Chem. Commun., 1997, 1231 RSC; (j) V. Reutrakul, V. Prapansiri and C. Panyachotipun, Tetrahedron Lett., 1984, 25, 1949 CrossRef CAS; (k) T. Satoh, T. Sato, T. Oohara and K. Yamakawa, J. Org. Chem., 1989, 54, 3973 CrossRef CAS; (l) S. Florio, L. Troisi, V. Capriati and G. Ingrosso, Tetrahedron Lett., 1999, 40, 6101 CrossRef CAS; (m) J. Sweeney, Eur. J. Org. Chem., 2009, 29, 4911 CrossRef; (n) G. B. Mullen, G. A. Bennett and St. V. Georgiev, Eur. J. Org. Chem., 1990, 1, 109 CrossRef; (o) G. Callebaut, T. Meiresonne, N. de Kimpe and S. Mangelinckx, Chem. Rev., 2014, 114, 7954 CrossRef CAS PubMed; (p) N. de Kimpe, P. Sulmon, R. Verhé, L. de Buyck and N. Schamp, J. Org. Chem., 1983, 48, 4320 CrossRef CAS; (q) N. de Kimpe, R. Verhé, L. de Buyck and N. Schamp, J. Org. Chem., 1980, 45, 5319 CrossRef CAS; (r) B. Denolf, E. Leemans and N. de Kimpe, J. Org. Chem., 2007, 72, 3211 CrossRef CAS PubMed; (s) N. de Kimpe, R. Verhé, L. de Buyck and N. Schamp, Synth. Commun., 1975, 5, 269 CrossRef CAS; (t) B. Denolf, S. Mangelinckx, K. W. Törnroos and N. de Kimpe, Org. Lett., 2006, 8, 3129 CrossRef CAS PubMed.
- (a) Q. Wang, Z. Zhang, X. Zhang, J. Zhang, Y. Kang and J. Peng, RSC Adv., 2015, 5, 4788 RSC; (b) D. M. B. Hickey, A. R. MacKenzie, C. J. Moody and C. W. Rees, J. Chem. Soc., Perkin Trans. 1, 1987, 921 RSC; (c) K. Buggle and B. Fallon, J. Chem. Res., Synop., 1988, 11, 349 Search PubMed.
- (a) X. Sun, Y. Lyu, D. Z. Negrerie, Y. Du and K. Zhao, Org. Lett., 2013, 15, 6222 CrossRef CAS PubMed; (b) X. Li, Y. Du, Z. Liang, X. Li, Y. Pan and K. Zhao, Org. Lett., 2009, 11, 2643 CrossRef CAS PubMed.
- (a) G. Feuer, Progress in Medicinal Chemistry, ed. G. P. Ellis and G. P. West, North-Holland Publishing Company, New York, 1974, vol. 10, pp. 85−158 Search PubMed; (b) F. M. Dean, Naturally Occurring Oxygen Ring Compounds, Butterworth-Heinemann, London, 1963, pp. 176−220 Search PubMed; (c) A. Goel and V. J. Ram, Natural and synthetic 2H-pyran-2-ones and their versatility in organic synthesis, Tetrahedron, 2009, 65, 7865–7913 CrossRef CAS.
- (a) S. Paul, K. Pradhan, S. Ghosh, S. K. de and A. R. Das, Adv. Synth. Catal., 2014, 6, 1301 CrossRef; (b) P. P. Ghosh and A. R. Das, J. Org. Chem., 2013, 78, 6170 CrossRef CAS PubMed; (c) P. P. Ghosh, G. Pal, S. Paul and A. R. Das, Green Chem., 2012, 14, 2691 RSC; (d) G. Pal, S. Paul, P. P. Ghosh and A. R. Das, RSC Adv., 2014, 4, 8300 RSC; (e) G. Pal, S. Paul and A. R. Das, New J. Chem., 2014, 38, 2787 RSC; (f) S. Paul and A. R. Das, Catal. Sci. Technol., 2012, 2, 1130 RSC; (g) K. Pradhan, S. Paul and A. R. Das, Catal. Sci. Technol., 2014, 4, 822 RSC; (h) P. P. Ghosh and A. R. Das, Tetrahedron Lett., 2012, 53, 3140 CrossRef CAS; (i) P. Bhattacharyya, K. Pradhan, S. Paul and A. R. Das, Tetrahedron Lett., 2012, 53, 4687 CrossRef CAS; (j) K. Pradhan, P. Bhattacharyya, S. Paul and A. R. Das, Tetrahedron Lett., 2012, 53, 5840 CrossRef CAS; (k) P. P. Ghosh, S. Paul and A. R. Das, Tetrahedron Lett., 2013, 54, 138 CrossRef CAS.
- (a) A. M. Shestopalov, Y. M. Emelianova and V. N. Nesterov, Russ. Chem. Bull., 2003, 52, 1164 CrossRef CAS; (b) A. Alizadeh, A. Rezvanian and F. Bayat, Helv. Chim. Acta, 2014, 97, 532 CrossRef CAS.
- J. Sun, D. Z. Negrerie, Y. Du and K. Zhao, J. Org. Chem., 2015, 80, 1200 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data; copies of 1H and 13C NMR spectra for all compounds. CCDC 1404567, 1404569, 1404581 and 1434523. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra24510a |
| This journal is © The Royal Society of Chemistry 2016 |