
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMulti-stimuli-responsive polymers enabled by bio-inspired dynamic equilibria of flavylium chemistry†
Yuxi
Liu
 ac,
Rico F.
Tabor
a,
Piotr
Pawliszak
bc,
David A.
Beattie
bc,
Marta
Krasowska
bc,
Benjamin W.
Muir
d,
San H.
Thang
ac and
Chris
Ritchie
*ac
ac,
Rico F.
Tabor
a,
Piotr
Pawliszak
bc,
David A.
Beattie
bc,
Marta
Krasowska
bc,
Benjamin W.
Muir
d,
San H.
Thang
ac and
Chris
Ritchie
*ac
aSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia. E-mail: yuxi.liu@monash.edu; rico.tabor@monash.edu; san.thang@monash.edu; chris.ritchie@monash.edu
bFuture Industries Institute, University of South Australia, Mawson Lakes, SA 5095, Australia. E-mail: piotr.pawliszak@unisa.edu.au; david.beattie@unisa.edu.au; marta.krasowska@unisa.edu.au
cARC Centre of Excellence for Enabling Eco-Efficient Beneficiation of Minerals, Australia
dCSIRO Manufacturing, Bag 10, Clayton South, VIC 3169, Australia
First published on 12th March 2025
Abstract
As part of a complex equilibria network with other chemical species, flavyliums, the chromophoric component of anthocyanins, hold great potential for use in functional polymers. This study presents the successful syntheses of polymers containing two distinct flavylium-structures, generated via post-modification of a parent polymer synthesised using reversible addition-fragmentation chain transfer (RAFT) polymerisation. The selective modification of acetophenone moieties enabled precise tuning of the polymers' properties, which are strongly influenced by the markedly different chemical characteristics of flavyliums and the other species in equilibria with them. The synthesised flavylium-containing polymers exhibit multi-stimuli responsiveness to variations in solvent, pH, light, and temperature, thereby introducing intricacy and viable functionality to the polymer system. The surface activity and critical aggregation concentrations (CAC) of the synthesised polymers were studied using profile analysis tensiometry (PAT), revealing distinct aggregation and self-assembly behaviours. Fractal-like aggregates formed by the flavylium-containing polymers were investigated using cryogenic electron microscopy (Cryo-EM) and small-angle X-ray scattering (SAXS). This research bridges the colourful dynamic equilibria of flavylium chemistry with polymer chemistry, paving the pathway for further investigations into flavylium–polymer interactions and the development of tuneable material properties of responsive polymers.
Introduction
Flavylium (C15H11O+), or 2-phenyl-1-benzopyrilium, is an omnipresent cationic chromophore found in the naturally occurring anthocyanin pigments of many flowers, fruits, and plants, where it plays a crucial role in creating a vibrant palette of colours, ranging from vivid red to deep blue.1,2 These colours are key for attracting pollinators, aiding seed dispersal, and providing photoprotection to the plant via responsiveness to environmental changes such as soil pH and bioavailable metal ions (e.g., Al3+).3 Anthocyanins also fulfill crucial biochemical roles as antioxidants in plants and are involved in both abiotic and biotic stresses, such as UV radiation, cold temperatures, drought, and in defence against pathogens and herbivores.4,5 These functions arise from their chemical complexity whereby they undergo significant chemical transformations and coexist with multiple species in a network of dynamic equilibria, such as hemiketals (via deprotonation and hydration), quinoidal bases (through deprotonation) cis-chalcones (via tautomerisation), and trans-chalcones (via isomerisation) as shown in Scheme 1.6 These transformations include shifts in charge state (positive, neutral, or negative), as well as hydration, protonation, and photochemical isomerisation, which all impact their biological functionality, while presenting as a broad spectrum of colours through changes in their spectral absorption profile. Intriguingly, these molecular switches are triggered by external stimuli, such as changes in pH,6 light exposure,7 and temperature8 that can be reproduced and modified in unnatural compounds via synthetic chemistry to create novel and smart chromophores and fluorophores.9–13 The tuneability of the oxonium positive charge via delocalisation by introducing conjugated electron donating or withdrawing substituents posits allowing for modifications to substituent groups at different positions by using various starting materials and following distinct synthetic routes.1 By leveraging flavylium's chemical tuneability and responsiveness, researchers can develop advanced materials that respond dynamically to environmental changes, paving the way for applications in drug delivery,14 probes,15,16 sensors17,18 and shortwave infrared imaging.19,20 | ||
| Scheme 1 A representative equilibria network for: flavylium, hemiketal, cis-chalcone and trans-chalcone. In some instances, the hemiketal and cis-chalcone are transient species that equilibrate rapidly and a clean isosbestic point and a pseudo pKa could be observed between flavylium and trans-chalcone species. R4′ = substituent group at 4′-position of the flavylium core structure, R7 = substituent group at 7-position of the flavylium core structure. These two positions of substitution are more widely investigated. In this work, there is no hydroxy group at R4′ or R7 positions, so quinoidal base as another known species in the flavylium equilibria is not presented. | ||
As synthetic approaches evolve, polymeric materials have become increasingly smart, adapting to the diverse demands of their applications. A significant factor in the development of these intelligent materials has been the advancement of techniques for incorporating responsive motifs and decades of extensive research into stimuli-responsive polymers.21–23 Stimuli such as temperature,24,25 pH26,27 and light28 have been widely studied in the design of these polymers. As the research area continues to mature, and increasingly complex polymers from the endless library of possibilities are isolated, the desire for multi-stimuli responsive materials grows.29 Realising these goals will endow these materials with properties that can be exquisitely controlled due to the integration of several cooperative stimuli-triggered transformations within one polymer.30,31 These multi-stimuli-responsive materials are posited to be applied in optical sensing,32 antimicrobial,33 controlled release,34 moisture capturing35 and nanomedicines.36–38
Flavylium's responsiveness to stimuli like pH, light, and temperature makes it an excellent candidate for designing multi-stimuli-responsive polymers. Advancing research on flavylium-functionalised polymers holds promise for developing innovative multi-stimuli-responsive systems, presenting a valuable avenue for future investigation.39,40 In this work, two flavylium structures were installed on a RAFT statistical copolymer via post-modification (Scheme 2). The flavylium-containing polymers showed multi-stimuli responsiveness profiles to solvent, pH, light and temperature, showcasing that the chemical processes observed for small molecules can be translated into the macromolecule domain. Moreover, the synthesised polymers presented surface activity and self-assembly behaviour, highlighting their potential to be developed as smart surface-active materials.
 | ||
| Scheme 2 Synthetic route of flavylium-containing polymers and a schematic analogy of the polymer synthesis process. | ||
Results and discussion
Synthesis of the flavylium-containing polymers
The syntheses of the flavylium-containing polymers and parent polymers followed the convergent synthetic route communicated in our previous work.40 We used reversible addition–fragmentation chain transfer (RAFT) polymerisation41–43 to build a parent polymer with acetophenone functionality that is post-synthetically modified to install flavylium moieties. The parent polymer composition screening is presented in the ESI (Fig. S1, Table S1 and S2†). The parent polymer (P1) with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :200 molar ratio in feed of N-(4-acetylphenyl)methacrylamide (NAPhMA) and N-isopropylmethacrylamide (NIPMAM) was selected as the candidate in this study, as it contains approximately 6.4% units with acetylphenyl moieties and has suitable water solubility at room temperature.
:200 molar ratio in feed of N-(4-acetylphenyl)methacrylamide (NAPhMA) and N-isopropylmethacrylamide (NIPMAM) was selected as the candidate in this study, as it contains approximately 6.4% units with acetylphenyl moieties and has suitable water solubility at room temperature.
P1 was converted into the flavylium-containing derivatives reported herein by reacting the parent polymer with an excess of the respective salicylaldehyde to yield polymers with R7 groups of either diethylamino or methoxy functionality at the flavylium core. This process resulted in quantitative conversion as confirmed by the disappearance of the methyl protons of the acetophenone at 2.5 ppm in the 1H NMR spectra with new broad and poorly resolved resonances in the aromatic region attributed to the protons of the flavylium structures (Fig. 1). The flavylium-containing polymers have distinctive colours (Et2N-FlavP1 – magenta; MeO-FlavP1 – orange) with bisulphate as the charge balancing anion in both examples. At room temperature, both polymers dissolve readily in deionised water, however, sonication was also used to ensure complete dissolution for stock solution preparation. In the view of the appearance of broad flavylium resonances, along with intense colouration of the samples, the synthesis of flavylium-containing polymers is comparable to a process that resembles the ‘blossoming’ of acetophenone ‘flower buds’ on a polymeric ‘branch’, which grows from the RAFT agent ‘sprout’ (Scheme 2).
 | ||
| Fig. 1 1H NMR spectra (400 MHz, MeOD-d4) of parent polymer P1; Et2N-FlavP1 and MeO-FlavP1. The disappearance of the methyl (–CH3) proton resonance of acetylphenyl groups indicates quantitative conversion. | ||
Equilibration of the flavylium moieties
The stability of flavylium moieties can be influenced by pH, concentration, and water content (in non-aqueous solutions). Upon dissolution, the equilibration of the flavylium component of the polymers involves hydration and deprotonation of flavylium (AH+) to yield the neutral hemiketal (B) as shown in Scheme 1, while subsequent tautomerisation affords cis-chalcone (Cc), which isomerises to yield trans-chalcones (Ct). As hydration is the initial step, the stability of the AH+ species in solution is significantly affected by both water content and pH, which we investigated via steady-state UV-vis spectroscopy under controlled conditions. Equilibration of both flavylium-containing polymers was investigated in methanol, deionised water and in Theorell and Stenhagen's universal buffer (pH 7)44 at the concentration of 0.1 g L−1 for 24 hours at 25 °C in the dark.Samples of flavylium-rich polymers were dissolved at a concentration of 10 g L−1 in methanol or deionised water. These stock solutions were then diluted to the appropriate concentrations for the equilibration study presented in Fig. 2. As anticipated, methanolic solutions present the simplest spectral profiles due to water being the limiting reagent required for the multiple chemical transformations described earlier. Over the 24-hour period an increase in the flavylium content of Et2N-FlavP1 is noted (Fig. 2A) while a decrease is observed for MeO-FlavP1 (Fig. 2D)—processes that are rationalised by the differing initial polymer compositions of the respective samples. Furthermore, the reduction in flavylium content in MeO-FlavP1 over the 24-hour period is indicative of this polymer being more susceptive to the formation of the colourless hemiketal product (Scheme 1) than Et2N-FlavP1 with subsequent tautomerisation being disfavoured.
 | ||
| Fig. 2 The spectral evolution of Et2N-FlavP1 (0.1 g L−1) at 25 °C in (A) methanol; (B) deionised water and (C) universal buffer (pH 7). The spectral evolution of MeO-FlavP1 (0.1 g L−1) at 25 °C in (D) methanol; (E) deionised water and (F) universal buffer (pH 7). | ||
On dissolution of both polymers in deionised water (0.1 g L−1), a pH of (∼3) was measured due to (i) dissociation of the bisulphate counterion, (ii) deprotonation of the flavylium species, and (iii) residual acid in the polymer precipitates. This acidic pH (∼3) partially stabilises the flavylium species (Fig. 2B and E), whereas the neutral pH (∼7) of the universal buffer solution favours hydration of the flavylium to transient, spectroscopically unobserved species B and Cc followed by the growth of the Ct spectral band (Et2N-Ct, 455 nm; MeO-Ct, 365 nm, Table 1). Comparatively, equilibration of the initial flavylium-rich polymers at pH 7 occurs more rapidly for MeO-FlavP1 than for Et2N-FlavP1 (Fig. 2C and F), with negligible flavylium remaining on reaching equilibrium for the methoxy derivative. This behaviour is in line with previously reported data for related small molecule analogues.45,46
| Polymer | AH+ absorption (nm) | Ct absorption (nm) | Ct− absorptiona (nm) | AH+ emission (nm) | Ct emission (nm) | Ct− emission (nm) |
|---|---|---|---|---|---|---|
| a The pH-dependent solution-state spectra were measured up to a pH of 12, where the Ct− absorption maxima were observed and summarised. At higher pH values, the bands may shift. | ||||||
| Et2N-FlavP1 | 530 | 455 | 485 | 615 | 550 | 570 |
| MeO-FlavP1 | 465 | 365 | 445 | 525 | 490/650 | 590 |
Spectra of each polymer were also recorded at the concentration of 1 g L−1 in deionized water over a 24-hour period (Fig. S2†), showing comparable rates and speciation for MeO-FlavP1. However, the diethylamino derivative Et2N-FlavP1 reached equilibrium with a significantly higher flavylium content compared to its behaviour at 0.1 g L−1. The underlying cause of this phenomenon is still under investigation but is likely linked to the initial pH upon dissolution of the polymer sample as it resembles the spectra of a pH 1-equilibrated sample (Fig. 3A).
 | ||
| Fig. 3 (A) UV-vis spectra of Et2N-FlavP1 solutions (0.1 g L−1) in universal buffer solutions from pH 1 to 12, after 24 hours equilibration in the dark (dashed vertical lines represent excitation wavelengths). Emission spectra of Et2N-FlavP1 (0.1 g L−1) at pH 1, 4, 7 and 11 at excitation wavelengths (B) λexc = 455 nm (Et2N-chalcone) and (C) λexc = 530 nm (Et2N-flavylium); (D) UV-vis spectra of the solutions in universal buffer from pH 1 to 12 of MeO-FlavP1 (0.1 g L−1) after 24 hours equilibration in the dark (Dashed vertical lines represent excitation wavelengths); emission spectra of MeO-FlavP1 (0.1 g L−1) at pH 1, 4, 7 and 11 at excitation wavelengths (E) λexc = 365 nm (MeO-chalcone) and (F) λexc = 465 nm (MeO-flavylium), The y-axis is displayed on a logarithmic scale; (G) pH-dependent chromism of polymer solutions (0.1 g L−1) after equilibrated for 24 hours at room temperature in the dark; (H) a fluorescence change observed in Et2N-FlavP1 and MeO-FlavP1 solution (1 g L−1, incubated at 40 °C overnight to increase the MeO-chalcone species concentration) with UV irradiation at 365 nm for 1 s, 5 s and 15 s. | ||
pH-dependent equilibria network and fluorochromism
Understanding the representative absorption peaks of each flavylium-containing polymers can help to assess another important characteristic of flavylium structures, which is their ability to respond to environmental pH changes. To investigate the pH responsiveness of the functionalised polymers, solutions were prepared by diluting the stock solutions (aq. 10 g L−1) into universal buffer at various pH levels, which were then allowed to equilibrate in the dark for 24 hours at room temperature. The equilibrated solutions displayed distinct colours that varied with pH (Fig. 3G). For Et2N-FlavP1, a magenta colour was observed at acidic pH, transitioning to a bright yellow hue as the pH increased, indicating a predominance of chalcone species in the equilibria. It is worth mentioning a pseudo-equilibrium from pH 1 to pH 8 of flavylium and trans-chalcone species and a pseudo-pKa of approximately pH 4 could be observed (Fig. 3A). This might be due to the intermediate species hemiketal and cis-chalcone, which exist in the equilibria between flavylium and trans-chalcone, being short-lived in these buffer environments. Notably, at pH 12, a shift in absorption maximum to approximately 485 nm was observed (Fig. 3A) likely resulting from the deprotonation of the phenolic proton of the chalcone to yield the ionic chalconate (also known as anionic chalcone or deprotonated chalcone) (Scheme 3). | ||
| Scheme 3 The pH- and photo-responsive processes of flavylium equilibria involved in polymeric system. Chalconates (trans- or cis-) are only observed at basic pH while the other species may exist in a range of different pHs. In amino-flavylium systems the hemiketal and cis-chalcone are in general minor species and a pseudo-equilibrium is normally established between the flavylium cation and trans-chalcone. | ||
The flavylium chromophore λmax of MeO-FlavP1 is blue shifted by 65 nm with respect to that of Et2N-FlavP1, highlighting the tuneability of the polymer's spectral properties. MeO-FlavP1 polymer displayed a comparable behaviour in its pH-dependent chromism (Fig. 3D). At acidic pH, the MeO-flavylium was the predominant species and the flavylium-dominant solutions appeared orange yellow. As the pH approached neutral, the absorption of the MeO-flavylium (λmax = 465 nm) gradually decreased. MeO-chalcone emerged and dominated the spectra, leading to a blue shift in absorption maximum (λmax = 365 nm). The increasing chalcone contents resulted in a more colourless solution because MeO-chalcone absorbs mainly in the UV region. The MeO-chalconate exhibited an intermediate absorption peak between the flavylium and chalcone bands and the population of this species increases from pH 9. When MeO-chalconate (λmax = 445 nm) formed in the basic environment, the solutions were pale yellow in colour.
The deprotonation and hydration of flavylium to form chalcones typically lead to water insolubility. Consequently, earlier studies on water-insoluble chalcones relied on buffer-ethanol mixtures for equilibria studies.47,48 However, equilibria observed in mixed solvents can differ from those in single-solvent systems due to the mixture's ability to solubilise water-insoluble species, which can influence the equilibrium dynamics. Notably, incorporating flavylium structures into hydrophilic polymers allows the chalcone species to be observed in pure water or buffer, as the hydrophilic polymer components improve their solubility.
To investigate the fluorescence properties of MeO-FlavP1 and Et2N-FlavP1, solutions of these two polymers were equilibrated for 24 hours in the dark at the respective pHs of interest (1, 4, 7 and 11). As the composition and spectra of each material is pH dependent, two excitation wavelengths were selected for each material that coincides with λmax for Ct and AH+ (Table 1, Fig. 3B and C). Red fluorescence (λem = 615 nm) was observed for Et2N-FlavP1 on excitation (λexc = 530 nm) of AH+ with decreasing emission intensity at elevated pHs in line with expectation due to conversion to Ct. Spectral overlap in the proximity of the Ct λmax resulted in more complex emission with weak AH+ emission observed at pH 1, due to excitation of the blue edge of the AH+ absorption band, with gradual diminishment of emission on increasing pH and emergence of green (λem = 550 nm) Ct emission. Deprotonation of the chalcone to yield anionic chalconate Ct− is evidenced by characteristic emission (λem = 570 nm) observed for both excitation wavelengths at pH 11. It is noted that the photochemical conversion of Ct to Cc and subsequently AH+ on excitation of Ct is inefficient with radiative relaxation via fluorescence dominating.
As for MeO-FlavP1, intense green fluorescence (λem = 525 nm) was observed on excitation (λexc = 465 nm) of MeO-AH+ at pH 1, however, significant quenching was observed at pH 4 relative to the change in AH+ concentration based on the acquired absorption spectra (Fig. 3E). Similar behaviour has been documented previously and is rationalised by the association of AH+ moieties with phenolic molecules to form co-pigmentation complexes.49,50 In this case, co-pigmentation complexes can happen between AH+ and phenolic chalcones, which are more prevalent at higher pHs. Since the co-pigmentation complexes are generally non-fluorescent,51,52 they are expected to account for the observed fluorescence quenching.53 At pH 7, all MeO-AH+ has been converted to other species due to the relative instability of MeO-AH+vs. Et2N-AH+ at pHs >4. The shorter excitation wavelength (λexc = 365 nm) was also explored to investigate the steady-state photochemistry of MeO-Ct (Fig. 3F), with chalcone emission (λem = 490 nm) dominating at pH 4 and 7 with a transition to Ct− emission (λem = 590 nm) at pH 11.
However, a detailed time-resolved spectroscopy campaign is required to fully rationalise and understand the influence of various factors on the photochemical behaviour of polymeric systems composed of flavyliums such as the impacts of ionic strength and concentration on polymer aggregation and self-assembly. Excitation spectra at pHs 1, 4, 7 and 11 for both polymers are provided in the ESI (Fig. S3 and S4).†
As shown in Fig. 3H, solutions of both flavylium-containing polymers (1 g L−1 in deionised water) were incubated overnight at 40 °C in a water bath to generate more thermodynamically stable Ct species. The solutions were then illuminated by UV light at 365 nm, where in Et2N-FlavP1, the red fluorescence remained relatively unchanged over time, whereas in MeO-FlavP1 where an obvious change in fluorescence was observed, the emission colour changed gradually from white to green and more bright green upon exposure to UV light, indicating the recovery of methoxy AH+ upon UV irradiation. Based on these observations, the following section discusses the photochemical reactivity of both polymers.
pH-dependent photo-responsiveness
The photochemistry of the complex flavylium equilibria network revolves around the photoisomerization of cis- and trans-chalcones, which may undergo further transformations throughout the equilibria (Scheme 3), a process that is also regulated by pH. To investigate this, polymer solutions were equilibrated for 24 hours in the dark using universal buffers at pH 4, 7, and 11 as intermediate stages for pH-dependent chromism tests. Following equilibration in the dark, the solutions were exposed to appropriate wavelengths to assess their photo-responsiveness using steady-state UV-vis spectroscopy.Illumination of Et2N-FlavP1 (λexc = 455 nm), at pH 4, caused subtle spectral change during the initial irradiation, attributed to the presence of a significant amount of flavylium at this pH (Fig. 4A and B). In contrast, illumination of solutions equilibrated at pH 7 and 11 revealed the consumption of Ct without the formation of flavylium. Instead, there was a commensurate increase in Cc and B2, along with potentially B4 hemiketals54–56 (Scheme S1†), which are spectroscopically challenging to assign. The presence of B4 hemiketals has been reported to reduce the photoconversion rate.57 Studies have shown that the photochemical reactions of neutral amino substituted trans-chalcone species to flavylium were difficult to achieve in aqueous solution.58,59 The photochemistry in Et2N-FlavP1 is likely driven by the 4′-amido-trans-chalcone (–NH–CO–) moiety, as a small-molecule 4′-amido-trans-chalcone derivative has been reported to exhibit low quantum yield of conversion for the photochemical conversion of Ct to Cc/B (0.09).59
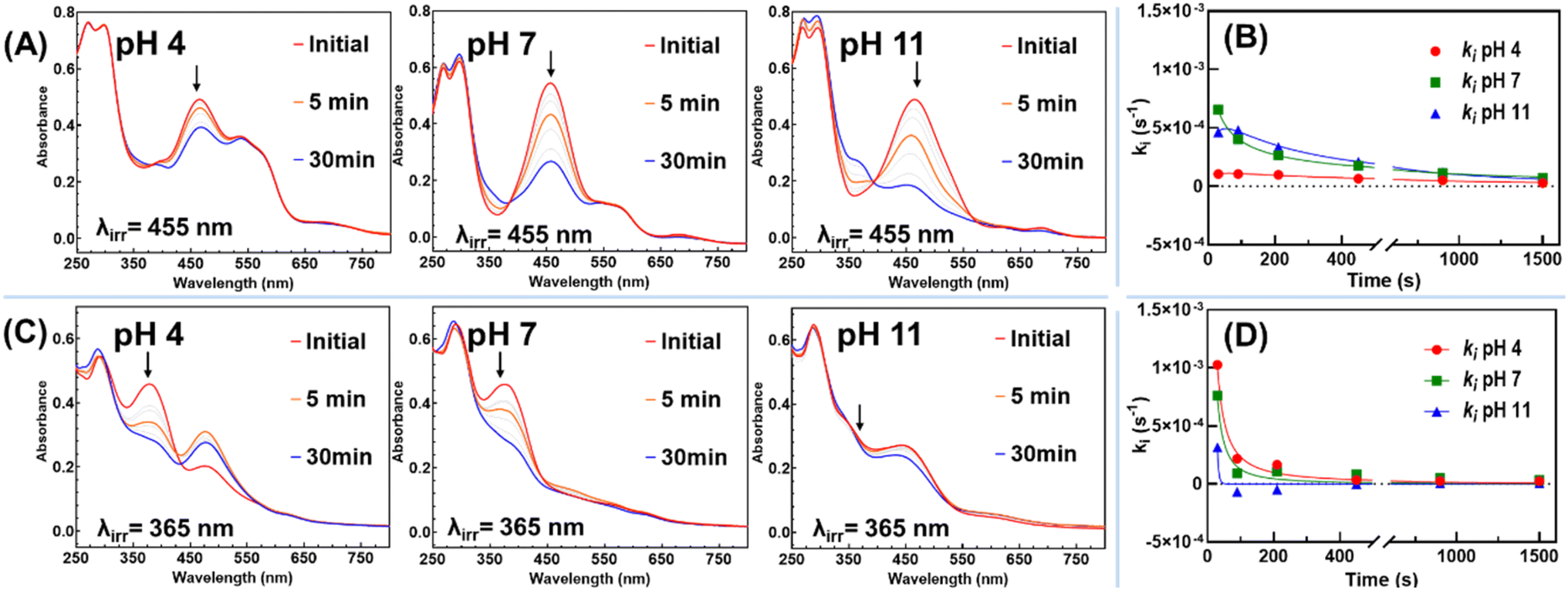 | ||
| Fig. 4 (A) UV-vis absorption spectral variations observed over time upon irradiation of equilibrated Et2N-FlavP1 solution (0.1 g L−1, equilibrated in the dark for 24 hours at pH 4, 7 and 11) with 455 nm light (P = 2.1 W); (B) the rates of the absorbance change at irradiation wavelength at different pH levels for Et2N-FlavP1 solution; (C) UV-vis absorption spectral variations observed upon irradiation of equilibrated MeO-FlavP1 solution (0.1 g L−1, equilibrated in the dark for 24 hours at pH 4, 7 and 11) with 365 nm light (P = 2.1 W); and (D) the rates of the absorbance change at irradiation wavelength at different pH levels for MeO-FlavP1 solution. | ||
Additionally, it has been reported that protonated tertiary amine Ct+ species are more conducive to convert to flavylium following illumination.60,61 To investigate whether this behaviour extends to polymeric systems, Et2N-FlavP1 solution was illuminated for 30 minutes, followed by the addition of 30 μL trifluoroacetic acid. Acid addition led to the disappearance of the Ct absorption band, yielding Ct+ (Fig. S5 and Scheme S2†), which could be subsequently photochemically converted to AH+ by UV illumination (λexc = 365 nm).60
In contrast, MeO-FlavP1 showed a faster photoconversion rate of Ct (Fig. 4C and D), with the production of flavylium following excitation (λexc = 365 nm) at pH 4. Prolonged illumination (30 minutes), however, resulted in a decrease in flavylium concentration indicating the possibility of a competing thermal process or partial photodecomposition that requires further investigation. Illumination of a solution equilibrated at pH 7 yielded only a trace of flavylium with the diminishment of the Ct electronic band the only notable spectral change. Due to spectral crowding, identification of Cc, B2 and B4 hemiketals is prohibited. At pH 11, the absorption spectra are dominated by the deprotonated MeO-chalconate Ct− with excitation resulting in minimal changes to the spectra, an observation consistent with the 4′-methoxyflavylium analogue reported by Pina et al.62 This pH-dependent photochemistry was also used to block the photo-responsive ring shuttling of alkoxyl-chalcone to form flavylium at higher pH levels.63
Thermo-responsiveness of polymers
PolyNIPMAM is a well-known thermo-responsive material that exhibits a lower critical solution temperature (LCST) of 44 °C,64,65 defining a thermal threshold for polymer solubility. Installation of flavylium moieties along with their inherent thermal properties endows these materials with complex thermo-responsive behaviour. As shown in Fig. 5A. The parent polymer P1 containing NAPhMA showed a rapid transition of the transmittance at 35 °C, representing a cloud point temperature lower than the LCST for NIPMAM homopolymers. We attribute this to the additional hydrophobicity of the polymer chains which is contributed by the NAPhMA. On cooling, hysteresis is observed with the solution not regaining 100% transparency until reaching 25 °C. This is typical behaviour for polyNIPMAM-based materials.66,67 | ||
| Fig. 5 Transmittance change at λ = 700 nm as a function of temperature of polymer solutions (1 g L−1) of (A) parent polymer P1; (B) Et2N-FlavP1 and (C) MeO-FlavP1 (solid line: heating; the dotted line: cooling); (D) states of polymeric solutions after heating: the heated MeO-FlavP1 solution shows increased turbidity (left of blue line) and larger aggregates at 60 °C (right of blue line). On cooling, solid arrows indicate full transmittance recovery; dashed arrows indicate partial recovery. | ||
In the case of Et2N-FlavP1, it is noted that the transmittance dropped steadily while heating the solution (Fig. 5B). This phenomenon was caused by increased absorption due to the thermal equilibration of Et2N-flavylium, rather than an increase in turbidity as observed for P1 (Fig. S6†). On cooling, the transmittance did not increase as observed for P1, again indicating it was due to speciation of Et2N-FlavP1. Thus, the thermal behaviour of Et2N-FlavP1 was more influenced by the diethylamino flavylium functionality, and doesn't exhibit pronounced LCST-type thermo-responsiveness from the polyNIPMAM components, a well-documented process where the LCST is strongly influenced by the nature of hydrophobic or hydrophilic comonomers.40,68–70 After cooling, the characteristic purple colour of the flavylium species was partially recovered (Fig. 5D), resulting from the reversion of the diethylamino flavylium species at lower temperatures due to the re-establishment of the thermal equilibria.
As for MeO-FlavP1, the transmittance of the solution decreased slowly when heating (Fig. 5C), but on reaching 55 °C, the solution rapidly became turbid, suggesting a process attributed to the LCST-type properties of the polyNIPMAM composition. The cloud point of MeO-FlavP1 is therefore higher than that of P1, which can be rationalised by the fact that methoxy flavylium shows greater hydrophilicity due to the change from a neutral acetophenone to a positively charged aromatic oxonium. Additionally, the dissolution of MeO-FlavP1 results in an increase of ionic strength, a decrease of the Debye length and hence the range of electrostatic repulsions, thus the polymer formed larger aggregates at 60 °C (Fig. 5D). On cooling, the solution became less turbid, and similarly to Et2N-FlavP1, not all flavylium was recovered on cooling to 20 °C. This suggests that in MeO-FlavP1, the thermo-responsive properties arose from a combination of the polyNIPMAM component's LCST behaviour and the thermodynamic equilibria of methoxy flavylium.
Surface activity and self-assembly properties of polymers
Both Et2N-FlavP1 and MeO-FlavP1 were investigated using 1H NMR spectra in D2O and MeOD to interrogate the impact of solvent on their self-assembly properties (Fig. S7 and S8†). Regardless of solvent, the isopropyl proton of the NIPMAM units presented sharp resonances consistent with effective solvation. A subtle upfield shift of the proton signal from δ 3.90 (MeOD) to δ 3.87 (D2O) likely arises from solvent-induced electronic effects in both cases of the polymers. In contrast, the aromatic protons of flavylium resonances broadened significantly in D2O compared to MeOD, indicating reduced molecular mobility due to potential aggregation of the flavylium moieties. To further investigate this observation, various experiments were conducted to assess the surface activity of these polymers as well as their ability to form aggregates/assemblies in aqueous solution.Profile analysis tensiometry (PAT) with emerging bubble geometry was utilised to study the surface activity of the polymer solutions.71,72 For the parent polymer P1, at the concentration of 0.001 g L−1, it took over 30 minutes to record a rapid decrease in the surface tension, indicating that polymer adsorption at the air–water interface was slow (Fig. 6A). The surface tension versus time plot exhibited an ‘S’ shape, with plateaux at both the initial and final equilibrium stages, which is characteristic of non-ionic small-molecule surfactants. This is consistent with the neutral structure of the parent polymer P1 in addition to the negligible zeta potential value (−1.7 mV) as shown in Table 2. As the polymer concentration was increased, the surface tension decreased more rapidly, indicating faster adsorption of the polymer at the air–water interface. At concentrations above 0.025 g L−1, the equilibrium value of surface tension was quickly established, indicating that the critical concentration required for polymer to aggregate/assemble had been reached. This concentration is generally referred to as the critical micellar concentration (CMC) in small-molecule surfactant solutions; however, since random polymers often form more complex aggregates rather than simple micelles, this concentration is therefore referred in this section as critical aggregation concentration (CAC),73 and it occurs at the point where the regression line of the linearly dependent surface tension region intersects with the straight line passing through the plateau. The CAC of P1 was found to be 0.013 g L−1 (Fig. 6B).
 | ||
| Fig. 6 (A) Surface tension change of P1 aqueous solutions as a function of time at different concentrations; (B) an illustration of finding the CAC of parent polymer P1 by plotting the equilibrium surface tension against concentration. | ||
| Polymer | Equilibrium surface tensionaσ (mN m−1) | CACb (g L−1) | Zeta potentialcζ (mV) |
|---|---|---|---|
| a Determined at the point where the regression line of the linearly dependent surface tension region intersects with the straight line passing through the plateau (Fig S9 and S10). b Calculated by finding the intersection of the two curves plotted at the decreasing stage and plateauing stage (Fig S11 and S12). c Determined in aqueous solutions (1 g L−1) from electrophoretic mobility measurements using Malvern NanoZetasizer. All polymers were dissolved in deionised water; however, due to different degree of ionisation, the ionic strength in these solutions was different. | |||
| P1 | 46.7 ± 0.1 | 0.013 | −1.7 |
| Et2N-FlavP1 | 49.6 ± 0.1 | 0.015 | +10.6 |
| MeO-FlavP1 | 47.9 ± 0.1 | 0.009 | +14.7 |
The surface tension was measured for the both flavylium-functionalised polymers' solutions, with CAC values of 0.015 g L−1 for Et2N-FlavP1 and 0.009 g L−1 for MeO-FlavP1 (Table 1 and Fig. S9–S12†). These CAC values are comparable to that of the parent polymer, as the overall polymer compositions, with 6.4% of the polymer chain comprising functional units, were not significantly altered by the post-modification, leaving their affinity towards air–water interface mostly unchanged. The CAC of Et2N-FlavP1 was slightly higher than that of P1, likely due to the increased hydrophilicity of the diethylamino-flavylium structure compared to the acetophenone structure, as the methacrylamide derivative is known for its water solubility. On the other hand, MeO-FlavP1 formed aggregates at a lower concentration, as evidenced by its lower CAC compared to the parent polymer. Despite the enhanced water solubility of the positively charged methoxy-flavylium structure compared to acetophenone, particularly in acidic environments, MeO-FlavP1 reached maximum air–water interface coverage and aggregate formation faster than P1 and Et2N-FlavP1, which suggested that the property of the R7 group might greatly influence both the interfacial behaviour and the in-solution aggregate formation of the flavylium-containing polymers. Due to the higher hydrophilicity of the positively charged flavylium structures and larger determined absolute values of zeta potential, which indicates stronger electrostatic repulsion between the molecules at the air–water interface, both modified polymers were found to adsorb to a lesser extent than P1, as reflected in their equilibrium surface tension of 49.6 mN m−1 for Et2N-FlavP1 and 47.9 mN m−1 for MeO-FlavP1 comparing to 46.7 mN m−1 of P1 (Table 2). Given the duration of the tensiometry measurements (>3 hours) in conjunction with the complex chemical equilibria of the FlavPs, it is reasonable to consider that the surface activity and CAC values are representative of chemically complex polymers approaching equilibrium.
The PAT with emerging bubble geometry allows one to minimise surfactant depletion effects. This is essential for surfactant solutions of low concentration and slow adsorption kinetics.66 The determined zeta potential values of polymer in aqueous solutions (1 g L−1) were +10.6 mV for Et2N-FlavP1 and +14.7 mV for MeO-FlavP1 (Table 2), confirming successful cationic modifications. However, these relatively low absolute values of the zeta potential (<|15| mV) suggest that these polymers in solution are not colloidally stable and there will be a tendency for aggregate formation/coagulation in solution (especially at higher ionic strengths).74,75
To reveal the size of aggregates formed by these polymers, dynamic light scattering (DLS) experiments were performed on aqueous solutions. Solutions of the parent polymer P1 showed scattering from particles present that had an average hydrodynamic diameter (Dh) of 44.5 nm with a polydispersity index (PDI) of 0.172 (Fig. 7A). Attempts were made to determine the Dh for flavylium-containing polymers on DLS; however, this was unsuccessful due to the strongly coloured and fluorescent compounds at the sampling concentration (1 g L−1).76 Small-angle X-ray scattering (SAXS) is not susceptible to these drawbacks and was therefore used to investigate the in situ morphology of the aggregates formed in aqueous media.76,77 The SAXS data profile of parent polymer P1 was fitted to a fractal model,78 with a primary particle radius (Rp) of 15 Å and a fractal dimension (df) of 2.22 (Fig. 7B, S13 and Table S3†). The fractal dimension was higher than a random walk mass-fractal structure in theta solvent (df = 2.00),79 suggesting the fractal aggregates were not simply solvated polymers, but larger fractal networks formed by individual primary polymer particles. The dimension of P1 was close to a reaction-limited cluster aggregation (RLCA),80 which indicates that not all collisions between particles result in aggregation, and these clusters are usually more compact than the aggregates formed by diffusion-limited cluster aggregation, which happens upon particle contact.81 Thus, we anticipate that the fractal clusters seen here are dynamic in nature and loosely aggregated by weak, physical interactions.
 | ||
| Fig. 7 (A) DLS result by intensity of parent polymer P1 (aq. 10 g L−1); (B) room-temperature SAXS profiles in log–log plots of P1, MeO-FlavP1 and Et2N-FlavP1. Solid curves are the fitted results achieved with SasView using a fractal core–shell model (the original SAXS data and tabulated fitting results are provided in ESI†). (C) Cryo-EM micrograph of aggregates formed in MeO-FlavP1 solution (aq. 10 g L−1) in vitrified ice in a Lacey carbon film copper grid, red dotted line: fractal aggregates; blue dashed line: individual primary polymer particles. The larger dark spots are crystalline ice contamination stemmed on the surface; (D) Cryo-EM micrographs of MeO-FlavP1 fractal clusters (aq. 10 g L−1) in ice vitrified in a Quantifoil copper grid. | ||
SAXS data from solutions of flavylium-containing polymers fitted better to a core–shell fractal model (Fig. 7B, S14, S15 and Table S4, S5†), with core Rp of 20 Å for MeO-FlavP1 and 26 Å for Et2N-FlavP1, and shell thickness of 30 Å and 46 Å, respectively, resulting in primary particles sizes (total diameter) of approximately 10.0 nm for MeO-FlavP1, and 14.5 nm for Et2N-FlavP1. This difference probably arises because the diethylamino groups of Et2N-FlavP1 result in a polymer that is more hydrophilic and thus particles were potentially more solvated than those formed from MeO-FlavP1. These individual primary particles then further aggregated into fractal structures with df of 2.78 for MeO-FlavP1 and 2.93 for Et2N-FlavP1, respectively. These fractal dimensions are extremely high, which indicates that comparatively strong attractive forces exist between the primary particles, forming dense and compact clusters.82 Relatively high dimensional polymer colloidal fractal aggregates (df = 2.40–2.60) were previously reported in long chain colloidal spheres of poly(methyl methacrylate) (PMMA) in liquids.83 The formation of primary particles and fractal cluster here is likely driven by pendant chain hydrophobic interactions and π–π stacking interactions of the acetophenone moiety in the parent polymer. Stable segmental associations have been previously found in some fractal aggregates formed by the in-plane stacking of phenylene or phenyl moiety in toluene.84
In the case of the two flavylium-containing polymers, the primary particles exhibited a core–shell structure. This information in conjunction with 1H NMR analysis supports our hypothesis that the broad resonances indicate the flavylium structures were more likely aggregated and denser within the core, while the polymer backbones and NIPMAM units were more concentrated in the shell. It is hypothesised that the sulphate counter anions are attracted to the positively charged, flavylium-rich core, leading to a higher electron density in the core when compared to the shell. This difference in electron density would provide a significant contrast for X-rays, resulting in the more pronounced core–shell features observed in the SAXS profiles of the primary particles formed by flavylium-containing polymers compared to their parent polymer P1.
The origin of the aggregated flavylium structures in solutions can be rationalised by: (i) strong π–π interactions between the flavyliums, which are known to induce self-assembly and liquid crystal formation.85,86 These π–π interactions between pendant flavylium moieties in these modified polymers could enable the formation of larger, higher-dimensional clusters, (ii) the intrinsic intermolecular self-association behaviour of flavyliums87–90 which might also have contributed to the formation of aggregated flavylium structures in the particle core, (iii) intramolecular and intermolecular co-pigmentation of the flavyliums and chalcones in the polymers as mentioned in previous sections cannot be ruled out as a possible cause to the primary particle formation.91 In those single polymer chains whose flavyliums moieties were partially converted into chalcones, intramolecular co-pigmentation could be more pronounced; (iv) moderately positive ζ-potentials in flavylium-modified polymers introduced repulsive forces that slowed aggregation kinetics, favouring RLCA and yielding compact, dense, and higher dimensional fractal aggregates. Within individual particles, aromatic flavylium moieties were preferentially buried in the core, while NIPMAM components were enriched in the corona/shell. Collectively, these interactions contributed to the formation of fractal aggregates with higher dimensions than typical colloidal systems by consolidating and reinforcing the interactions as discussed. Insights from the spontaneous formation of these aggregates driven by π-π interactions, flavylium self-association and co-pigmentation on the pendant side chains can inform the design of novel self-assembled polymersomes.70,92,93
A solution of MeO-FlavP1 (10 g L−1) was interrogated using cryogenic electron microscopy (Cryo-EM), which revealed fractal-like aggregates. This observation agrees with the modelled SAXS data. The difference in the size of individual particles (8–12 nm) and fractal aggregates of MeO-FlavP1 is evident and presented in Fig. 7C and D. The agreement between SAXS and Cryo-EM confirm the aggregation behaviour of MeO-FlavP1 in solution. Attempts to image the aggregates under conventional transmission electron microscopy (TEM) with negative staining by uranyl acetate resulted in varying morphologies for both MeO-FlavP1 and Et2N-FlavP1 (Fig. S16†) and were deemed non-representative of the solvated aggregates reflected by the SAXS results. The change in the aggregate morphology was likely caused by the sample preparation procedures, particularly during the drying process on the copper grids, where the individual droplets merged when water evaporated, and the structures presented in this case might be driven by capillary force and rendered globular domains on the carbon film of the copper grid. Similar morphological change was observed in solution state during the process of evaporation evidenced by SAXS.94 These findings emphasised the limitations of conventional TEM in studying solvated polymer aggregates, as the drying process can alter the true aggregate morphology. In contrast, Cryo-EM preserves the sample in vitrified ice and offers a clear advantage by maintaining the native state of the aggregates, especially for these fractal aggregates formed by random copolymers.95
Conclusions
Water-soluble statistical poly(NAPhMA-co-NIPMAM) was successfully synthesised using RAFT polymerisation. Post-modification of acetophenone moieties on the polymer side chains produced two flavylium derivatives: 7-methoxy flavylium (MeO-FlavP1) and 7-diethylamino flavylium (and Et2N-FlavP1). The selective modification of these acetophenone moieties introduced tuneable properties to the polymers, which are significantly influenced by the chemical nature of the flavylium moieties. The modified polymers demonstrated a dynamic ability to switch between various chemical states in response to stimuli including solvent, pH, heat, and light. The pH-dependent chromism, fluorescence and photo-chemical interconversions showcase the versatility of these flavylium-modified polymers. These features underscore the inclusion of flavylium components within polymers as an innovative functionalisation strategy to yield multi-responsive luminescent materials.96,97By optimising the composition of the flavylium-containing polymers to enhance water solubility, the intricate pH-dependent equilibria of flavylium and the photochemical transitions of chalcone moieties can be investigated directly in pure water or buffer conditions. This approach demonstrated that covalently integrating flavylium structures into polymer matrices created a synergistic platform, where concepts from both flavylium chemistry and polymer chemistry enrich each other, offering insights and advancing both fields. Studying the equilibria of flavylium moieties on the polymer chains also brings insights to the intramolecular complexation between of flavyliums with other structures such as co-pigmentation.98
The surface activity and CACs of the polymers were systematically investigated. The polymers were found to self-assemble into fractal-like aggregates, as confirmed by SAXS. The aggregates formed by MeO-FlavP1 were revealed using Cryo-EM. These findings demonstrate that modifying the flavylium structures can influence polymer aggregate formation in solution. Although the complex dynamic equilibria and intricate behaviour of flavylium-containing polymers in aqueous systems made it difficult to pinpoint the precise origin of their self-assembly, the investigations on the surface activity of flavylium-containing polymers provided valuable insights for designing novel stimuli-responsive and surface-active materials. Gaining a deeper understanding of how substituent groups affect flavylium equilibria, and consequently polymer self-assembly will be critical for optimising these aggregates to respond to specific environmental stimuli. Concurrently, the π–π stacking interactions, self-association and co-pigmentation between flavylium moieties offer inspiration for the design of self-assembling flavylium-containing polymers and novel trans-chalcone crosslinked single-chain nanoparticles.99
Finally, this work presents a significant advancement in the development of multi-stimuli-responsive polymers by integrating bio-inspired flavylium chemistry100 into polymer systems by covalent incorporation, creating pathways for novel research on interactions between flavylium moieties and polymer matrices. Understanding how substituent groups influence flavylium equilibria and polymer self-assembly is crucial for optimising these systems to design fluorescent materials with tailored responses to environmental stimuli. The resulting materials have significant potential for advanced applications in smart chromophores, responsive surfactants, and fluorescent biochemical sensors.
Abbreviations
| NIPMAM | N-Isopropylmethacrylamide |
| RLCA | Reaction-limited cluster aggregation |
| RAFT | Reversible addition-fragmentation chain transfer |
| NAPhMA | N-(4-Acetylphenyl)methacrylamide |
| CDTPA | 4-Cyano-4-[(dodecylsulfanylthiocarbonyl)sulfanyl]pentanoic acid |
| AIBN | 2,2′-Azobis(isobutyronitrile) |
| MeO-FlavP1 | 7-Methoxy flavylium-containing polymer |
| SAXS | Small-angle X-ray scattering |
| CMC | Critical micellization concentration |
| CAC | Critical aggregation concentration |
| GPC | Gel permeation chromatography |
| DLS | Dynamic light scattering |
| TEM | Transmission electron microscopy |
| Cryo-EM | Cryogenic electron microscopy |
| NMR | Nuclear magnetic resonance spectroscopy |
| d f | Fractal dimension |
Data availability
All the data supporting this article have been included in the main text and the electronic (ESI).†Author contributions
Conceptualisation: Y. L., C. R.; investigation, methodology, data acquisition, and curation: Y. L.; P .P.; R. F. T., B. W. M.; writing-original draft preparation: Y. L.; writing-review and editing: all authors; funding acquisition: D. A. B., M. K., C. R., S. H. T. The manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
Y.L. thanks Monash University for the award of his CF-MGS scholarship and extends his gratitude to Dr Tina Hsia for her assistance with the GPC, Dr Celesta Fong for her assistance in Chemspeed, Dr Simon Crawford and Dr Sylvain Trépout for providing the inductions to Cryo-TEM, A.Prof. Alison Funston and A. Prof. Toby Bell for fluorimeter access and Dr Tzong-Hsien Lee for Zetasizer access. The authors acknowledge the use of instruments and assistance at the Monash Ramaciotti Centre for Cryo-Electron Microscopy, a Node of Microscopy Australia. This research was undertaken in part using the SAXS/WAXS beamline at the Australian Synchrotron, part of ANSTO, and the help from the beamline scientists is highly appreciated. C.R., S.H.T, D.A.B. and M.K. all acknowledge the funding support from the Australian Research Council for the ARC Centre of Excellence for Enabling Eco-Efficient Beneficiation of Minerals, grant number CE200100009.References
- L. Cruz, N. Basilio, N. Mateus, V. de Freitas and F. Pina, Natural and Synthetic Flavylium-Based Dyes: The Chemistry Behind the Color, Chem. Rev., 2022, 122(1), 1416–1481, DOI:10.1021/acs.chemrev.1c00399.
- F. Pina, M. J. Melo, C. A. Laia, A. J. Parola and J. C. Lima, Chemistry and applications of flavylium compounds: a handful of colours, Chem. Soc. Rev., 2012, 41(2), 869–908, 10.1039/c1cs15126f.
- M. C. Moncada, S. Moura, M. J. Melo, A. Roque, C. Lodeiro and F. Pina, Complexation of aluminum(III) by anthocyanins and synthetic flavylium salts: A source for blue and purple color, Inorg. Chim. Acta, 2003, 356, 51–61, DOI:10.1016/S0020-1693(03)00394-3.
- N. Tena, J. Martin and A. G. Asuero, State of the Art of Anthocyanins: Antioxidant Activity, Sources, Bioavailability, and Therapeutic Effect in Human Health, Antioxidants, 2020, 9(5), 451, DOI:10.3390/antiox9050451.
- F. Cappellini, A. Marinelli, M. Toccaceli, C. Tonelli and K. Petroni, Anthocyanins: From Mechanisms of Regulation in Plants to Health Benefits in Foods, Front. Plant Sci., 2021, 12, 748049, DOI:10.3389/fpls.2021.748049.
- F. Pina, J. Oliveira and V. de Freitas, Anthocyanins and derivatives are more than flavylium cations, Tetrahedron, 2015, 71(20), 3107–3114, DOI:10.1016/j.tet.2014.09.051.
- F. Pina, V. Petrov and C. A. T. Laia, Photochromism of flavylium systems. An overview of a versatile multistate system, Dyes Pigm., 2012, 92(2), 877–889, DOI:10.1016/j.dyepig.2011.03.033.
- M. A. Spirache, P. Marrec, A. J. Dias Parola and C. A. Tonicha Laia, Reversible thermochromic systems based on a new library of flavylium spirolactone leuco dyes, Dyes Pigm., 2023, 214, 111208, DOI:10.1016/j.dyepig.2023.111208.
- P. Correia, P. Araújo, A. Plácido, A. R. Pereira, L. J. Bessa, N. Mateus, V. de Freitas, J. Oliveira and I. Fernandes, Light-activated amino-substituted dyes as dual-action antibacterial agents: Bio-efficacy and AFM evaluation, Dyes Pigm., 2024, 224, 111975, DOI:10.1016/j.dyepig.2024.111975.
- C. Tian and K. Burgess, Flavylium- and Silylrhodapolymethines In Excitation Multiplexing, ChemPhotoChem, 2021, 5(8), 702–704, DOI:10.1002/cptc.202000287.
- O. Uranga-Barandiaran, D. Casanova and F. Castet, Flavylium Fluorophores as Near-Infrared Emitters, ChemPhysChem, 2020, 21(20), 2243–2248, DOI:10.1002/cphc.202000544.
- D. Svechkarev and A. M. Mohs, Organic Fluorescent Dye-based Nanomaterials: Advances in the Rational Design for Imaging and Sensing Applications, Curr. Med. Chem., 2019, 26(21), 4042–4064, DOI:10.2174/0929867325666180226111716.
- L. Wang, M. He, Y. Sun, L. Liu, Y. Ye, L. Liu, X. C. Shen and H. Chen, Rational engineering of biomimetic flavylium fluorophores for regulating the lysosomal and mitochondrial localization behavior by pH-induced structure switch and application to fluorescence imaging, J. Mater. Chem. B, 2022, 10(20), 3841–3848, 10.1039/d2tb00181k.
- D. Moreira, O. Regev, N. Basílio and E. F. Marques, Light and pH responsive catanionic vesicles based on a chalcone/flavylium photoswitch for smart drug delivery: from molecular design to the controlled release of doxorubicin, J. Colloid Interface Sci., 2023, 650, 2024–2034, DOI:10.1016/j.jcis.2023.07.129.
- I. Crnolatac, L. Giestas, G. Horvat, A. J. Parola and I. Piantanida, Flavylium Dye as pH-Tunable Fluorescent and CD Probe for Double-Stranded DNA and RNA, Chemosensors, 2020, 8(4), 129, DOI:10.3390/chemosensors8040129.
- T. Pewklang, S. Wet-Osot, S. Wangngae, U. Ngivprom, K. Chansaenpak, C. Duangkamol, R. Y. Lai, P. Noisa, M. Sukwattanasinitt and A. Kamkaew, Flavylium-Based Hypoxia-Responsive Probe for Cancer Cell Imaging, Molecules, 2021, 26(16), 4938, DOI:10.3390/molecules26164938.
- A. S. Pires, K. Droguett Munoz, V. de Freitas, N. Basilio and L. Cruz, Host-Guest Chemosensor Ensembles based on Water-Soluble Sulfonated Calix[n]arenes and a Pyranoflavylium Dye for the Optical Detection of Biogenic Amines, J. Agric. Food Chem., 2024, 72(7), 3719–3729, DOI:10.1021/acs.jafc.3c08695.
- Y. Sun, P. Sun, Z. Li, L. Qu and W. Guo, Natural flavylium-inspired far-red to NIR-II dyes and their applications as fluorescent probes for biomedical sensing, Chem. Soc. Rev., 2022, 51, 7170–7205, 10.1039/d2cs00179a.
- E. D. Cosco, J. R. Caram, O. T. Bruns, D. Franke, R. A. Day, E. P. Farr, M. G. Bawendi and E. M. Sletten, Flavylium Polymethine Fluorophores for Near- and Shortwave Infrared Imaging, Angew. Chem., Int. Ed., 2017, 56(42), 13126–13129, DOI:10.1002/anie.201706974.
- E. D. Cosco, B. A. Arús, A. L. Spearman, T. L. Atallah, I. Lim, O. S. Leland, J. R. Caram, T. S. Bischof, O. T. Bruns and E. M. Sletten, Bright Chromenylium Polymethine Dyes Enable Fast, Four-Color In Vivo Imaging with Shortwave Infrared Detection, J. Am. Chem. Soc., 2021, 143(18), 6836–6846, DOI:10.1021/jacs.0c11599.
- P. Theato, B. S. Sumerlin, R. K. O'Reilly and T. H. Epps, 3rd. Stimuli responsive materials, Chem. Soc. Rev., 2013, 42(17), 7055–7056, 10.1039/c3cs90057f.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Future perspectives and recent advances in stimuli-responsive materials, Prog. Polym. Sci., 2010, 35(1), 278–301, DOI:10.1016/j.progpolymsci.2009.10.008.
- S. Dolui, B. Sahu and S. Banerjee, Stimuli-Responsive Functional Polymeric Materials: Recent Advances and Future Perspectives, Macromol. Chem. Phys., 2025, 2400472, DOI:10.1002/macp.202400472.
- M. I. Gibson and R. K. O'Reilly, To aggregate, or not to aggregate? considerations in the design and application of polymeric thermally-responsive nanoparticles, Chem. Soc. Rev., 2013, 42(17), 7204–7213, 10.1039/c3cs60035a.
- S. Qiao and H. Wang, Temperature-responsive polymers: Synthesis, properties, and biomedical applications, Nano Res., 2018, 11(10), 5400–5423, DOI:10.1007/s12274-018-2121-x.
- G. Kocak, C. Tuncer and V. Bütün, pH.-Responsive polymers, Polym. Chem., 2017, 8(1), 144–176, 10.1039/c6py01872f.
- S. Dai, P. Ravi and K. C. Tam, pH-Responsive polymers: synthesis, properties and applications, Soft Matter, 2008, 4(3), 435–449, 10.1039/b714741d.
- O. Bertrand and J.-F. Gohy, Photo-responsive polymers: synthesis and applications, Polym. Chem., 2017, 8(1), 52–73, 10.1039/c6py01082b.
- C. Boyer and R. Hoogenboom, Multi-responsive polymers, Eur. Polym. J., 2015, 69, 438–440, DOI:10.1016/j.eurpolymj.2015.06.032.
- P. Schattling, F. D. Jochum and P. Theato, Multi-stimuli responsive polymers – the all-in-one talents, Polym. Chem., 2014, 5(1), 25–36, 10.1039/c3py00880k.
- S. Guragain, B. P. Bastakoti, V. Malgras, K. Nakashima and Y. Yamauchi, Multi-Stimuli-Responsive Polymeric Materials, Chem.–Eur. J., 2015, 21(38), 13164–13174, DOI:10.1002/chem.201501101.
- T. Shu, L. Hu, H. Hunter, N. Balasuriya, C. Fang, Q. Zhang and M. J. Serpe, Multi-responsive micro/nanogels for optical sensing, Adv. Phys.:X, 2022, 7(1), 2043185, DOI:10.1080/23746149.2022.2043185.
- J. Udabe, S. Bongiovanni Abel, M. S. Orellano and M. Calderon, Multiresponsive Nanogels for the Selective Delivery of Antimicrobial Drugs to Mucosal Tissues, Biomacromolecules, 2024, 25(9), 5968–5978, DOI:10.1021/acs.biomac.4c00633.
- A. Ali, S. P. Nagumantri, T. Rakshit and S. Pal, Control of Glucose-Induced Degradation and Cargo Release in Multi-Responsive Polymer Hydrogels, Macromol. Chem. Phys., 2021, 222(16), 2100121, DOI:10.1002/macp.202100121.
- L. Chen, G. Yang, Z. Wang, Y. Liu, X. Dou, Y. Li, H. Zhu and X. Yuan, Photothermal-Responsive Sponge with Switchable Wettability for Intelligent and Sustainable Moisture Capture and Release, ACS Appl. Polym. Mater., 2023, 5(9), 7666–7674, DOI:10.1021/acsapm.3c01734.
- J. Xu, M. Chen, M. Li, S. Xu and H. Liu, Integration of chemotherapy and phototherapy based on a pH/ROS/NIR multi-responsive polymer-modified MSN drug delivery system for improved antitumor cells efficacy, Colloids Surf., A, 2023, 663, 131015, DOI:10.1016/j.colsurfa.2023.131015.
- Y. Zhang, J. Li and K. Pu, Recent advances in dual- and multi-responsive nanomedicines for precision cancer therapy, Biomaterials, 2022, 291, 121906, DOI:10.1016/j.biomaterials.2022.121906.
- F. Wang, Z. Wu, Y. Zhang, M. Li, P. Wei, T. Yi and J. Li, Semiconducting polymer nanoprodrugs enable tumor-specific therapy via sono-activatable ferroptosis, Biomaterials, 2025, 312, 122722, DOI:10.1016/j.biomaterials.2024.122722.
- N. Jordão, R. Gavara and A. J. Parola, Flavylium-Supported Poly(N-isopropylacrylamide): A Class of Multistimuli Responsive Polymer, Macromolecules, 2013, 46(22), 9055–9063, DOI:10.1021/ma401497q.
- Y. Liu, P. W. McDonald, S. H. Thang and C. Ritchie, Flavylium-Containing Stimuli-Responsive RAFT Polymers: Synthesis and Enhanced Stability, Macromolecules, 2024, 57(7), 3451–3461, DOI:10.1021/acs.macromol.3c02358.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad and G. Moad, et al., Living free-radical polymerization by reversible addition - Fragmentation chain transfer: The RAFT process, Macromolecules, 1998, 31(16), 5559–5562, DOI:10.1021/ma9804951.
- G. Moad, E. Rizzardo and S. H. Thang, Living radical polymerization by the RAFT process, Aust. J. Chem., 2005, 58(6), 379–410, DOI:10.1071/CH05072.
- G. Moad, E. Rizzardo and S. H. Thang, Living radical polymerization by the RAFT process–a third update, Aust. J. Chem., 2012, 65(8), 985–1076, DOI:10.1071/CH12295.
- F. Küster and A. Thiel, Tabelle Per Le Analisi Chimiche e Chimico-Fisiche, 12th edn, Milano, 1982, pp. 157–160 Search PubMed.
- J. Avó, V. Petrov, N. Basílio, A. Jorge Parola and F. Pina, Evidence against the Twisted Intramolecular Charge Transfer (TICT) model in 7-aminoflavylium derivatives, Dyes Pigm., 2016, 135, 86–93, DOI:10.1016/j.dyepig.2016.06.007.
- V. Petrov, R. Gomes, A. J. Parola and F. Pina, Flash photolysis and stopped flow studies of the 2′-methoxyflavylium network in aq. acidic and alkaline solution, Dyes Pigm., 2009, 80(1), 149–155, DOI:10.1016/j.dyepig.2008.06.005.
- P. Araujo, J. Oliveira, N. Basilio, A. J. Parola, V. de Freitas and F. Pina, Modulating the thermodynamics, kinetics and photochemistry of 7-diethylamino-4'-dimethylaminoflavylium in water/ethanol, SDS and CTAB micelles, Phys. Chem. Chem. Phys., 2022, 24(29), 17593–17604, 10.1039/d2cp01966c.
- J. Mendoza, F. Pina, N. Basílio, M. Guimarães, V. de Freitas and L. Cruz, Extending the stability of red and blue colors of malvidin-3-glucoside-lipophilic derivatives in the presence of SDS micelles, Dyes Pigm., 2018, 151, 321–326, DOI:10.1016/j.dyepig.2018.01.007.
- P. Ferreira da Silva, J. C. Lima, F. H. Quina and A. L. Maçanita, Excited-State Electron Transfer in Anthocyanins and Related Flavylium Salts, J. Phys. Chem. A, 2004, 108(46), 10133–10140, DOI:10.1021/jp047300d.
- Chapter 5 - Polyphenols. in Enological Chemistry, ed. Moreno, J. and Peinado, R. edn. Academic Press, 2012, pp. 53–76 Search PubMed.
- P. Ferreira da Silva, J. C. Lima, A. A. Freitas, K. Shimizu, A. L. Maçanita and F. H. Quina, Charge-Transfer Complexation as a General Phenomenon in the Copigmentation of Anthocyanins, J. Phys. Chem. A, 2005, 109(32), 7329–7338, DOI:10.1021/jp052106s.
- R. F. Rodrigues, P. Ferreira da Silva, K. Shimizu, A. A. Freitas, S. A. Kovalenko, N. P. Ernsting, F. H. Quina and A. Maçanita, Ultrafast Internal Conversion in a Model Anthocyanin–Polyphenol Complex: Implications for the Biological Role of Anthocyanins in Vegetative Tissues of Plants, Chem.–Eur. J., 2009, 15(6), 1397–1402, DOI:10.1002/chem.200801207.
- V. O. Silva, A. A. Freitas, A. L. Maçanita and F. H. Quina, Chemistry and photochemistry of natural plant pigments: the anthocyanins, J. Phys. Org. Chem., 2016, 29(11), 594–599, DOI:10.1002/poc.3534.
- P. Araújo, A. Borges, J. Oliveira, A. Seco, M. Outis, J. C. Lima, N. Basílio, V. de Freitas and F. Pina, The impressive colors of the bis-aminostyrylbenzopyrylium system. Evidence by stopped-flow for the B4 formation at pH>13, Dyes Pigm., 2024, 229, 112280, DOI:10.1016/j.dyepig.2024.112280.
- S. Gago, V. Petrov, A. M. Diniz, A. J. Parola, L. Cunha-Silva and F. Pina, Unidirectional switching between two flavylium reaction networks by the action of alternate stimuli of acid and base, J. Phys. Chem. A, 2012, 116(1), 372–380, DOI:10.1021/jp209913f.
- D. Sousa, N. Basilio, J. Oliveira, V. de Freitas and F. Pina, A New Insight into the Degradation of Anthocyanins: Reversible versus the Irreversible Chemical Processes, J. Agric. Food Chem., 2022, 70(2), 656–668, DOI:10.1021/acs.jafc.1c06521.
- R. Gavara, V. Petrov, V. López and F. Pina, Photochromism of naphthoflavylium. On the role of 4-OH hemiketal in flavylium network, J. Photochem. Photobiol., A, 2011, 220(1), 4–10, DOI:10.1016/j.jphotochem.2011.03.002.
- P. Yang, N. Basílio, X. Liao and F. Pina, Enabling the Photochromism of 4’-Aminoflavylium Compounds in Water by Host-Guest Interaction with β-Cyclodextrin, Dyes Pigm., 2024, 234, 112526, DOI:10.1016/j.dyepig.2024.112526.
- L. Giestas, F. Folgosa, J. C. Lima, A. J. Parola and F. Pina, Bio-Inspired Multistate Networks Responsive to Light, pH and Thermal Inputs – An Example of a Multistate System Operating Through Different Algorithms, Eur. J. Org Chem., 2005, 2005(19), 4187–4200, DOI:10.1002/ejoc.200500318.
- A. Roque, C. Lodeiro, F. Pina, M. Maestri, S. Dumas, P. Passaniti and V. Balzani, Multistate/Multifunctional Systems. A Thermodynamic, Kinetic, and Photochemical Investigation of the 4‘-Dimethylaminoflavylium Compound, J. Am. Chem. Soc., 2003, 125(4), 987–994, DOI:10.1021/ja0287276.
- F. Pina, J. Parola, R. Gomes, M. Maestri and V. Balzani, Multistate/Multifunctional Molecular-Level Systems: Photochromic Flavylium Compounds, Mol. Switches, 2011, 181–226 CAS.
- F. Pina, M. J. Melo, M. Maestri, R. Ballardini and V. Balzani, Photochromism of 4‘-Methoxyflavylium Perchlorate. A “Write−Lock−Read−Unlock−Erase” Molecular Switching System, J. Am. Chem. Soc., 1997, 119(24), 5556–5561, DOI:10.1021/ja9704646.
- A. Zubillaga, P. Ferreira, A. J. Parola, S. Gago and N. Basilio, pH-Gated photoresponsive shuttling in a water-soluble pseudorotaxane, Chem. Commun., 2018, 54(22), 2743–2746, 10.1039/c8cc00688a.
- M. Cors, L. Wiehemeier, J. Oberdisse and T. Hellweg, Deuteration-Induced Volume Phase Transition Temperature Shift of PNIPMAM Microgels, Polymers, 2019, 11(4), 620, DOI:10.3390/polym11040620.
- I. Berndt, J. S. Pedersen and W. Richtering, Temperature-Sensitive Core–Shell Microgel Particles with Dense Shell, Angew. Chem., Int. Ed., 2006, 45(11), 1737–1741, DOI:10.1002/anie.200503888.
- Z. Zhang, S. Maji, A. B. d. F. Antunes, R. De Rycke, Q. Zhang, R. Hoogenboom and B. G. De Geest, Salt Plays a Pivotal Role in the Temperature-Responsive Aggregation and Layer-by-Layer Assembly of Polymer-Decorated Gold Nanoparticles, Chem. Mater., 2013, 25(21), 4297–4303, DOI:10.1021/cm402414u.
- S. Fujishige, K. Kubota and I. Ando, Phase transition of aqueous solutions of poly(N-isopropylacrylamide) and poly(N-isopropylmethacrylamide), J. Phys. Chem., 1989, 93(8), 3311–3313, DOI:10.1021/j100345a085.
- H. Feil, Y. H. Bae, J. Feijen and S. W. Kim, Effect of comonomer hydrophilicity and ionization on the lower critical solution temperature of N-isopropylacrylamide copolymers, Macromolecules, 1993, 26(10), 2496–2500, DOI:10.1021/ma00062a016.
- X. Yin, A. S. Hoffman and P. S. Stayton, Poly(N-isopropylacrylamide-co-propylacrylic acid) Copolymers That Respond Sharply to Temperature and pH, Biomacromolecules, 2006, 7(5), 1381–1385, DOI:10.1021/bm0507812.
- C. K. Wong, A. F. Mason, M. H. Stenzel and P. Thordarson, Formation of non-spherical polymersomes driven by hydrophobic directional aromatic perylene interactions, Nat. Commun., 2017, 8(1), 1240, DOI:10.1038/s41467-017-01372-z.
- P. Pawliszak, J. Zawala, V. Ulaganathan, J. K. Ferri, D. A. Beattie and M. Krasowska, Interfacial characterisation for flotation: 2. Air-water interface, Curr. Opin. Colloid Interface Sci., 2018, 37, 115–127, DOI:10.1016/j.cocis.2018.07.002.
- P. Pawliszak, B. H. Bradshaw-Hajek, C. Greet, W. Skinner, D. A. Beattie and M. Krasowska, Interfacial Tension Sensor for Low Dosage Surfactant Detection, Colloids Interfaces, 2021, 5(1), 9, DOI:10.3390/colloids5010009.
- N. Jain, S. Trabelsi, S. Guillot, D. McLoughlin, D. Langevin, P. Letellier and M. Turmine, Critical Aggregation Concentration in Mixed Solutions of Anionic Polyelectrolytes and Cationic Surfactants, Langmuir, 2004, 20(20), 8496–8503, DOI:10.1021/la0489918.
- R. J. Hunter, Zeta Potential In Colloid Science: Principles And Applications, Academic press, 2013 Search PubMed.
- K. Pate and P. Safier, Chemical metrology methods for CMP quality. in Advances In Chemical Mechanical Planarization (CMP), 2016, pp. 299–325 Search PubMed.
- S. Bhattacharjee, DLS and zeta potential - What they are and what they are not?, J. Controlled Release, 2016, 235, 337–351, DOI:10.1016/j.jconrel.2016.06.017.
- D. Geißler, C. Gollwitzer, A. Sikora, C. Minelli, M. Krumrey and U. Resch-Genger, Effect of fluorescent staining on size measurements of polymeric nanoparticles using DLS and SAXS, Anal. Methods, 2015, 7(23), 9785–9790, 10.1039/c5ay02005k.
- J. Teixeira, Small-angle scattering by fractal systems, J. Appl. Crystallogr., 1988, 21(6), 781–785, DOI:10.1107/S0021889888000263.
- G. Beaucage, Small-Angle Scattering from Polymeric Mass Fractals of Arbitrary Mass-Fractal Dimension, J. Appl. Crystallogr., 1996, 29(2), 134–146, DOI:10.1107/S0021889895011605.
- S. Lazzari, L. Nicoud, B. Jaquet, M. Lattuada and M. Morbidelli, Fractal-like structures in colloid science, Adv. Colloid Interface Sci., 2016, 235, 1–13, DOI:10.1016/j.cis.2016.05.002.
- J. C. Berg, An Introduction to Interfaces and Colloids: The Bridge to Nanoscience, 2010, DOI:10.1142/7579.
- S. Shinkai, T. Nozaki, K. Maeshima and Y. Togashi, Bridging the dynamics and organization of chromatin domains by mathematical modeling, Nucleus, 2017, 8(4), 353–359, DOI:10.1080/19491034.2017.1313937.
- P. J. Lu, J. C. Conrad, H. M. Wyss, A. B. Schofield and D. A. Weitz, Fluids of Clusters in Attractive Colloids, Phys. Rev. Lett., 2006, 96(2), 028306, DOI:10.1103/PhysRevLett.96.028306.
- Y.-C. Li, K.-B. Chen, H.-L. Chen, C.-S. Hsu, C.-S. Tsao, J.-H. Chen and S.-A. Chen, Fractal Aggregates of Conjugated Polymer in Solution State, Langmuir, 2006, 22(26), 11009–11015, DOI:10.1021/la0612769.
- R. Forschner, J. A. Knöller, A. Zens, W. Frey, Y. Molard and S. Laschat, Luminescent liquid crystals: from supramolecular plant dyes to emissive flavylium salts, Liq. Cryst., 2023, 50(7–10), 1310–1323, DOI:10.1080/02678292.2023.2179122.
- R. Forschner, J. Knelles, K. Bader, C. Muller, W. Frey, A. Kohn, Y. Molard, F. Giesselmann and S. Laschat, Flavylium Salts: A Blooming Core for Bioinspired Ionic Liquid Crystals, Chem. - Eur. J., 2019, 25(56), 12966–12980, DOI:10.1002/chem.201901975.
- A. Fernandes, N. F. Brás, N. Mateus and V. d. Freitas, A study of anthocyanin self-association by NMR spectroscopy, New J. Chem., 2015, 39(4), 2602–2611, 10.1039/C4NJ02339K.
- A. Pagan, J. I. Lee and J. Kang, Concentration-Dependent Association of Flavylium Chloride with Differential Hydroxy Moieties in Ethanol, Colorants, 2021, 1(1), 20–37, DOI:10.3390/colorants1010004.
- T. Hoshino, Anthocyanin self-aggregates. Part 7. Self-association of flavylium cations of anthocyanidin 3,5-diglucosides studied by circular dichroism and proton NMR, Phytochem, 1992, 31(2), 647–653 CrossRef CAS.
- P. Trouillas, J. C. Sancho-Garcia, V. De Freitas, J. Gierschner, M. Otyepka and O. Dangles, Stabilizing and Modulating Color by Copigmentation: Insights from Theory and Experiment, Chem. Rev., 2016, 116(9), 4937–4982, DOI:10.1021/acs.chemrev.5b00507.
- P. Yang, N. Basilio, X. Liao, Z. Xu, O. Dangles and F. Pina, Influence of Acylation by Hydroxycinnamic Acids on the Reversible and Irreversible Processes of Anthocyanins in Acidic to Basic Aqueous Solution, J. Agric. Food Chem., 2024, 72(7), 3719–3729, DOI:10.1021/acs.jafc.4c06847.
- C. K. Wong, A. D. Martin, M. Floetenmeyer, R. G. Parton, M. H. Stenzel and P. Thordarson, Faceted polymersomes: a sphere-to-polyhedron shape transformation, Chem. Sci., 2019, 10(9), 2725–2731, 10.1039/c8sc04206c.
- S. Berruee, J. M. Guigner, T. Bizien, L. Bouteiller, L. Sosa Vargas and J. Rieger, Spontaneous formation of polymeric nanoribbons in water driven by pi-pi interactions, Angew. Chem., Int. Ed., 2024, e202413627, DOI:10.1002/anie.202413627.
- J. Zhang, P. Song, Z. Zhu, Y. Li, G. Liu, M. J. Henderson, J. Li, W. Wang, Q. Tian and N. Li, Evaporation-induced self-assembly of Janus pyramid molecules from fractal network to core-shell nanoclusters evidenced by small-angle X-ray scattering, J. Colloid Interface Sci., 2024, 674, 437–444, DOI:10.1016/j.jcis.2024.06.156.
- S. Shin, M. L. Gu, C. Y. Yu, J. Jeon, E. Lee and T. L. Choi, Polymer Self-Assembly into Unique Fractal Nanostructures in Solution by a One-Shot Synthetic Procedure, J. Am. Chem. Soc., 2018, 140(1), 475–482, DOI:10.1021/jacs.7b11630.
- E. Blasco, M. B. Sims, A. S. Goldmann, B. S. Sumerlin and C. Barner-Kowollik, 50th Anniversary Perspective: Polymer Functionalization, Macromolecules, 2017, 50(14), 5215–5252, DOI:10.1021/acs.macromol.7b00465.
- D. Xu, M. Wang, R. Huang, J. F. Stoddart and Y. Wang, A Dual-Pathway Responsive Mechanophore for Intelligent Luminescent Polymer Materials, J. Am. Chem. Soc., 2025, 147(5), 4450–4458, DOI:10.1021/jacs.4c15655.
- P. F. da Silva, L. Paulo, A. Barbafina, F. Eisei, F. H. Quina and A. L. Macanita, Photoprotection and the photophysics of acylated anthocyanins, Chem.–Eur. J., 2012, 18(12), 3736–3744, DOI:10.1002/chem.201102247.
- M. Nagao, K. Mundsinger and C. Barner-Kowollik, Photoinduced Energy/Electron Transfer within Single-Chain Nanoparticles, Angew. Chem., Int. Ed., 2025, e202419205, DOI:10.1002/anie.202419205.
- F. H. Quina and E. L. Bastos, Chemistry Inspired by the Colors of Fruits, Flowers and Wine, An. Acad. Bras. Cienc., 2018, 90(1), 681–695, DOI:10.1590/0001-3765201820170492.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc00977d |
| This journal is © The Royal Society of Chemistry 2025 |