
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA nearly perfect icosahedral Ir@Au12 superatom with superior photoluminescence obtained by ligand engineering†
Katsuya
Mutoh
 *a,
Teppei
Yahagi
a,
Shinjiro
Takano
b,
Sonomi
Kawakita
a,
Takeshi
Iwasa
cd,
Tetsuya
Taketsugu
cd,
Tatsuya
Tsukuda
*b and
Takuya
Nakashima
*a
*a,
Teppei
Yahagi
a,
Shinjiro
Takano
b,
Sonomi
Kawakita
a,
Takeshi
Iwasa
cd,
Tetsuya
Taketsugu
cd,
Tatsuya
Tsukuda
*b and
Takuya
Nakashima
*a
aDepartment of Chemistry, Graduate School of Science, Osaka Metropolitan University, Sumiyoshi-ku, Osaka 558-8585, Japan. E-mail: mutoh@omu.ac.jp; takuya.nakashima@omu.ac.jp
bDepartment of Chemistry, Graduate School of Science, The University of Tokyo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: tsukuda@chem.s.u-tokyo.ac.jp
cDepartment of Chemistry, Faculty of Science, Hokkaido University, North 10 West 5, Sapporo, Hokkaido 060-0810, Japan
dWPI-ICReDD, Hokkaido University, Sapporo, Hokkaido 060-0810, Japan
First published on 28th March 2025
Abstract
Heterometal doping and the introduction of surface ligands drastically alter the optical and photophysical properties of gold-based superatoms by modulating their electronic structures and the excited state dynamics. In this study, we investigate how the structures and the optical properties of an Ir@Au12 superatom capped by a diphosphine ligand, bis[benzo[b]phosphindole]ethane (bbpe), in which the rotation of the phenyl groups is prohibited, differ from those capped by the conventional diphosphine ligands, such as 1,2-bis(diphenylphosphino)ethane (dppe) and bis(diphenylphosphino)methane (dppm). The co-reduction of Ir(III)- and Au(I)-precursors under mild reaction conditions yielded homoleptically capped [IrAu12(bbpe)6]3+ clusters (IrAu12-b) as the primary product. Single crystal X-ray diffraction analysis of IrAu12-b revealed the formation of a nearly perfect icosahedral Ir@Au12 superatomic core, in which the central Ir atom is equidistant from each vertex Au atom. The energy gap between occupied 1P and unoccupied 1D superatomic orbitals of IrAu12-b was larger than that of its dppm-capped counterpart, [IrAu12(dppm)6]3+ as evidenced by a blue shift (140 nm) of the photoluminescence (PL) wavelength and DFT calculations. IrAu12-b exhibited PL at 596 nm with a high quantum yield of 87% in deaerated CH2Cl2 due to the expanded 1P–1D energy gap and the restricted molecular motions of the bbpe ligands.
Introduction
Atomically precise metal clusters possess size-specific electronic structures characterized by discrete energy levels, just like atoms and molecules. As a result, they exhibit novel physicochemical properties that differ from their bulk and nanoparticle counterparts and have attracted attention not only for fundamental research but also for various applications.1–12 For example, gold clusters with clear energy gaps between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) have been demonstrated to exhibit attractive photofunctions, including photoluminescence (PL), circularly polarized luminescence (CPL), triplet sensitization, and photocatalysis.13–20 The widely reported icosahedral Au13 cluster has been considered a representative superatom.21–25 Direct stabilization of the Au13 superatom with phosphines and N-heterocyclic carbenes (NHCs), without forming Au(I)-ligand staple motifs, often leads to pronounced PL in the red to near-infrared region.21,26–30 The heteroatom doping further perturbs the superatomic orbitals, resulting in the drastic modulation of the HOMO–LUMO energy gaps (HL gaps).30–38 The diphosphine-protected Au13 clusters such as [Au13(dppm)6]5+ (dppm: bis(diphenylphosphino)-methane) and [Au13(dppe)5Cl2]3+ (dppe: 1,2-bis(diphenyl-phosphino)ethane) serve as a platform for the heteroatom doping.33–40 The latter has been further modified by the exchange reaction of the Cl ligands with other halogens, alkynyl and subsequently isocyanide ligands.41–43The ligand shell is not only involved in controlling the geometry of the superatomic cores, but also affects their electronic structures and photophysical processes.44–47 More specifically, the interactions of the ligand with the superatomic core and the adjacent ligands are crucial for controlling the HL gap and the excited state dynamics of the cores. For example, Lu et al. reported a ligand-induced contraction of the Au13 core leading to an expansion of the HL gap and a significant increase in the PL quantum yield (QY).48 The use of N,N′-dibenzyl-substituted benzimidazolium NHC ligands rigidifies the Au13 superatom through multiple CH–π and π–π interactions in the ligand layer, also resulting in high PLQYs.45,49 In contrast to the recent successful applications of various NHC ligands,50–53 less systematic studies have been conducted for the diphosphine molecules as capping ligands for the Au13 superatoms.
Bis[benzo[b]phosphindole]ethane (bbpe) is a derivative of dppe in which the two phenyl rings are fused by a chemical bond (Fig. 1).45 Since the benzo[b]phosphindole (bp) framework of bbpe rigidly links the phenyl rings into a planar structure, the degree of freedom of the aromatic units is significantly restricted. This simple chemical modification suppresses the vibrational and rotational modes that can have a significant impact on the relaxation process of the photo-excited state of the Au13 superatoms. Motivated by our recent development of highly luminescent [IrAu12(dppe)5Cl2]+ cluster (IrAu12-e-Cl),35 in this study, we used bbpe as a ligand to stabilize Ir@Au12 superatoms with the intention of further enhancing their PL property (Fig. 1). The cluster synthesis with bbpe unexpectedly yielded a Cl-free cluster [IrAu12(bbpe)6]3+ (IrAu12-b) as the main product. The homoleptic capping of the Ir@Au12 core with six bbpe ligands resulted in an almost perfect icosahedral structure. Due to the expanded HL gap as well as the suppressed non-radiative relaxation pathways of the excited state, IrAu12-b exhibited bright PL at 596 nm with PLQY as high as 0.87. The remarkable roles of the bbpe ligands on the PL properties of IrAu12-b are discussed by comparing the geometry and optical properties of [IrAu12(dppm)6]3+ (IrAu12-m) as a reference (Fig. 1).
 | ||
| Fig. 1 The molecular structures of the diphosphine ligands and the corresponding Ir@Au12 clusters for (a) [IrAu12(dppe)5Cl2]+ (IrAu12-e-Cl), (b) [IrAu12(dppm)6]3+ (IrAu12-m) and (c) [IrAu12(bbpe)6]3+ (IrAu12-b). | ||
Results and discussion
Synthesis and structure analysis
The detailed synthetic procedure of IrAu12-b is given in the ESI.† The bbpe ligand and the corresponding gold complex, Au2(bbpe)Cl2, were synthesized according to the literature procedure.54 The mixture of Au2(bbpe)Cl2 and [Ir(COD)Cl]2 (COD: 1,5-cyclooctadiene) was co-reduced in CH2Cl2. While a typical reduction condition using NaBH4 at room temperature did not produce any luminescent species, the milder condition using borane-tert-butylamine complex26,55,56 at 273 K yielded a luminescent solution. The larger agglomerates in the as-synthesized solution were removed by a reverse-phase column chromatography (Wakosil 100C18, Wako-Fuji Film) using a methanol solution containing 0.1 vol% trifluoroacetic acid (TFA) and 0.1 vol% diethylamine (DEA) as an eluent.57 The pretreated sample was then analyzed by HPLC using a C30 reverse-phase column (GL Sciences Inc.) as shown in Fig. S1a.† Five well-separated peaks were observed, and three fractions with retention times of 11, 25, and 29 min (peaks 1, 3, and 4 in Fig. S1a,† respectively) contained luminescent species. Electrospray ionization (ESI) mass spectrometry of the HPLC fractions (Fig. S1b†) showed that the major fraction (peak 1 in Fig. S1a†) contained the target cluster (IrAu12-b) and that another minor fraction (peak 4 in Fig. S1†) was contaminated by other species such as [IrAu12(bbpe)5Cl2]+ (Fig. S2†). The PL maxima for peaks 1, 3, and 4 were 598, 632, and 642 nm, respectively (Fig. S1c†). It should be noted that gold clusters with visible PL could not be obtained without [Ir(COD)Cl]2 in the synthesis.Since IrAu12-b showed the brightest PL among the species obtained, the main fraction (peak 1) was subjected to isolation by a preparative HPLC cycle, followed by recrystallization after treating the solution with NaPF6. The geometrical structure of IrAu12-b was determined by single crystal X-ray diffraction (SCXRD) analysis (Fig. 2a). The Ir@Au12 superatomic core was protected by six bbpe ligands with a coordination pattern, similar to that of IrAu12-m.34 However, the superatomic core of IrAu12-b has a higher symmetry than that of IrAu12-m. A singlet peak in the 31P{1H} NMR spectrum of IrAu12-b indicates that Ir atom is located at the center of the Ir@Au12 core (Fig. S3†).35 The symmetric coordination of bbpe ligands to the Ir@Au12 core of IrAu12-b was also suggested by a simple peak pattern in the 1H NMR spectrum (Fig. S4†). The bond lengths in the Ir@Au12 core were compared between IrAu12-b and IrAu12-m (Fig. 2b, c and Table 1). Surprisingly, all the Ir–Au distances in IrAu12-b are identical (2.7397 ± 0.0003 Å), and the distribution of the Au–Au distances is remarkably narrower than that of IrAu12-m. Since the average Ir–Au and Au–Au bond lengths of IrAu12-b and IrAu12-m are very similar (Table 1), the narrower bond length distributions for IrAu12-b indicate the formation of a higher symmetric icosahedron of Ir@Au12 by the protection with bbpe. The formation of a nearly ideal icosahedral Ir@Au12 core in IrAu12-b was also confirmed by a much smaller continuous symmetry measure (CSM) value than that of IrAu12-m (Table 1). To the best of our knowledge, the CSM value of 0.014 is comparable to the smallest one (0.013) for the Pd@Au12 core of [PdAu12(dppe)5(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)2]2+ (PdAu12-e-PA)43 among the series of M@Au12 superatoms reported to date (Table S1†). Nonetheless, the dopant Pd atom is not situated at the very center of icosahedron in PdAu12-e-PA (Pd–Au: 2.731 ± 0.020 Å).43 The ethylene bridge between the coordinative phosphorous atoms in bbpe may relax the structural strain observed in IrAu12-m where the less flexible methylene bridge imposes constraints incompatible with the surface Au–Au bonds in the Ir@Au12 core. In the case of bbpe, the more rigid bp unit may affect the ethylene bride conformation, which further influences the distance and orientation of phosphorous atoms on the surface of the Ir@Au12 core. The coordination of six bbpe forms a cuboidal (hexahedral) cage surrounding the icosahedral core (Fig. 2a).
CPh)2]2+ (PdAu12-e-PA)43 among the series of M@Au12 superatoms reported to date (Table S1†). Nonetheless, the dopant Pd atom is not situated at the very center of icosahedron in PdAu12-e-PA (Pd–Au: 2.731 ± 0.020 Å).43 The ethylene bridge between the coordinative phosphorous atoms in bbpe may relax the structural strain observed in IrAu12-m where the less flexible methylene bridge imposes constraints incompatible with the surface Au–Au bonds in the Ir@Au12 core. In the case of bbpe, the more rigid bp unit may affect the ethylene bride conformation, which further influences the distance and orientation of phosphorous atoms on the surface of the Ir@Au12 core. The coordination of six bbpe forms a cuboidal (hexahedral) cage surrounding the icosahedral core (Fig. 2a).
 | ||
| Fig. 2 (a) The structure of IrAu12-b determined by SCXRD. (b and c) Bond length distributions in (b) IrAu12-b and (c) IrAu12-m. Color code: yellow, Au; dark blue, Ir; orange, P; gray, C. | ||
| Ir–Au (Å) | Au–Au (Å) | Au–P (Å) | CSM | |
|---|---|---|---|---|
| IrAu12-b | 2.740 ± 0.000 | 2.881 ± 0.028 | 2.285 ± 0.002 | 0.014 |
| IrAu12-m (ref. 40) | 2.741 ± 0.014 | 2.883 ± 0.072 | 2.301 ± 0.013 | 0.091 |
Optical properties
The ligand effect on the electronic structure of the Ir@Au12 core was investigated by UV-vis absorption and luminescence spectroscopy. Fig. 3 shows the absorption, PL, and excitation spectra of IrAu12-b and IrAu12-m in CH2Cl2. The absorption onset and PL peak (λmax) of IrAu12-b were blue-shifted by ∼100 and ∼139 nm, respectively, from those of IrAu12-m.34 These results indicate that the HL gap of IrAu12-b is larger than that of IrAu12-m. The PL was assigned to the excited triplet state-based phosphorescence since the PL intensity decreases in the presence of molecular oxygen. Notably, the slight modification of the ligand structure from dppm to bbpe drastically enhanced the PL efficiency. The PLQY of IrAu12-b was 0.870 in deaerated CH2Cl2 determined by the absolute method (JASCO, ILF-135), while that of IrAu12-m was reported to be 0.14.40 This PLQY is among the highest reported for the icosahedral M@Au12 clusters.30 The PLQY did not show a clear dependence on the solvent polarity (Table S2 and Fig. S5†). The PL lifetime of IrAu12-b in CH2Cl2 was determined to be 4.18 μs (Table 2 and Fig. S6†), which is slightly longer than that of IrAu12-m (3.6 μs).40 The rate constants for the PL (kr) and the nonradiative process (knr) of IrAu12-b were calculated to be 2.1 × 105 and 3.1 × 104 s−1, respectively, by assuming that the intersystem crossing (ISC) efficiency is close to unity for a simpler estimation.40,58 | ||
| Fig. 3 UV-vis absorption, photoluminescence and excitation spectra of (a) IrAu12-b and (b) IrAu12-m in CH2Cl2. | ||
| λ max (nm) | τ (μs) | PLQY | k r (s−1) | k nr (s−1) | |
|---|---|---|---|---|---|
| IrAu12-b | 596 | 4.18 | 0.870 | 2.1 × 105 | 3.1 × 104 |
| IrAu12-m (ref. 40) | 736 | 3.6 | 0.135 | 3.7 × 104 | 2.4 × 105 |
The non-radiative relaxation pathway of the excited state of IrAu12-b is significantly suppressed compared to that of IrAu12-m due to the rigid molecular structure of the bbpe ligand. The enlarged PL energy also contributes to the increase in kr. The highly luminescent property of IrAu12-b was achieved by both (1) accelerating the radiative process and (2) suppressing the non-radiative deactivation process. First, the acceleration of the radiative process is ascribed to the blue shift of the PL maximum, since the phosphorescence rate constant (kp) is approximately proportional to the square of the radiation energy 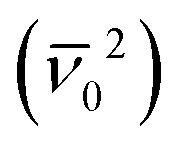 according to the relationship of
according to the relationship of  where fT is the transition probability from T1 to S0. The fT value depends on the transition probability from S0 to S1, the magnitude of spin–orbit coupling constant, and the S1–T1 splitting.59,60 The absorption probabilities for IrAu12-b and IrAu12-m were comparable, due to the similar absorbance (Fig. S7†). The fT value for IrAu12-b should be increased due to the negligible S1–T1 splitting. The more symmetrical Ir@Au12 core of IrAu12-b likely enhances the orthogonality between the 1D to 1P superatomic orbitals, thereby strengthening the spin–orbit coupling.61 The smaller S1–T1 splitting and the strengthened spin–orbit coupling thus contribute to the moderate increase in the kr value.
where fT is the transition probability from T1 to S0. The fT value depends on the transition probability from S0 to S1, the magnitude of spin–orbit coupling constant, and the S1–T1 splitting.59,60 The absorption probabilities for IrAu12-b and IrAu12-m were comparable, due to the similar absorbance (Fig. S7†). The fT value for IrAu12-b should be increased due to the negligible S1–T1 splitting. The more symmetrical Ir@Au12 core of IrAu12-b likely enhances the orthogonality between the 1D to 1P superatomic orbitals, thereby strengthening the spin–orbit coupling.61 The smaller S1–T1 splitting and the strengthened spin–orbit coupling thus contribute to the moderate increase in the kr value.
Second, the knr value of IrAu12-b was drastically reduced, although the spin–orbit coupling also promotes the non-radiative deactivation.60 Considering the independence of PLQY on the solvent polarity (Table S1†), the electrostatic interaction between the cationic Ir@Au12 core and the counter anion (PF6−) has little effect on the PLQY. Therefore, specific interactions between the ligands are expected. Fig. 4 shows the orientation of the aromatic rings in the ligand layers for IrAu12-b and IrAu12-m. The bbpe ligands are oriented so that each end faces the benzene ring of the adjacent one (Fig. 4a). The distances between the hydrogen atoms of a bbpe ligand and an aromatic ring of the adjacent one were estimated to be less than 3.1 Å, which is within the typical distance (2.7–3.2 Å) for the CH–π interaction of the edge-to-face aromatic conformation.62 The restricted rotation of phenyl rings fixes the orientation of the bp units, which is advantageous for the bp ring to serve as both a CH donor and π acceptor. As a result, all the bp rings participate in the CH–π interaction network found in the outermost of IrAu12-b. Three bp units form a triangular CH–π interaction network in a complementary manner, and eight stable triangles are formed at all the corners of the cuboidal cluster. The formation of the CH–π interaction network was also confirmed in the solution state by the 1H NMR spectral change. Two broad peaks at 6.67 and 7.11 ppm could be assigned to the protons at the 3- and 4-positions of bp-units in IrAu12-b (Fig. S4†). Those peaks were shifted upfield by more than 0.8 ppm relative to corresponding protons in the Au2(bbpe)Cl2 precursor due to the ring current effect by the bp unit of the adjacent ligand whereas other two peaks remain unchanged. Thus, the packing of bbpe ligands is solidified, which may also rigidify the central Ir@Au12 core, contributing to the suppression of non-radiative pathways and excited state structural relaxations, and the similar geometries of S1 and T1 states. On the other hand, such regular network interactions are absent in IrAu12-m. While both CH–π and π–π interactions between densely packed phenyl groups are occasionally found, the avoidance of steric repulsions between phenyl rings with their rotational freedom seems to be prioritized.
 | ||
| Fig. 4 Configuration of aromatic rings on the surface of (a) IrAu12-b and (b) IrAu12-m. | ||
Electrochemistry and electronic structures
The electrochemical measurement was carried out to experimentally determine the energy levels of HOMO and LUMO. Differential pulse voltammetry (DPV) clarified the first reduction (R1) and oxidation (O1) potentials (vs. Fc/Fc+, Fc: ferrocene) for IrAu12-b as −1.88 V and 0.76 V, and for IrAu12-m as −1.94 V and 0.34 V, respectively (Fig. S8†). Although doping with a low-valent metal atom has been shown to decrease the O1 potential of MAu12 clusters,35,40 the O1 potential of 0.76 V for IrAu12-b is higher than those (0.4 and 0.74 V vs. Fc/Fc+, respectively) of undoped [Au13(dppm)6]5+ and [Au13(dppe)5Cl2]3+.35,39 The electrochemical HL gaps (HLe) for IrAu12-b and IrAu12-m were estimated to be 2.29 and 2.05 eV, by considering the charging energies (0.35 and 0.24 eV, respectively) estimated from the potential gaps between O1 and O2.63 The HLe values are consistent with the optical gap (HLo) as shown in Fig. S8 and S9.† Thus, the large increase in the oxidation potential for IrAu12-b results in the larger HL gap (2.29 eV). While the HLo and PL energies are comparable with those of highly luminescent IrAu12-e-Cl, the much larger O1 potential makes IrAu12-b more tolerant toward the chemical oxidation of the Ir@Au12 core.35The lowest singlet excited state (S1) of most gold clusters undergoes rapid intersystem crossing to excited triplet states (Tn).61 The energy differences between the S1 and T1 states (ΔEST) of IrAu12-m and IrAu12-Cl-e have been reported to be smaller than those of the undoped counterparts, probably because of the small structural difference between the S1 and T1 states34,35 and the small exchange integral due to the presence of the heteroatom at the center of the superatom.61 The ΔEST values estimated by DFT calculations for IrAu12-b and IrAu12-m were 0.007 and 0.08 eV, respectively (Fig. S10†). The smaller ΔEST value of IrAu12-b should enhance the rate of the intersystem crossing,35 further supporting our assumption of the quantitative ISC efficiency. The calculated T1–S0 gaps of IrAu12-b and IrAu12-m were 2.11 and 1.54 eV, respectively, which well reproduced the experimental PL energies (2.08 and 1.68 eV, respectively). The large PL blue-shift of IrAu12-b is thus considered to originate from the expanded HL gap. The HOMO and LUMO of IrAu12-b can be assigned to the superatomic 1P and 1D orbitals, respectively (Fig. 5 and S11†). The electronic structures of the gold clusters have been qualitatively explained by a jellium model in which the energy levels of the superatomic orbitals vary depending on the potential width and depth formed by the metal core.64 Fig. S12† shows the DFT-optimized structures and the natural charges of IrAu12-b and IrAu12-m. The optimized structure of IrAu12-b is highly symmetric (T-symmetry), while the symmetry of IrAu12-m is reduced to S6-symmetry by Jahn–Teller distortion. The natural charges for whole Ir@Au12 cores of IrAu12-b and IrAu12-m were estimated to be +0.53 and +0.57, respectively (Fig. S12†). The similar natural charges between the cores of IrAu12-b and IrAu12-m suggest the similar depth of the jellium potentials, which, in turn, cannot explain the difference in their HOMO levels.
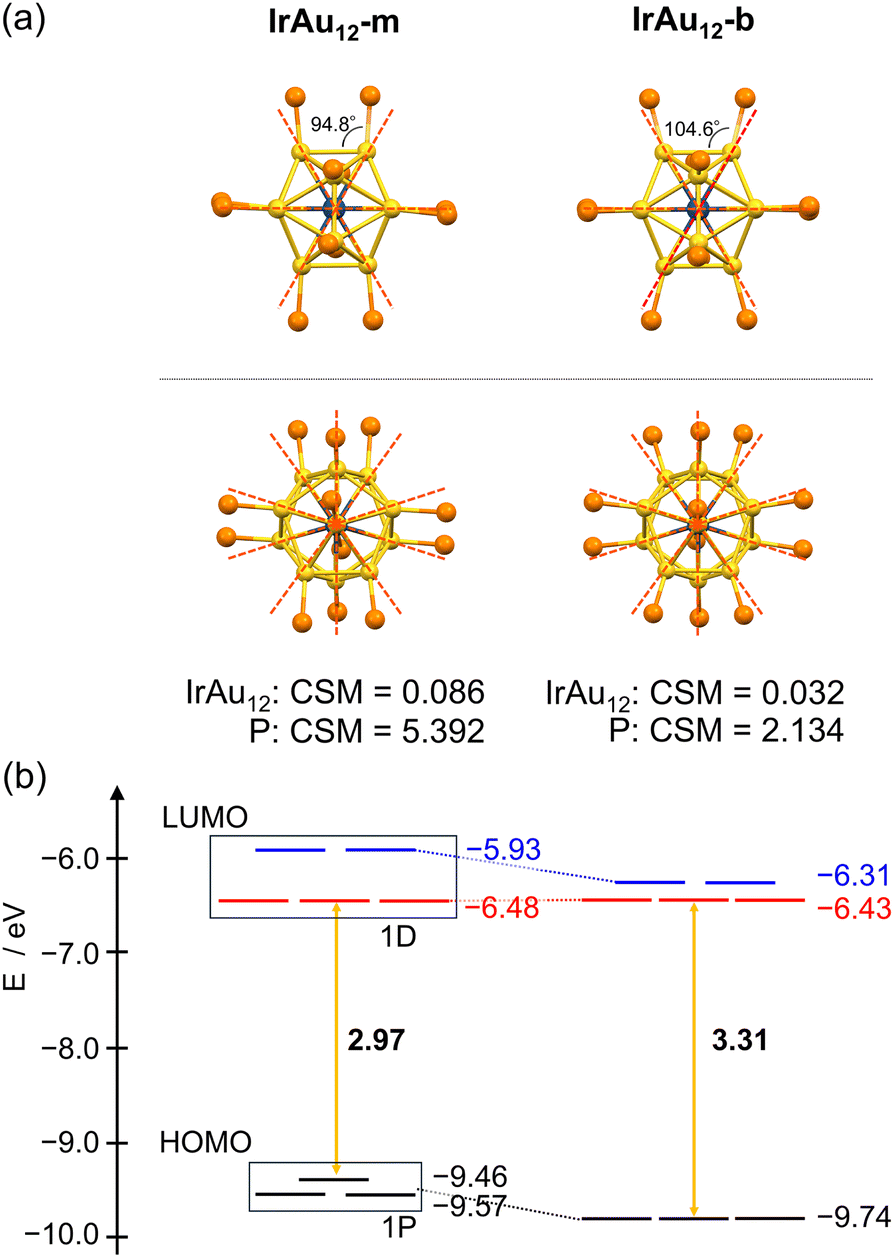 | ||
| Fig. 5 (a) The positions of Au, Ir, and P atoms in the optimized structures of IrAu12-m (left) and IrAu12-b (right). The top and bottom panels show the view from an edge and a vertex direction of icosahedron, respectively. Color code: yellow, Au; dark blue, Ir; orange, P. (b) Energy levels of the Kohn–Sham orbitals of IrAu12-m and IrAu12-b estimated by DFT calculation. | ||
The 1P and 1D superatomic orbitals of IrAu12-b are more degenerated compared to those of IrAu12-m (Fig. 5b). This trend can be explained by the higher symmetry of the Ir@Au12 core of IrAu12-b (CSM = 0.032 for the optimized structure) and the higher symmetry of the phosphine coordination in IrAu12-b, as described below. Five occupied orbitals below the 1P orbitals have the distinct contribution of the Ir 5d orbitals in a similar manner to the electronic structure of IrAu12-Cl-e.33Fig. 5a shows the positions of Ir, Au, and P atoms in the DFT-optimized structures of IrAu12-m and IrAu12-b. The superimposed orange dotted lines are the extension of the Ir–Au radial bonds and can be used as a measure to estimate the symmetry of the phosphine ligation. Clearly, the ligation symmetry in IrAu12-b is higher than that in IrAu12-m, since the P atoms of the former are located closer to the guide lines than those of the latter. Indeed, the Au–Au–P angle in IrAu12-b was 104.6°, close to that expected for a perfect Ih coordination (120°), while that in IrAu12-m was 94.8° (Fig. 5a). In addition, the CSM value for the P12 cage of IrAu12-b (2.134) was smaller than that of IrAu12-m (5.392), supporting that the phosphine ligands in IrAu12-b coordinate more symmetrically to the Ir@Au12 core. As a result, the ligand field in IrAu12-b is more spherical compared to IrAu12-m, leading to the less energy splitting of the occupied 1P and unoccupied 1D superatomic orbitals (Fig. 5b).48,65–67 Although further theoretical and experimental studies are needed to explain the change in the energy levels of the superatomic orbitals, we suggest that the higher Ih symmetry of the Ir@Au12 core and the more uniform surface ligation contribute to the larger HL gap for IrAu12-b.
Conclusions
We used bis[benzo[b]phosphindole]ethane (bbpe) with a rigid molecular framework to stabilize the Ir@Au12 superatom. The use of bbpe ligands resulted in a hierarchically symmetric cluster: a nearly perfect Ir@Au12 core is capped by eight units of three bbpe ligands, circularly linked by CH–π interactions, arranged at the vertices of a cuboid. We observed interesting PL properties, including a relatively large PL energy (ca. 2.1 eV) and large PLQY (0.87), due to the symmetric structure with the rigidified core. These results demonstrate that a slight modification of the ligand structure results in a remarkable improvement of the PL properties of gold superatoms. The [IrAu12(bbpe)6]3+ cluster with high T1 energy together with the large oxidation potential, i.e. deep HOMO level, will find application as a photoredox catalyst.35,37Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
K. Mutoh: conceptualization, data curation, validation, visualization, funding acquisition, writing–original draft, writing–review & editing. T. Yahagi & S. Kawakita: investigation, data curation. S. Takano: data curation, validation, methodology, writing–review & editing. T. Iwasa & T. Taketsugu: data curation, validation, writing–review & editing. T. Tsukuda: funding acquisition, data curation, methodology, resources, writing–review & editing. T. Nakashima: conceptualization, data curation, funding acquisition, resources, supervision, writing–review & editing. Each author contributed substantially to the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JST CREST (Grant Number: JPMJCR20B2), JSPS KAKENHI grant numbers JP22H05134 (TN) for Transformative Research (A) “Revolution of Chiral Materials Science using Helical Light”, JP23H01938 (TN) and JP24K01458 (KM) for Scientific Research (B), and by Tokyo Ohka Foundation for The Promotion of Science and Technology and Kansai Research Foundation for Technology Promotion (KM). A part of this work was conducted in Institute for Molecular Science, supported by “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” of the Ministry of Education, Culture, Sports, Science and Technology (MEXT). Proposal Number JPMXP1224MS0001. A part of calculations was performed using the Research Center for Computational Science, Okazaki, Japan.Notes and references
- M. Zhou, K. Li, Y. Pei, S. Jin and M. Zhu, J. Phys. Chem. Lett., 2018, 14, 11715–11724 CrossRef PubMed.
- Q. Tang, G. Hu, V. Fung and D. Jiang, Acc. Chem. Res., 2018, 51, 2793–2802 CrossRef CAS.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS.
- Y. Li and R. Jin, J. Phys. Chem. C, 2021, 125, 15773–15784 CrossRef CAS.
- Y. Saito, C. Murata, M. Sugiuchi, Y. Shichibu and K. Konishi, Coord. Chem. Rev., 2022, 470, 214713 CrossRef CAS.
- X. Zou, X. Kang and M. Zhu, Chem. Soc. Rev., 2023, 52, 5892–5967 RSC.
- J. Chen, P. Gu, G. Ran, Y. Zhang, M. Li, B. Chen, H. Lu, Y.-Z. Han, W. Zhang, Z. Tang, Q. Yan, R. Sun, X. Fu, G. Chen, Z. Shi, S. Wang, X. Liu, J. Li, L. Wang, Y. Zhu, J. Shen, B. Z. Tang and C. Fan, Nat. Mater., 2024, 23, 271–280 CrossRef CAS.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- S. Bonacchi, S. Antonell, T. Dainese and F. Maran, Chem.–Eur. J., 2021, 27, 30–38 CrossRef CAS PubMed.
- Y. Shichibu, Y. Ogawa, M. Sugiuchi and K. Konishi, Nanoscale Adv., 2021, 3, 1005–1011 RSC.
- W.-D. Si, C. Zhang, M. Zhou, W.-D. Tian, Z. Wang, Q. Hu, K.-P. Song, L. Feng, X.-Q. Huang, Z.-Y. Gao, C.-H. Tung and D. Sun, Sci. Adv., 2023, 9, eadg3587 CrossRef CAS PubMed.
- W.-D. Si, C. Zhang, M. Zhou, Z. Wang, L. Feng, C.-H. Tung and D. Sun, Sci. Adv., 2024, 10, eadm6928 CrossRef CAS.
- H. Yi, S. Song, S. M. Han, J. Lee, W. Kim, E. Sim and D. Lee, Angew. Chem., Int. Ed., 2023, 62, e202302591 CrossRef CAS PubMed.
- M. Mitsui, D. Arima, Y. Kobayashi, E. Lee and Y. Niihori, Adv. Opt. Mater., 2022, 10, 2200864 CrossRef CAS.
- S. Hossain, D. Hirayama, A. Ikeda, M. Ishimi, S. Funaki, A. Samanta, T. Kawawaki and Y. Negishi, Aggregate, 2023, 4, e255 CrossRef CAS.
- P. Luo, X.-J. Zhai, S. Bai, Y.-B. Si, X.-Y. Dong, Y.-F. Han and S.-Q. Zang, Angew. Chem., Int. Ed., 2023, 62, e202219017 CrossRef CAS PubMed.
- M. Zhou, K. Li, Y. Pei, S. Jin and M. Zhu, J. Phys. Chem. Lett., 2023, 14, 11715–11724 CrossRef CAS.
- E. L. Albright, S. Malola, S. I. Jacob, H. Yi, S. Takano, K. Mimura, T. Tsukuda, H. Häkkinen, M. Nambo and C. M. Crudden, Chem. Mater., 2024, 36, 1279–1289 CrossRef CAS.
- T. I. Levchenko, H. Yi, M. D. Aloisio, N. K. Dang, G. Gao, S. Sharma, C.-T. Dinh and C. M. Crudden, ACS Catal., 2024, 14, 4155–4163 CrossRef CAS.
- K. Isozaki, K. Iseri, R. Saito, K. Ueda and M. Nakamura, Angew. Chem., Int. Ed., 2024, 63, e202312135 CrossRef CAS PubMed.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- R. Jin, C. Liu, S. Zhao, A. Das, H. Xing, C. Gayathri, Y. Xing, N. L. Rosi, R. R. Gil and R. Jin, ACS Nano, 2015, 9, 8530–8536 CrossRef CAS PubMed.
- Y. Yang, Q. Zhang, Z.-J. Guan, Z.-A. Nan, J.-Q. Wang, T. Jia and W.-W. Zhan, Inorg. Chem., 2019, 58, 3670–3675 CrossRef CAS.
- B. Jin, Y. Wang, C. Jin, J. J. De Yoreo and R. Tang, J. Phys. Chem. Lett., 2021, 12, 5938–5943 CrossRef CAS PubMed.
- J.-S. Yang, Y.-J. Zhao, X.-M. Li, X.-Y. Dong, Y.-B. Si, L.-Y. Xiao, J.-H. Hu, Z. Yu and S.-Q. Zang, Angew. Chem., Int. Ed., 2024, 63, e202318030 CrossRef CAS PubMed.
- P. H. Woodworth, M. F. Bertino, A. Ahmedb, A. Anwar, M. R. Shah, D. S. Wijesinghe and J. M. Pettibone, Nano-Struct. Nano-Objects, 2016, 7, 32–40 CrossRef CAS.
- M. R. Narouz, S. Takano, P. A. Lummis, T. I. Levchenko, A. Nazemi, S. Kaappa, S. Malola, G. Yousefalizadeh, L. A. Calhoun, K. G. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2019, 141, 14997–15002 CrossRef CAS PubMed.
- H. Yi, K. M. Osten, T. I. Levchenko, A. J. Veinot, Y. Aramaki, T. Ooi, M. Nambo and C. M. Crudden, Chem. Sci., 2021, 12, 10436–10440 RSC.
- X. Yuan, Z. Ye, S. Malola, O. Shekhah, H. Jiang, X. Hu, J.-X. Wang, H. Wang, A. Shkurenko, J. Jia, V. Guillerm, O. F. Mohammed, X. Chen, N. Zheng, H. Häkkinen and M. Eddaoudi, Chem. Sci., 2024, 15, 16112–16117 RSC.
- D. A. Buschmann, H. Hirai and T. Tsukuda, Inorg. Chem. Front., 2024, 11, 6694–6710 RSC.
- H. Qian, D. Jiang, G. Li, C. Gayathri, A. Das, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2012, 134, 16159–16162 CrossRef CAS.
- S. Chandra, A. Sciortino, S. Shandilya, L. Fang, X. Chen, N. H. Jiang, L.-S. Johansson, M. Cannas, J. Ruokolainen, R. H. A. Ras, F. Messina, B. Peng and O. Ikkala, Adv. Opt. Mater., 2023, 11, 2201901 CrossRef CAS.
- H. Hirai, S. Takano, T. Nakamura and T. Tsukuda, Inorg. Chem., 2020, 59, 17889–17895 CrossRef CAS.
- S. Takano and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 1683–1698 CrossRef CAS.
- H. Hirai, S. Takano, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, Angew. Chem., Int. Ed., 2022, 61, e202207290 CrossRef CAS PubMed.
- H. Hirai, T. Nakashima, S. Takano, Y. Shichibu, K. Konishi, T. Kawai and T. Tsukuda, J. Mater. Chem. C, 2023, 11, 3095–3100 RSC.
- H. Hirai, S. Takano, S. Masuda and T. Tsukuda, ChemElectroChem, 2024, 11, e202300669 CrossRef CAS.
- W. Pei, L. Hou, J. Yang, S. Zhou and J. Zhao, Nanoscale, 2024, 16, 14081–14088 RSC.
- S.-S. Zhang, L. Feng, R. D. Senanayake, C. M. Aikens, X.-P. Wang, Q.-Q. Zhao, C.-H. Tung and D. Sun, Chem. Sci., 2018, 9, 1251–1258 RSC.
- S. Takano, H. Hirai, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 10560–10564 CrossRef CAS.
- M. Sugiuchi, Y. Shichibu, T. Nakanishi, Y. Hasegawa and K. Konishi, Chem. Commun., 2015, 51, 13519–13522 RSC.
- Z.-H. Gao, J. Dong, Q.-F. Zhang and L.-S. Wang, Nanoscale Adv., 2020, 2, 4902–4907 RSC.
- Y. Fukumoto, T. Omoda, H. Hirai, S. Takano, K. Harano and T. Tsukuda, Angew. Chem., Int. Ed., 2024, 63, e202402025 CrossRef CAS PubMed.
- Y. Shichibu, M. Zhang, T. Iwasa, Y. Ono, T. Taketsugu, S. Omagari, T. Nakanishi, Y. Hasegawa and K. Konishi, J. Phys. Chem. C, 2019, 123, 6934–6939 CrossRef CAS.
- X. Wang, R. Liu, L. Tian, J. Bao, C. Zhao, F. Niu, D. Cheng, Z. Lu and K. Hu, J. Phys. Chem. C, 2022, 126, 18374–18382 CrossRef CAS.
- L. Zeng, M. Zhou and R. Jin, ChemPhysChem, 2024, 25, e202300687 CrossRef CAS.
- N. L. Smith and K. L. Knappenberger Jr, J. Phys. Chem. A, 2024, 128, 7620–7627 CrossRef CAS PubMed.
- C. Yao, C.-Q. Xu, I.-H. Park, M. Zhao, Z. Zhu, J. Li, X. Hai, H. Fang, Y. Zhang, G. Macam, J. Teng, L. Li, Q.-H. Xu, F.-C. Chuang, J. Lu, C. Su, J. Li and J. Lu, Angew. Chem., Int. Ed., 2020, 59, 8270–8276 CrossRef CAS PubMed.
- P. Luo, X.-J. Zhai, S. Bai, Y.-B. Si, X.-Y. Dong, Y.-F. Han and S.-Q. Zang, Angew. Chem., Int. Ed., 2023, 62, e202219017 CrossRef CAS PubMed.
- E. L. Albright, T. I. Levchenko, V. K. Kulkarni, A. I. Sullivan, J. F. DeJesus, S. Malola, S. Takano, M. Nambo, K. Stamplecoskie, H. Häkkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2024, 146, 5759–5780 CrossRef CAS.
- X.-L. Pei, P. Zhao, H. Ube, Z. Lei, K. Nagata, M. Ehara and M. Shionoya, J. Am. Chem. Soc., 2022, 144, 2156–2163 CrossRef CAS PubMed.
- Z. Lei, M. Endo, H. Ube, T. Shiraogawa, P. Zhao, K. Nagata, X.-L. Pei, T. Eguchi, T. Kamachi, M. Ehara, T. Ozawa and M. Shionoya, Nat. Commun., 2022, 13, 4288 CrossRef CAS.
- X.-L. Pei, P. Zhao, H. Ube, Z. Lei, M. Ehara and M. Shionoya, Nat. Commun., 2024, 15, 5024 CrossRef CAS.
- B. M. Sutton, G. R. Girard, D. S. Eggleston, P. W. Baures, L. F. Faucette, R. K. Johnson and D. T. Hill, J. Heterocycl. Chem., 1990, 27, 1123–1126 CrossRef CAS.
- M. F. Bertino, Z.-M. Sun, R. Zhang and L.-S. Wang, J. Phys. Chem. B, 2006, 110, 21416–21418 CrossRef CAS.
- Z. Wu, M. A. MacDonald, J. Chen, P. Zhang and R. Jin, J. Am. Chem. Soc., 2011, 133, 9670–9673 CrossRef CAS PubMed.
- K. Mutoh, S. Kawakita, T. Yahagi and T. Nakashima, Nanoscale, 2024, 16, 21776–21782 RSC.
- Z. Liu, M. Zhou, L. Luo, Y. Wang, E. Kahng and R. Jin, J. Am. Chem. Soc., 2023, 145(36), 19969–19981 CrossRef CAS.
- K. Schmidt, S. Brovelli, V. Coropceanu, D. Beljonne, J. Cornil, C. Bazzini, T. Caronna, R. Tubino, F. Meinardi, Z. Shuai and J.-L. Brédas, J. Phys. Chem. A, 2007, 111(42), 10490–10499 CrossRef CAS.
- N. J. Turro , V. Ramamurthy and J. C. Scaiano, Modern Molecular Photochemistry of Organic Molecules, U Science, Sausalito, CA, 2010 Search PubMed.
- M. Mitsui, J. Phys. Chem. Lett., 2024, 15, 12257–12268 CrossRef CAS PubMed.
- H. Suezawa, T. Yoshida, Y. Umezawa, S. Tsuboyama and M. Nishio, Eur. J. Inorg. Chem., 2002, 3148–3155 CrossRef CAS.
- D. Lee, R. L. Donkers, G. Wang, A. S. Harper and R. W. Murray, J. Am. Chem. Soc., 2004, 126, 6193–6199 CrossRef CAS PubMed.
- E. Janssens, S. Neukermans and P. Lievens, Curr. Opin. Solid State Mater. Sci., 2004, 8, 185–193 CrossRef CAS.
- S.-Y. Kang, Z.-A. Nan and Q.-M. Wang, J. Phys. Chem. Lett., 2022, 13, 291–295 CrossRef CAS PubMed.
- C. Liao, M. Zhu, D. Jiang and X. Li, Chem. Sci., 2023, 14, 4666–4671 RSC.
- J.-X. Zhang, F. K. Sheong and Z. Lin, Inorg. Chem., 2020, 59, 8864–8870 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis, 1H NMR spectrum, PLQY and lifetime measurements, DPV measurement, NBO analysis, and the other experimental and DFT calculation results. X-ray crystallographic data, CCDC 2415728. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5sc00561b |
| This journal is © The Royal Society of Chemistry 2025 |