
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFrom two-component enzyme complex to nanobiohybrid for energy-efficient water–gas shift reaction†
Thomas
Pichon
a,
Claudio
Righetti
 b,
Julien
Pérard
a,
Alan
Le Goff
*b and
Christine
Cavazza
*a
b,
Julien
Pérard
a,
Alan
Le Goff
*b and
Christine
Cavazza
*a
aUniv. Grenoble Alpes, CEA, CNRS, IRIG, CBM, F-38000 Grenoble, France. E-mail: christine.cavazza@cea.fr
bUniv. Grenoble Alpes, CNRS, DCM, F-38000 Grenoble, France. E-mail: alan.le-goff@univ-grenoble-alpes.fr
First published on 28th January 2025
Abstract
The water–gas shift reaction (WGSR, CO + H2O ⇔ CO2 + H2) is widely used for the upgrading of syngas, a key substrate for various chemical processes. However, the industrial WGSR requires high pressure and temperature, and has low selectivity. Here, we have designed a biohybrid catalyst by combining CODH from Rhodospirillum rubrum, which catalyzes CO-to-CO2 conversion and a bioinspired nickel bisdiphosphine complex, which catalyzes the hydrogen evolution reaction, immobilized on carbon nanotubes. Carbon nanotubes enable the dual functioning of both catalysts providing efficient electrical conductivity and allowing electroless CO-to-CO2 conversion and H2 evolution. Owing to CO tolerance of the Ni complex, this bioinspired nanohybrid catalyst shows high performance by reaching 100% conversion yield and maximum TOF of 30 s−1 towards WGSR at ambient temperature and pressure in the presence of either pure CO or syngas.
1 Introduction
The syngas market has continuously grown in the last few years and is projected to reach 406![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 860 MWth by 2025. Syngas is a cheap and versatile substrate composed mainly of N2, CO2, H2 and CO, used for the synthesis of a wide range of products (liquid fuels, methanol, etc.).1 However, the current challenge is that this gas is mainly produced from fossil fuels. Concomitantly, the generation of syngas from a diverse range of biomass materials is increasing thanks to the development of biomass or waste feedstock gasification. However, while gasification and purification of gas from fossil raw materials is a well-established technology, relevant differences still exist when using biomass as feedstock, and specific challenges still must be addressed. It is noteworthy that achieving the precise H2/CO ratio is a significant prerequisite for the synthesis of fuels and chemicals. For instance, a H2-to-CO ratio of 2 is required for the synthesis of methanol. Given that the H2/CO ratio in syngas from slagging gasifiers typically ranges from 0.3 to 1, substantial H2-enrichment processes are imperative. Furthermore, the impurity levels are higher in derived biomass gas.2 The key reaction for the upgrading of syngas is the water–gas shift reaction or WGSR (CO + H2O ⇔ CO2 + H2). However, the current industrial processes occur at high pressure and temperature. Furthermore, the presence of contaminant species in the inlet gas can result in catalyst poisoning and a reduction in the overall process efficiency. Another significant limitation of the reaction is the initial H2/CO ratio, which must be relatively high to prevent unwanted side reactions.3 Optimizing performances and energy efficiency of WGSR is therefore of great interest. In nature, several microorganisms offer great potential as sustainable alternatives to fossil-based synthesis of chemicals and fuels. In acetogens, hydrogen-dependent CO2 reductases (HDCR) directly use H2 to reduced CO2 to formate: this is the first reaction in the Wood–Ljungdhal pathway, an ancient pathway for CO2 fixation, but also for energy conservation. The discovery of hydrogenogenic carboxydotrophic bacteria (e.g. Rhodospirillum rubrum) capable of using CO as their sole energy source, thanks to a biological analogue of WGSR (bio-WGSR) has the potential to offer a promising avenue for the design of green alternatives to the industrial catalysts.4 This is because the thermodynamically favorable bio-WGSR is less sensitive to syngas impurities and operates at room temperature and atmospheric pressure. In these microorganisms, the WGSR is catalyzed by a multiprotein complex using two redox nickel enzymes: a carbon monoxide dehydrogenase (CODH), which oxidizes CO to CO2 and an energy-conserving [NiFe] hydrogenase, which catalyzes the proton reduction to H2.4 In addition, a ferredoxin transfers the electron from the CODH to the hydrogenase.
860 MWth by 2025. Syngas is a cheap and versatile substrate composed mainly of N2, CO2, H2 and CO, used for the synthesis of a wide range of products (liquid fuels, methanol, etc.).1 However, the current challenge is that this gas is mainly produced from fossil fuels. Concomitantly, the generation of syngas from a diverse range of biomass materials is increasing thanks to the development of biomass or waste feedstock gasification. However, while gasification and purification of gas from fossil raw materials is a well-established technology, relevant differences still exist when using biomass as feedstock, and specific challenges still must be addressed. It is noteworthy that achieving the precise H2/CO ratio is a significant prerequisite for the synthesis of fuels and chemicals. For instance, a H2-to-CO ratio of 2 is required for the synthesis of methanol. Given that the H2/CO ratio in syngas from slagging gasifiers typically ranges from 0.3 to 1, substantial H2-enrichment processes are imperative. Furthermore, the impurity levels are higher in derived biomass gas.2 The key reaction for the upgrading of syngas is the water–gas shift reaction or WGSR (CO + H2O ⇔ CO2 + H2). However, the current industrial processes occur at high pressure and temperature. Furthermore, the presence of contaminant species in the inlet gas can result in catalyst poisoning and a reduction in the overall process efficiency. Another significant limitation of the reaction is the initial H2/CO ratio, which must be relatively high to prevent unwanted side reactions.3 Optimizing performances and energy efficiency of WGSR is therefore of great interest. In nature, several microorganisms offer great potential as sustainable alternatives to fossil-based synthesis of chemicals and fuels. In acetogens, hydrogen-dependent CO2 reductases (HDCR) directly use H2 to reduced CO2 to formate: this is the first reaction in the Wood–Ljungdhal pathway, an ancient pathway for CO2 fixation, but also for energy conservation. The discovery of hydrogenogenic carboxydotrophic bacteria (e.g. Rhodospirillum rubrum) capable of using CO as their sole energy source, thanks to a biological analogue of WGSR (bio-WGSR) has the potential to offer a promising avenue for the design of green alternatives to the industrial catalysts.4 This is because the thermodynamically favorable bio-WGSR is less sensitive to syngas impurities and operates at room temperature and atmospheric pressure. In these microorganisms, the WGSR is catalyzed by a multiprotein complex using two redox nickel enzymes: a carbon monoxide dehydrogenase (CODH), which oxidizes CO to CO2 and an energy-conserving [NiFe] hydrogenase, which catalyzes the proton reduction to H2.4 In addition, a ferredoxin transfers the electron from the CODH to the hydrogenase.
For several decades now, these families of enzymes have been interfaced with electrodes, and have demonstrated respective reversible CO oxidation and proton reduction at near zero-overpotential requirement.5–13 This is the reason why Armstrong and Reisner were able to achieve enzymatic WGSR by mixing these enzymes with graphite pellets in a test tube.14 Owing to the conductivity of graphite pellets and the addition of CO, both wired enzymes were able to drive the WGSR at room temperature and pressure, without any external energy output. However, this first attempt was limited by the low specific surface area of graphite pellets and the well-known inhibition of hydrogenases by CO. We and others have recently developed the use of carbon nanotubes (CNTs) as high-performance electrode material for the wiring of such enzymes.15–19 We have also demonstrated that specific molecular functionalization can improve their immobilization and electrical interfacing with carbon nanotubes. In the case of hydrogenases, a series of mononuclear nickel complexes have been first designed by D. L. DuBois and coworkers and further refined to approach the exceptional performances of hydrogenases towards both hydrogen evolution and uptake.20–24 Again, carbon nanotubes have demonstrated their ability to immobilize Ni complex and integrate these bio-inspired catalysts in high performance devices.25–27 In addition, these Ni complexes have demonstrated a much better tolerance toward CO than hydrogenases, making them great candidates for WGSR.28 While many nanobiohybrids have been designed to combine enzymes and/or molecular complexes on surfaces to perform catalytic reactions via a cascade strategy,29–33 only few examples have employed the nano-object support as the electronic bridge between the two catalysts operating in tandem.
In this work, we investigate the use of carbon nanotubes for the co-immobilization of two oxygen-sensitive catalysts: the recombinant CODH from Rhodospirillum rubrum overproduced in Escherichia coli (Rec-RrCODH) and a bio-inspired Ni complex to achieve WGSR. Their ability to act as nanowires for the intimate connection between CODH and Ni complex is aimed at achieving bioinspired electroless WGSR under CO without external driving force. The influence of pH, catalyst ratio and type of organic functionalization of CNTs on the performances of bioinspired WGSR was investigated and tested with pure CO or syngas (Fig. 1).
 | ||
| Fig. 1 Bioinspired WGSR principle. | ||
2 Results and discussion
2.1 Functionalization and electrochemistry of MWCNT films
First, multi-walled carbon nanotubes (MWCNT) electrodes were fabricated by filtration of a homogenous dispersion of 2 mg MWCNTs in 50:50 water and ethanol (2 mL) either on a carbon-cloth electrode (for electrochemical analysis) or on a microporous polytetrafluoroethylene (PTFE) membrane (for WGSR experiments) (Fig. S1†). This technique has been previously developed to afford highly homogenous CNT films with controlled thickness and morphology.25,34
The accessible carbon-cloth surface was reduced to 0.07 cm−2 for electrochemical analysis. Fig. 2 shows the optimized response of Rec-RrCODH immobilized at a MWCNT electrode. As previously described, the functionalization of MWCNTs has a significant impact on the surface concentration of immobilized enzymes and bioinspired catalysts. MWCNTs were previously modified with pyrene-adamantane (ADAMWCNTs), demonstrating the superior attachment of Rec-RrCODH on these functionalized MWCNTs owing to hydrophobic interactions.17 On the other hand, diazoniumnaphtoate tetrafluoroborate was employed to promote the functionalization of MWCNTs (NAMWCNTs) with the [NiII(PCy2NArg2)2]7+ complex (NiPNPArg) owing to ionic interactions.26,27 CV of the Rec-RrCODH-modified electrode in the presence of CO exhibits an irreversible electrocatalytic wave corresponding to the CO-to-CO2 electroenzymatic conversion (blue curve, Fig. 2A) with maximum current density of 2.2 mA cm−2 in CO-saturated solution at pH 7.0.
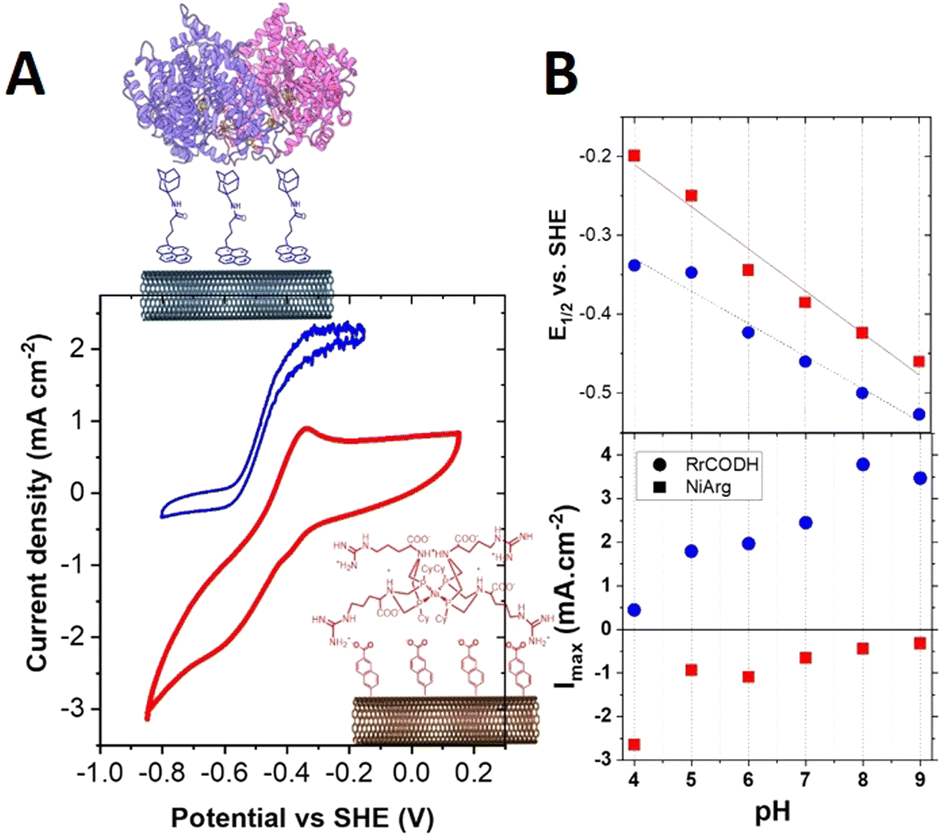 | ||
| Fig. 2 (A) Cyclic voltammetry (CV) of NiPNPArg-modified NAMWCNT electrode (red) under Ar, and Rec-RrCODH modified ADAMWCNT electrode (blue) under CO in 50 mM Bis–Tris propane buffer pH 7.0 (v = 5 mV s−1): (B) half-wave potential and Imaxversus pH for NiPNPArg-modified MWCNT electrode (red) under Ar, and Rec-RrCODH-modified MWCNT electrode (blue) under CO in 50 mM Bis–Tris propane buffer pH 7.0. | ||
The NiPNPArg-modified NAMWCNT electrode exhibits a reversible system at Ered1/2 = −0.32 V vs. SHE corresponding to the electroactivity of the immobilized NiPNPArg accompanied with an electrocatalytic wave corresponding to the hydrogen evolution reaction (HER) activity of the complex (curve red, Fig. 2A). Despite the fact that NiPNPArg is known to be more efficient for H2 oxidation (10 s−1vs. 0.5 s−1 for hydrogen evolution),22,27,35 this is still a high-performance catalyst for HER over a wide range of pH. Maximum current densities and half-wave potentials for both electrocatalytic response were investigated as a function of pH (Fig. 2B). As expected, these two electrocatalytic reactions follow a classic one-proton/one electron ratio potential–pH dependence. These measurements unambiguously demonstrate that the utilization of these two catalysts for CO oxidation and proton reduction provides a thermodynamic driving force for the WGSR within a pH range of 4.0 to 9.0, with an average ΔE of 80 mV.
MWCNT films were then modified with both Rec-RrCODH and NiPNPArg by two successive soaking steps. CV under CO is shown in Fig. 3 for this bifunctionalized MWCNT electrode.
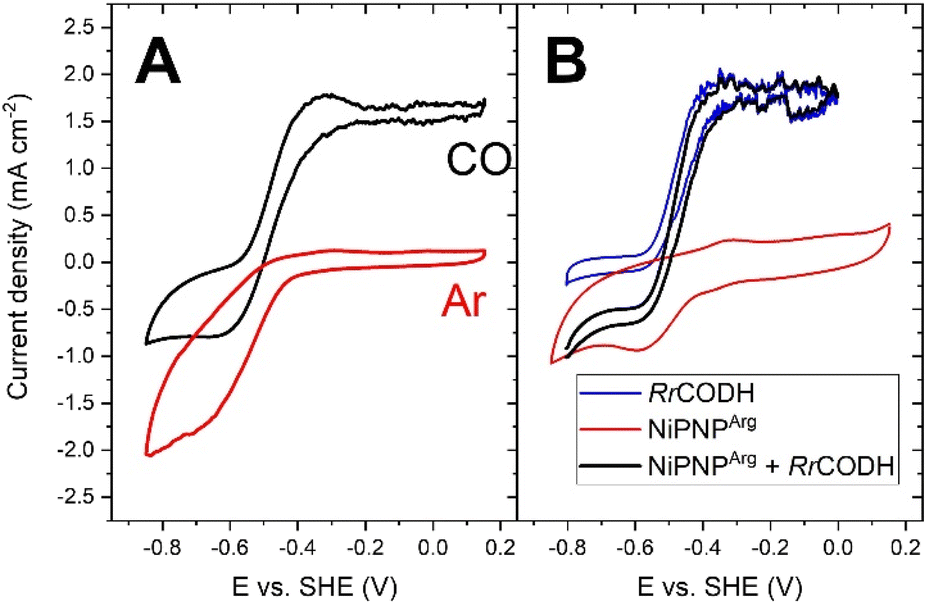 | ||
| Fig. 3 (A) CV of the bifunctionalized MWCNT electrode under Ar and CO in 50 mM Bis–Tris propane buffer pH 7.0 (v = 5 mV s−1); (B) CV of the Rec-RrCODH-modified MWCNT electrode (blue) and NiPNPArg-modified MWCNT electrode (red) under CO in 50 mM Bis–Tris propane buffer pH 7.0 (v = 5 mV s−1) and the corresponding sum of both CV (black, capacitive contribution of one CV was removed). | ||
When comparing the CV of the bifunctionalized electrode under CO (red curve, Fig. 3A) and CV of the NiPNPArg-modified MWCNT electrode under CO (red curve, Fig. 3B), a decrease of the HER catalytic current is observed corresponding to about 58% of the initial current and attributed to the partial inhibition of the HER of NiPNPArg in high CO concentration. This inhibition has already been observed for the complex in solution under high CO concentration and might be caused by weak coordination of CO to the NiPNPArg.22,36 Interesting, the CV response of the bifunctionalized electrodes corresponds to the sum of the responses of both catalysts, when tested independently (corresponding black curve from Fig. 3A and B). This electrocatalytic response shows that both catalysts can be effectively and stably immobilized at the surface of MWCNTs while keeping their electrocatalytic performances.
2.2 Bioinspired WGSR
MWCNT films deposited on PTFE membranes were then modified with both Rec-RrCODH and NiPNPArg for WGSR experiments under anaerobic conditions in a glove box ([O2] < 2 ppm). It is noteworthy that the deposition of the enzyme prior to the complex or the opposite, as well as mixing both catalysts in the soaking solution, has no noticeable effect on the WGSR performances. Surface concentrations, 0.9 nmol cm−2 and 110 nmol cm−2 for Rec-RrCODH and NiPNPArg respectively, were calculated from the total amount of each catalyst deposited on the MWCNT. This bifunctionalized MWCNT film was then introduced into a gas-tight vial with a minimum amount of buffer solution to immerse the MWCNT film (0.6 mL) (Fig. S1†). The active surface of the film was set to 1.77 cm−2. WGSR was started by adding CO using a gastight syringe. The vial was then placed outside the glove box in a temperature-controlled chamber at 150 rpm at 25 °C. The gas phase in the vial was analyzed at regular intervals by sampling 50 μL and injecting into gas chromatography to evaluate the CO2 and H2 formation and CO consumption over time. No traces of O2 were detected at any stage of the experiment. Fig. 4A shows the representative gas measurement (CO, CO2, H2) over time for a MWCNT film modified with RrCODH and NiPNPArg after injection of 5 mL of CO in the cell. | ||
| Fig. 4 (A) Quantity of gas in headspace during WGSR experiment determined by GC over time for H2, CO and CO2 (200 μmol CO injected at t0). (B) Quantity of H2 measured after 24 h at various CO concentration in 50 mM Bis–Tris propane buffer, pH 7.0 at 25 °C (pristine MWCNT film, 0.9 nmol per cm2Rec-RrCODH and 110 nmol per cm2NiPNPArg) (C) WGSR conversion yield for a nonmodified film, film modified with NiPNPArg, film modified with Rec-RrCODH and film modified with both NiPNPArg and Rec-RrCODH; (D) quantity of H2 in headspace of WGSR experiment determined by GC over time for pristine MWCNT, NAMWCNT and ADAMWCNT in 50 mM Bis–Tris propane buffer pH 7.0 at 25 °C (2 nmol Rec-RrCODH and 240 nmol NiPNPArg, 280 μmol injected CO). | ||
CO consumption over time, accompanied by the evolution of both CO2 and H2, confirms the WGSR activity of the bifunctionalized nanohybrid film (Fig. 4A). As illustrated in Fig. 4B, increasing the quantity of CO injected resulted in corresponding increase in H2 production with near 100% conversion yield at low CO concentration and 75% at 400 μmol CO. Under these conditions, no evidence of catalyst inhibition by CO was observed. The unmodified MWCNT films and MWCNT films modified only with the Ni complex do not demonstrate any CO consumption or H2/CO2 evolution (Fig. 4C). Conversely, a MWCNT film modified with only Rec-RrCODH displays slight amounts of H2 and CO2 evolution corresponding to a 2.5% conversion yield. The low activity of Rec-RrCODH-modified MWCNT films may be attributed to an inherent low HER activity when the enzyme is immobilized on MWCNTs, as previously described.37
The influence of surface modification of MWCNTs was also investigated to evaluate the effect of immobilization promoters such as pyreneadamantylamide or naphthoate groups. In the case of NiPNPArg, surface loadings were estimated from the integration of the charge under the NiPNPArg redox system obtained by CV in nonturnover conditions at pH 7. This pH was taken in order to partially avoid contribution of the catalytic activity of the complex towards both hydrogen evolution and H2 oxidation at this pH. It is noteworthy that surface loadings measured by CV corresponds to 6%, 18% and 63% of the amount of NiPNPArg in the soaking solution for pristine MWCNT, ADAMWCNT and NAMWCNT, respectively. This result underlines the efficiency of NAMWCNT to immobilize the catalyst. While MWCNT and ADAMWCNTs exhibits lower surface loadings, the CV of the Ni complex at ADAMWCNT electrodes shows that the adamantane functionalization inhibits HER current density (Fig. S2†). This inhibition also correlates with WGSR performance in which the use of pyreneadamantylamide reduces the WGSR activity resulting in a WGSR conversion yield of 40% (Fig. 4D). WGSR experiments show that both pristine MWCNT and naphtoate groups led to a 100% conversion yield, indicating NAMWCNTs do not provide a significant WGSR improvement. Despite the fact that NAMWCNT promotes NiPNPArg immobilization efficiency, it does not increase HER significantly as compared to pristine MWCNTs.
Optimization of the WGSR performance was also investigated over a range of pH and upon catalyst loadings. The optimum pH range is between 7.5 and 9 (Fig. 5A). As previously described, lower pHs have a drastic effect on the activity of the enzyme.17 On the contrary, at high pH, the HER efficiency of NiPNPArg is reduced22,38 and limits the overall WGSR efficiency.
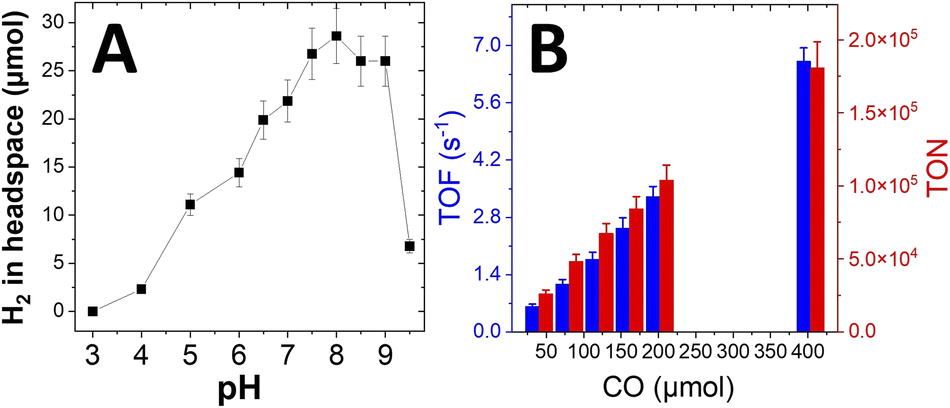 | ||
| Fig. 5 (A) Quantity of H2 in headspace of WGSR experiment determined by GC after 24 h versus pH (1 mL CO injected at t0), 0.7 nmol per cm2Rec-RrCODH and 14 nmol per cm2NiPNPArg, (buffer used: 50 mM citric acid pH 3, 50 mM sodium acetate pH 4 to 5.5, 50 mM Bis–Tris propane buffer pH 6 to 9.5); (B) turnover frequency (TOF) measured after 1 h WGSR, according to the total amount of Rec-RrCODH, and turnover number (TON) measured after 24 h versus starting amount of injected CO (50 mM Bis–Tris propane buffer pH 7, 0.9 nmol per cm2Rec-RrCODH and 110 nmol per cm2NiPNPArg). | ||
Optimum surface coverage was also investigated for both catalysts in terms of H2 production and overall turnover number (TON) by changing the soaking concentration of each catalyst (Fig. S3†). Surface concentrations for Rec-RrCODH is difficult to measure by CV considering the low amount of enzyme as compared to the Ni complex (almost two order of magnitude), the high background current arising from MWCNT capacitance contribution and the overlapping catalytic enzyme activity from the reduction of residual dissolved CO2. In order to estimate the amount of immobilized CODH, the UV-visible spectra of the remaining enzyme solution before and after the incubation step was performed (Fig. S4†). Following the absorbance spectra at 280 nm, between 70 and 80% of CODHs in solution are immobilized on CNT electrodes from high to low concentration range respectively.
WGSR performances were measured at difference concentrations of both catalysts. Performances level off for Rec-RrCODH concentrations above 0.1 nmol cm−2 and NiPNPArg concentrations above 14 nmol cm−2 (Fig. S3†). Optimum concentrations underline the fact that more than 100 times less enzyme is required as compared to NiPNPArg to maximize WGSR performances. This mostly arises from the fact that there are several orders of magnitude between the catalytic rate of CO oxidation by Rec-RrCODH and the HER catalytic rate by NiPNPArg. Interestingly, when performing WGSR at low NiPNPArg concentration, CO inhibition is observed at 100 μmol CO (Fig. S5†). This might arise from partial CO inhibition of HER which correlates with what is observed on CV from Fig. 3. The optimal compromise for 24 hours experiments was determined to be a concentration of 0.7 nmol cm−2 of Rec-RrCODH and 14 nmol cm−2 of NiPNPArg at which no instability of the catalyst is observed. In addition, this concentration is sufficiently high for preventing CO inhibition to limit WGSR performances. More Ni catalysts is therefore required to counterbalance its lower catalytic activity and its CO sensitivity. In optimized conditions, the more CO is injected the more H2 is produced with maximum TON of 180000 after 24 hours and maximum TOF of 6.6 s−1 (Fig. 5B). It is noteworthy that a higher TOF value of 30 s−1 can be reached by using less enzyme (Fig. S6†). Stability of the WGSR was also investigated over time: after three injections of CO at 0, 25 and 50 h, WGSR performance decreased by 30% after 150 h. After injection at 50 h, the biohybrid catalyst is still active but operates at a slower rate of 38% of the initial apparent TOF (i.e. 0.48 s−1) (Fig. S7A†). This is expected from the stability of CODH over time observed in solution and on electrode.15,17 Furthermore, oxygen injection was also performed. The injection of 0.5% of oxygen has little effect on the WGSR performance (Fig. S7B†). When 2.5% of oxygen were added during CO injection or added after 3 h, a drastic decrease of the WGSR performance (42% and 34% respectively) is observed. This instability over time or towards oxygen injection mostly relates to the instability of CODHs investigated in solution or on electrode.9,15,17,39
This places our bio-inspired WGSR device as a top performing catalyst at room temperature, in agreement with the fact that CODHs catalyze the CO oxidation reaction in a thermodynamically efficient way, i.e. by a direct electronic pathway between both redox catalysts (without the need of a natural or synthetic redox partner acting as an electron relay). Industrial iron oxide based catalysts and other types of metal-oxide-based catalysts reaches TOF values between 10−3 and 4 s−1 above 300 °C.40–42 In addition, the only example of the combination of a CODH and a hydrogenase at graphite pellets reaches TOF of 2.5 s−1 at 30 °C. This strategy is also limited by the low H2 production. Only low amounts of CO can be used as it inhibits hydrogenase activity and impact WGSR performances.
Surface characterization was performed by Scanning Electron Microscopy (SEM) and X-ray photoelectron spectroscopy (XPS) (Fig. 6). SEM underlines the large surface and porosity of the functionalized MWCNT film (Fig. 6A). XPS analysis was performed before and after 24 h WGSR run (Fig. 6B).
 | ||
| Fig. 6 (A) SEM micrograph and (B) XPS spectra of Ni 2p core energy levels for MWCNT film modified with both NiPNPArg- and Rec-RrCODH before (a) and after (b) 24 h WGSR accompanied with XPS simulation (red line). | ||
The presence of the intact NiPNPArg complex is confirmed at the Ni 2p core level by the sharp peaks at 856.4 and 874.1 eV accompanied with broad satellite peaks at 863.1 and 880.9 eV characteristics of NiII species.25 A 32% decrease in the intensity of the Ni signal after 24 h WGSR indicates that partial demetallation of the NiPNPArg complex is observed. This is supported by the fact that N 1s and P 1s core energy levels, respectively observed at 400.9 and 133.2 eV are not impacted as compared to Ni 2p levels (Fig. S8†). Unfortunately, the small amount of Rec-RrCODH compared to that of NiPNPArg precludes its characterization by XPS in either the Fe 2p or Ni 2p region.
2.3 Optimized bio-inspired WGSR for syngas upgrading
Finally, a WGSR set-up was performed by using a representative syngas mixture containing 30% CO, 30% H2, 20% CO2, 10% CH4 and 10% N2 with the Rec-RrCODH/NiPNPArg-modified MWCNT system using previously optimized concentrations. The proportions of H2 and CO in the mixture were subsequently determined by GC. Over time, the 7 mL of injected syngas was upgraded to a H2-rich and CO-poor mixture, with a final H2 to CO ratio of 4.4 after 24 h, and only 10% of CO remaining. The presence of methane, a typical component of syngas, had no discernible effect on the catalytic performance of the system, with a maximum TOF of 6.6 s−1 towards H2 production. Notably, a H2 to CO ratio of 2.1 was reached after 5 h, under mild conditions and without the necessity of process gas–catalyst interface optimization. In upgrading syngas systems, this ratio corresponds to that required for future applications such as the synthesis of methanol or the synthesis of liquid fuels by the Fischer–Tropsch process (Fig. 7). | ||
| Fig. 7 Evolution of the proportions of H2 and CO detected by GC in the headspace of a WGSR experiment over time and its corresponding H2/CO ratio (vial flushed with syngas mix (30% CO, 30% H2, 20% CO2, 10% CH4, 10% N2 at t0, corresponding to 7 mL gas mix at ambient pressure and 25 °C, 0.9 nmol per cm2Rec-RrCODH and 110 nmol per cm2NiPNPArg)). | ||
3 Experimental
3.1 Electrode preparation
Gas diffusion electrodes were modified with MWCNT. A dispersion of MWCNT in 50% H2O and 50% ethanol solution was prepared by 4 h sonication of 1 mg mL−1 MWCNT in solution. 2 mL of dispersion were drop casted on a 15 mm diameter GDE, and the solvent was removed under vacuum. The GDE was rinsed with ethanol, and reduced to 3 mm diameter before using.WGSR was started by adding CO or syngas mix using a gastight syringe. The vial was then put in a temperature-controlled chamber under agitation at 25 °C. The gas phase in the vial was analyzed at regular interval by sampling 50 μL and injecting in gas chromatography.
4 Conclusions
This work demonstrates the coupling of a high-performance enzyme for CO-to-CO2 oxidation with a Ni complex catalyzing efficiently the hydrogen evolution reaction, at MWCNTs. This dual catalytic nanohybrid system achieves efficient WGSR with CO or syngas as substrate, working over a period of 24 hours, with TOF reaching a maximum of 30 s−1 under mild conditions. This makes it a proof of concept for the development of a bioinspired WGSR catalyst. Notably, the isolated CODH:Ferredoxin:Hydrogenase complex from Carboxydothermus hydrogenoformans, reaches a WGSR activity of 1700 s−1 at 70 °C.40 This underlines the fact that there is still room for improvement if such TOF values could be achieved in a nanostructured nanohybrid system. For example, the development of WGSR with more robust catalysts towards oxygen or temperature via protein engineering or enzyme selection is one possible strategy. It is noteworthy that the two catalysts used in this study are bidirectional, allowing their potential use in reverse water–gas shift reaction (rWGSR, CO2 + H2 ⇔ CO + H2O) with applications in power-to-syngas processes.
In the future, the system developed here could therefore be optimized for WGSR in its direct or reverse way for its integration in operational devices.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
T. Pichon: investigation, formal analysis, C. Righetti: investigation, formal analysis, J. Pérard: investigation, formal analysis, A. Le Goff: conceptualization, funding acquisition, supervision, writing original draft, writing review and editing, C. Cavazza: conceptualization, funding acquisition, supervision, writing original draft, writing review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Agence Nationale de la Recherche through the LabEx ARCANE program (ANR-11-LABX-0003-01) and the Graduate School on Chemistry, Biology and Health of Univ Grenoble Alpes CBH-EUR-GS (ANR-17-EURE-0003). This work was supported by “the NTE PhD program, CEA” for TP's PhD funding. This work has benefited from French State aid managed by the Agence Nationale de la Recherche under France 2030 plan, bearing the reference code ANR-22-PESP-0010: Projet ciblé “POWERCO2” within the PEPR project SPLEEN. We kindly thank Dr Bertrand Reuillard for his help for the synthesis of the NiPNPArg complex. Valérie Flaud and Eddy Petit from Institut Charles Gerhardt (University of Montpellier 2) are gratefully acknowledged for XPS analysis.Notes and references
- A. Galadima and O. Muraza, J. Nat. Gas Sci. Eng., 2015, 25, 303–316 CrossRef CAS.
- Ö. Tezer, N. Karabağ, A. Öngen, C. Ö. Çolpan and A. Ayol, Int. J. Hydrogen Energy, 2022, 47, 15419–15433 CrossRef.
- E. Baraj, K. Ciahotný and T. Hlinčík, Fuel, 2021, 288, 119817 CrossRef CAS.
- B. Soboh, D. Linder and R. Hedderich, Eur. J. Biochem., 2002, 269, 5712–5721 CrossRef CAS PubMed.
- V. C.-C. Wang, S. W. Ragsdale and F. A. Armstrong, ChemBioChem, 2013, 14, 1845–1851 CrossRef CAS PubMed.
- M. Can, F. A. Armstrong and S. W. Ragsdale, Chem. Rev., 2014, 114, 4149–4174 CrossRef CAS PubMed.
- A. Parkin, J. Seravalli, K. A. Vincent, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2007, 129, 10328–10329 CrossRef CAS PubMed.
- T. W. Woolerton, S. Sheard, E. Reisner, E. Pierce, S. W. Ragsdale and F. A. Armstrong, J. Am. Chem. Soc., 2010, 2132–2133 CrossRef CAS PubMed.
- E. C. Wittenborn, C. Guendon, M. Merrouch, M. Benvenuti, V. Fourmond, C. Léger, C. L. Drennan and S. Dementin, ACS Catal., 2020, 10, 7328–7335 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- M. G. Allan, T. Pichon, J. A. McCune, C. Cavazza, A. Le Goff and M. F. Kühnel, Angew. Chem., Int. Ed., 2023, 62, e202219176 CrossRef CAS PubMed.
- N. Lalaoui, A. de Poulpiquet, R. Haddad, A. Le Goff, M. Holzinger, S. Gounel, M. Mermoux, P. Infossi, N. Mano, E. Lojou and S. Cosnier, Chem. Commun., 2015, 51, 7447–7450 RSC.
- L. Domnik, M. Merrouch, S. Goetzl, J.-H. Jeoung, C. Leger, S. Dementin, V. Fourmond and H. Dobbek, Angew. Chem., Int. Ed., 2017, 56, 15466–15469 CrossRef CAS PubMed.
- O. Lazarus, T. W. Woolerton, A. Parkin, M. J. Lukey, E. Reisner, J. Seravalli, E. Pierce, S. W. Ragsdale, F. Sargent and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14154–14155 CrossRef CAS PubMed.
- U. Contaldo, M. Curtil, J. Pérard, C. Cavazza and A. Le Goff, Angew. Chem., Int. Ed., 2022, 134, e202117212 CrossRef.
- S. Gentil, S. M. Che Mansor, H. Jamet, S. Cosnier, C. Cavazza and A. Le Goff, ACS Catal., 2018, 3957–3964 CrossRef CAS.
- U. Contaldo, B. Guigliarelli, J. Perard, C. Rinaldi, A. Le Goff and C. Cavazza, ACS Catal., 2021, 5808–5817 CrossRef CAS.
- M. A. Alonso-Lomillo, O. Rüdiger, A. Maroto-Valiente, M. Velez, I. Rodríguez-Ramos, F. J. Muñoz, V. M. Fernández and A. L. De Lacey, Nano Lett., 2007, 7, 1603–1608 CrossRef CAS PubMed.
- A. Le Goff and M. Holzinger, Sustain. Energy Fuels, 2018, 2, 2555–2566 RSC.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358–366 CrossRef CAS PubMed.
- S. Raugei, S. Chen, M.-H. Ho, B. Ginovska-Pangovska, R. J. Rousseau, M. Dupuis, D. L. DuBois and R. M. Bullock, Chem. Commun., 2012, 6493–6506 CAS.
- A. Dutta, J. A. S. Roberts and W. J. Shaw, Angew. Chem., Int. Ed., 2014, 53, 6487–6491 CrossRef CAS PubMed.
- A. Dutta, D. L. DuBois, J. A. S. Roberts and W. J. Shaw, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16286–16291 CrossRef CAS.
- A. A. Oughli, A. Ruff, N. P. Boralugodage, P. Rodríguez-Maciá, N. Plumeré, W. Lubitz, W. J. Shaw, W. Schuhmann and O. Rüdiger, Nat. Commun., 2018, 9, 864 CrossRef PubMed.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Metaye, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS.
- N. Coutard, B. Reuillard, T. N. Huan, F. Valentino, R. T. Jane, S. Gentil, E. Andreiadis, A. Le Goff, T. Asset, F. Maillard, B. Jousselme, A. Morozan, S. Lyonnard, V. Artero and P. Chenevier, Chem Catal., 2021, 1(1), 88–105 CrossRef CAS.
- S. Gentil, N. Lalaoui, A. Dutta, Y. Nedellec, S. Cosnier, W. J. Shaw, V. Artero and A. Le Goff, Angew. Chem., Int. Ed., 2017, 56, 1845–1849 CrossRef CAS PubMed.
- P. D. Tran, A. Le Goff, J. Heidkamp, B. Jousselme, N. Guillet, S. Palacin, H. Dau, M. Fontecave and V. Artero, Angew. Chem. Int. Ed., 2011, 50, 1371–1374 CrossRef CAS PubMed.
- F. C. Macazo and S. D. Minteer, Curr. Opin. Electrochem., 2017, 5, 114–120 CrossRef CAS.
- C. Agnès, B. Reuillard, A. Le Goff, M. Holzinger and S. Cosnier, Electrochem. commun., 2013, 105–108 CrossRef.
- B. Reuillard, A. Le Goff and S. Cosnier, Anal. Chem., 2014, 86, 4409–4415 CrossRef CAS PubMed.
- B. Reuillard, A. Le Goff and S. Cosnier, Chem. Commun., 2014, 50, 11731–11734 RSC.
- M. Yuan, M. J. Kummer and S. D. Minteer, Chem.–Eur. J., 2019, 25, 14258–14266 CrossRef CAS PubMed.
- Z. Wu, Z. Chen, X. Du, J. M. Logan, J. Sippel, M. Nikolou, K. Kamaras, J. R. Reynolds, D. B. Tanner, A. F. Hebard and A. G. Rinzler, Science, 2004, 305, 1273–1276 CrossRef CAS PubMed.
- B. Reuillard, C. Costentin and V. Artero, Angew. Chem., Int. Ed., 2023, 62, e202302779 CrossRef CAS PubMed.
- A. D. Wilson, K. Fraze, B. Twamley, S. M. Miller, D. L. DuBois and M. Rakowski DuBois, J. Am. Chem. Soc., 2008, 130, 1061–1068 CrossRef CAS PubMed.
- S. Menon and S. W. Ragsdale, Biochemistry, 1996, 35, 15814–15821 CrossRef CAS PubMed.
- S. Gentil, N. Lalaoui, A. Dutta, Y. Nedellec, S. Cosnier, W. J. Shaw, V. Artero and A. Le Goff, Angew. Chem., Int. Ed., 2017, 56, 1845–1849 CrossRef CAS PubMed.
- M. Merrouch, J. Hadj-Saïd, L. Domnik, H. Dobbek, C. Léger, S. Dementin and V. Fourmond, Chem.–Eur. J., 2015, 21, 18934–18938 CrossRef CAS PubMed.
- B. Soboh, D. Linder and R. Hedderich, Eur. J. Biochem., 2002, 269, 5712–5721 CrossRef CAS PubMed.
- R. B. King, J. Organomet. Chem., 1999, 586, 2–17 CrossRef CAS.
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Pérez, Science, 2007, 318, 1757–1760 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc06394e |
| This journal is © The Royal Society of Chemistry 2025 |