
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRevealing the dissolution mechanism of organic carbonyl electrodes in lithium–organic batteries†
Shu
Zhang
,
Weiwei
Xie
 ,
Zhuo
Yang
,
Shuo
Xu
,
Qi
Zhao
,
Yong
Lu
,
Kai
Zhang
,
Zhenhua
Yan
and
Jun
Chen
*
,
Zhuo
Yang
,
Shuo
Xu
,
Qi
Zhao
,
Yong
Lu
,
Kai
Zhang
,
Zhenhua
Yan
and
Jun
Chen
*
Frontiers Science Center for New Organic Matter, Key Laboratory of Advanced Energy Materials Chemistry (Ministry of Education), State Key Laboratory of Advanced Chemical Power Sources, College of Chemistry, Nankai University, Tianjin 300071, China. E-mail: chenabc@nankai.edu.cn
First published on 25th January 2025
Abstract
Organic carbonyl electrode materials (OCEMs) have shown great promise for high-performance lithium batteries due to their high capacity, renewability, and environmental friendliness. Nevertheless, the severe dissolution of these materials in conventional electrolytes results in poor cycling stability, which hinders their practical application. Herein, a unified model considering the effects of both ion-solvation structures and electrolyte solvents is proposed to elucidate the dissolution mechanism of OCEMs in electrolytes. In this new model, dissolution is driven by the interactions of OCEMs with ion-solvation structures and free (uncoordinated) solvents in electrolytes. In non-polar electrolytes, the strong interactions between OCEMs and Li-anion aggregates accelerate the dissolution of OCEMs, leading to anomalously high solubility of OCEMs. Conversely, the high dissolution in strongly polar electrolytes is dominated by the interaction with free solvents. This unified model transcends the conventional perspective that dissociation solely depends on the solute–solvent interactions. Based on this model, we propose that tuning the effects of ion-solvation structures and free solvents by altering solvent polarity could be an effective strategy for inhibiting the dissolution of organic electrodes to achieve long-cycle Li–organic batteries.
1 Introduction
The increasing demand for lithium-ion batteries, driven by the popularity of electronics and electric vehicles, has prompted continuous optimization and innovation of cathode materials.1–3 However, commercialized inorganic cathodes, such as LiCoO2, LiNixCoyMn1−x−yO2 and LiFePO4, face challenges related to the use of non-renewable resources and environmental incompatibility.4–7 Therefore, there is an urgent need to develop renewable cathodes for lithium batteries. Organic electrode materials, especially carbonyl cathodes, have emerged as viable alternatives due to their high elemental abundance, environmental friendliness and structural design flexibility.8–12 One of the main bottlenecks impeding the application of organic cathodes in large-scale energy storage is their high solubility in conventional electrolytes. In contrast to the strong ionic bonding in conventional inorganic cathodes, organic molecules are bound by van der Waals forces. These weak intermolecular interactions result in the severe dissolution of organic electrodes in conventional organic liquid electrolytes, leading to the loss of active materials and capacity fading during cycles.13,14Hitherto, various strategies have been implemented to suppress the dissolution of carbonyl cathodes, including the polymerization of the redox-active organic carbonyl molecules, the salification of active sites, and composite formation with mesoporous solid supports (e.g., advanced conductive carbon).15–21 However, these methods are often plagued by complex synthesis processes, high manufacturing costs and a sacrifice of initial battery capacity. As an alternative to cathode regulation, electrolyte optimization has become increasingly attractive due to its flexible design and minimal impact on the manufacturing process and energy density of batteries.22–28 However, debates and conflicting results persist in the literature regarding whether dissolution is suppressed or enhanced in high-concentration electrolytes. Recent studies have shown that high-concentration and localized high-concentration electrolytes can effectively inhibit the dissolution of organic cathodes. For instance, the dissolution of the N,N′-dimethylphenazine cathode is suppressed with increasing salt concentration of LiPF6 in the ethylene carbonate/diethyl carbonate electrolyte.29 The pillar[5]quinone shows a high capacity of 310 mA h g−1 after 900 cycles using the 4 mol L−1 lithium bis(trifluoromethanesulfonyl)amide (LiTFSI)/acetonitrile electrolyte.30 In contrast, Vlad et al. found that the dissolution of the 2,5-diamino-1,4-benzoquinone cathode becomes more pronounced with increasing concentration of LiTFSI in tetraethylene glycol dimethyl ether/1,3-dioxolane electrolyte.31 These controversial findings demonstrate that the origin of organic cathode dissolution in electrolytes is not yet fully understood.
The dissolution of organic compounds is reminiscent of the well-known ‘like dissolves like’ rule, a widely used guideline for predicting the solubility of organic compounds. It states that polar molecules tend to dissolve in polar phases, while non-polar molecules prefer non-polar solvents. This traditional model (e.g., Hansen solubility parameters32) has been widely used in industry to regulate the solubility of polymers in solvents. However, it is worth noting that this model only considers solute–solvent interactions and lacks a description of interactions between solutes and ion-solvation structures in electrolytes, which can significantly impact the dissolution of organic compounds. Therefore, there is an urgent need to establish a unified model that incorporates the effects of both ion-solvation structures and solvents on the dissolution of organic electrodes.
Herein, we propose a unified picture of the dissolution of organic electrodes in electrolytes, including the effects of both solvents and ion-solvation structures. Considering a series of representative ether solvents derived from 1,2-diethoxyethane (DEE) as the prototype molecule (Fig. 1), we systematically investigate the dissolution of carbonyl cathodes in different concentrations of these electrolytes by combining theoretical calculations with experimental spectroscopic characterization. The results reveal that the dissolution of the carbonyl cathode is driven by the competitive solvation of organic electrode molecules by ion-solvation structures and free (uncoordinated) solvents in electrolytes. Increasing the concentration of the electrolyte reduces the number of free solvent molecules, resulting in lower solubility of carbonyl cathodes. Simultaneously, the number of ion-solvation structures increases with concentration, leading to higher static permittivity33,34 and thus enhancing the dissolution of carbonyl molecules. We found that this competition can be tuned by altering the solvent polarity. In electrolytes with strongly polar solvents (e.g., ethylene carbonate), the decrease in free solvents is more important than the increase in ion-solvation structures, leading to suppressed dissolution of organic electrodes in high-concentration electrolytes. Conversely, in electrolytes with non-polar solvents (e.g., diethyl ether), the increase in ion-solvation structures dominates the dissolution mechanism, accelerating the dissolution of organic electrodes. Guided by this model, we developed an electrolyte (1,2-dibutoxyethane) that balances the effects of free solvents and ion-solvation structures. When paired with the pyrene-4,5,9,10-tetraone (PTO) cathode, it demonstrates a high initial capacity of 350 mA h g−1 and an excellent capacity retention of 76% after 300 cycles at 1C.
 | ||
| Fig. 1 Design logic of the ether solvent family derived from DEE. | ||
2 Results and discussion
2.1 Dissolution of organic electrode materials in electrolytes
The DEE molecule, a widely used electrolyte solvent in lithium–metal batteries,35,36 was used as a prototype electrolyte solvent. By adjusting the number of terminal/side alkyl chains, the coordination modes with Li+ and the number of functional groups, a series of ether electrolyte solvent molecules were derived (Fig. 1), including 1,2-dimethoxyethane (DME), 1,2-dipropoxyethane (DPE), 1,2-dibutoxyethane (DBE), diethoxymethane (DMOE), 1,3-diethoxypropane (DEOP), 1,4-diethoxybutane (DEOB), 1,2-diethoxypropane (IDEOP), 1,2-diethoxybutane (IDEOB), 1,2-diethoxypentane (DEOPT), diethyl ether (EE), diethylene glycol diethyl ether (DEGDEE) and triethylene glycol diethyl ether (TEGDEE). These solvents were paired with LiTFSI to form electrolytes for Li–organic batteries, with PTO, selected as the carbonyl cathode material.The solvation free energy is a powerful metric for describing solute–solvent interactions and has been widely used to predict protein–ligand binding energies37 and the dissolution of small molecules in water for drug discovery.38 In this work, we calculated the solvation free energy by performing classical MD simulations to evaluate the dissolution behaviour of the PTO cathode in different electrolytes. Fig. S1a† indicates the absolute solvation free energies of PTO in various electrolytes at different concentrations. Overall, the absolute solvation free energies increase with increasing electrolyte concentration from 0 m to 2.5 m, indicating that the addition of lithium salt enhances the dissolution of PTO. It is noteworthy that the solvation free energy (ΔG) can be decomposed into the contributions from electrostatic interactions (ΔGes) and van der Waals interactions (ΔGvdw), and it includes the cavitation energy required to create a cavity for PTO by pushing aside solvent molecules.39 We observe that ΔGvdw is independent of electrolyte concentration (Fig. S1†), suggesting that the solvation free energy is primarily governed by the PTO-solvent electrostatic interaction (Fig. 2a). In the following, we use the electrostatic component, ΔGes, to describe the dissolution of PTO in electrolytes, which will be referred to as ‘solvation free energy’.
 | ||
| Fig. 2 (a) Computed solvation free energies of PTO in electrolytes with different concentrations obtained from MD simulations. (b) Solubility by UV-vis spectroscopy of PTO dissolved in different electrolytes. (c) Solvation free energies of PTO in pure solvent as a function of dielectric constant. (d) Variation of solvation free energies from 0 m to 0.5 mol kg−1 LiTFSI electrolytes as a function of dielectric constant. | ||
To examine the accuracy of the calculated solvation free energy, we performed UV-vis spectroscopy characterization on the dissolution behaviors of PTO in several representative electrolytes (DME, DEE, DEGDEE, DBE and EE) at varying concentrations of LiTFSI. Fig. 2b displays the natural logarithm of the maximum absorbance at around 260 nm, corresponding to the n → π* transition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond of PTO.40 It is evident that higher concentration leads to higher dissolution of PTO in electrolytes, consistent with the solvation free energy calculations. Moreover, we observed a red shift in the absorption peak with increasing electrolyte concentration (Fig. S2†). This phenomenon can be attributed to the formation of the PTO–Li+ complex, as confirmed by the red shift observed in the calculated UV-vis spectra of PTO compared to that of the PTO–Li+ complex (Fig. S3†). Optical photographs also demonstrate a notable color difference between PTO dissolved in pure solvents and those in electrolytes, indicating a significant change in the solvation environment of the dissolved PTO (Fig. S4†).
O bond of PTO.40 It is evident that higher concentration leads to higher dissolution of PTO in electrolytes, consistent with the solvation free energy calculations. Moreover, we observed a red shift in the absorption peak with increasing electrolyte concentration (Fig. S2†). This phenomenon can be attributed to the formation of the PTO–Li+ complex, as confirmed by the red shift observed in the calculated UV-vis spectra of PTO compared to that of the PTO–Li+ complex (Fig. S3†). Optical photographs also demonstrate a notable color difference between PTO dissolved in pure solvents and those in electrolytes, indicating a significant change in the solvation environment of the dissolved PTO (Fig. S4†).
To gain further insights into the salt-promoting effects on the dissolution of PTO, we compared the solvation free energies of PTO in pure solvents and electrolytes. Fig. 2c shows the solvation free energy of PTO in solvents with varying dielectric constants. The dielectric constant (ε) is a widely used descriptor for measuring molecule polarity and ion–dipole interactions.41 We find that the dissolution of PTO increases with solvent polarity, consistent with the well-known ‘like dissolves like’ principle, where polar molecules tend to dissolve in polar phases. The intermolecular interactions energies between PTO and solvents such as EE, DBE and DME are −41.66 kJ mol−1, −43.43 kJ mol−1 and −46.18 kJ mol−1, respectively (Fig. S5†). The variation also proves that the interaction with the organic cathode strengthens with increased solvent polarity. However, this rule-of-thumb can break down with the addition of lithium salt. Based on the variation of solvation energy upon the addition of LiTFSI (Fig. 2d), the electrolyte solvents can be approximately categorized into two groups: non-polar solvents (ε < 3) and weakly polar solvents (ε > 3). The addition of LiTFSI has a minor influence on the solubility of PTO in weakly polar solvents (e.g., DME), while the mixture of LiTFSI and non-polar solvents (e.g., EE) significantly enhances the dissolution of PTO. This anomalous dissolution behavior contrasts with the predictions of the ‘like dissolves like’ rule, highlighting the need to consider the effect of ion-solvation structures on the dissolution of organic electrode materials in electrolytes. In the following, we select three characteristic solvents – EE, DBE and DME – to represent the non-polar and weakly polar solvents, respectively.
2.2 Effects of free solvents and ion-solvation structures on the dissolution
An electrolyte consists of ion-solvation structures, uncoordinated anions and uncoordinated solvents. We calculated the radial distribution functions (RDFs) and coordination numbers (CNs) of dissolved PTO with Li+, solvents and TFSI− anions in DME, DBE and EE electrolytes, respectively. As shown in Fig. S6,† the CN of PTO–Li+ is significantly larger than the CNs of PTO–Li+ and PTO–solvent, indicating that the PTO–Li+ interaction predominates among the species in all electrolytes. Notably, the CN of PTO–TFSI− increases from DME to DBE, to EE electrolyte. This increasing PTO–anion interaction is attributed to the weaker interaction between lithium ions and solvents, which leads to a higher number of lithium-anion aggregates (Fig. S12a and b†) and lithium–PTO clusters, as evidenced by the increased CN of PTO–Li+ (Fig. S6†). Note that this is not due to a stronger interaction between PTO and uncoordinated anions. Therefore, the interactions between PTO and electrolytes can divided into two components: interactions with ion-solvation structures and interactions with free solvents. It is therefore straightforward to decompose the solvation free energy into the contributions from ion-solvation structures and free solvents (Fig. 3a–c). As the electrolyte concentration increases, the contribution of free solvents to the solvation free energy gradually decreases, while the contribution of ion-solvation structures increases. This trend is attributed to the reduced number of free solvents (Fig. S7†) and the increased presence of ion-solvation structures at higher electrolyte concentrations. The dissolution of organic electrodes in electrolytes is driven by the competition between the interactions of PTO with ion-solvation structures and free solvents. | ||
| Fig. 3 Components of free solvents and ion-solvation structures contributing to the solvation free energies of PTO in (a) LiTFSI/EE, (b) LiTFSI/DBE, (c) LiTFSI/DME and (d) LiTFSI/EC electrolytes with different concentrations. | ||
In non-polar solvents (ε < 3), the contribution from ion-solvation structures increases dramatically with increasing electrolyte concentration, whereas the contribution from free solvents decreases slowly, leading to an ion-solvation-dominated dissolution mechanism (Fig. 3a and b). The addition of lithium salts significantly enhances the dissolution of PTO in these electrolytes. In weakly polar solvents (ε > 3), the decrease in the free solvent fraction primarily compensates for the increase in the fraction of ion-solvation structures, resulting in a weak dependence of PTO dissolution on electrolyte concentration (Fig. 3c). Notably, the decline in the free-solvent contribution exhibits a steeper concentration dependence as solvent polarity increases (Fig. S8†). This suggests that in strongly polar solvents (e.g., carbonate esters), the contribution of free solvents may surpass that of ion-solvation structures, leading to a free-solvent-dominated dissolution mechanism. To verify this hypothesis, we selected ethylene carbonate (EC), a commercial solvent used in lithium-ion batteries, as a representative strongly polar solvent (Fig. S9†). As shown in Fig. 3d, the reduction in the free-solvent component is significantly faster than the increase in the component of the ion-solvation structures, resulting in a decline in dissolution with increasing electrolyte concentration. This observation is further confirmed by the UV-vis absorption spectra (Fig. S10†) and aligns with reports indicating that high-concentration electrolytes can effectively inhibit the dissolution of organic electrodes.42–44
To investigate the influence of different ion-solvation structures on the dissolution of PTO in electrolytes, we define the solvation structures according to the distribution of Li+ solvates: solvent-surrounded Li+ (SSL), Li+-anion single pair (LASP) and Li+-anion cluster (LAC), each of which has the number of coordinating anions of 0, 1 and ≥2 in the solvation sheath,45,46 respectively (Fig. S11†). It is worth noting that this Li+-based definition differs from the conventional anion-based classification of solvation structures (i.e., solvent-separated ion pairs (SSIPs)), compact ion pairs (CIPs) and aggregated ion clusters (AGG).47 In the EE electrolyte, the solvation structures are predominantly LACs (Fig. S12a†). As the solvent polarity increases, the solvation structures show a mixture of LASP- and LAC-type structures (DBE, Fig. S12b†). In DME and EC electrolytes, SSL becomes the dominant Li+ solvate species, although its proportion decreases with increasing electrolyte concentration (Fig. S12c and d†). Meanwhile, the solvation structure of the 1 m electrolytes was also measured by Raman spectroscopy (Fig. S13†). The results are highly consistent with MD simulations. We further calculated the interaction energies between PTO and different ion-solvation structures in electrolytes. In EE electrolyte, the interaction energy between PTO and LAC structures is significantly larger than that of the other species (Fig. S14a†), indicating that aggregate ion clusters govern the PTO dissolution in the EE electrolyte. In DBE electrolyte, the PTO–LASP interaction dominates at low concentrations (Fig. S14b†), while the increase in LAC content leads to its dominant interaction with PTO at high concentrations. For DME and EC electrolytes, the interaction energies between SSL and PTO are notably larger compared to the LASP–PTO and LAC–PTO interaction energies (Fig. S14c and d†). This also indicates that the differences in solvation structures caused by different molecular polarity are the direct factors influencing the different dissolution behaviours.
2.3 Solvation of dissolved organic electrode molecules
The state of the dissolved PTO was investigated using MD simulations. Dissolved PTO can exist in two states: PTO surrounded by solvents (free state) or PTO coordinated with Li+ (coordination state). As the solvent polarity increases, the percentage of free PTO increases (Fig. S15†). This trend can be visualized in the MD snapshots (Fig. 4a–d). The interaction between PTO and solvated ion-clusters dominates in EE electrolyte, causing the dissolved PTO to participate in the formation of ion-cluster structures in the electrolyte (Fig. 4a). Accompanied by the evolution of molecular polarity and the dominant solvation structure, more PTO molecules are observed in the free state from EE to EC electrolytes (Fig. 4b–d), which reveals that free solvents gradually become the dominant factor affecting dissolution. The primary solvation structures of PTO are illustrated in Fig. 4e–h, corresponding to the dominant structures observed in these electrolytes, respectively. The changes in the solvation structures of the electrolyte can be supported by the variation in the number of uncoordinated TFSI− ions before and after the dissolution of PTO (Fig. S16†). Moreover, the radial distribution function (RDF) and coordination number of Li–O(PTO) for different electrolytes, obtained from MD simulations, confirm that the number of PTO molecules entering the first solvation shell decreases significantly with increasing solvent polarity (Fig. S17†).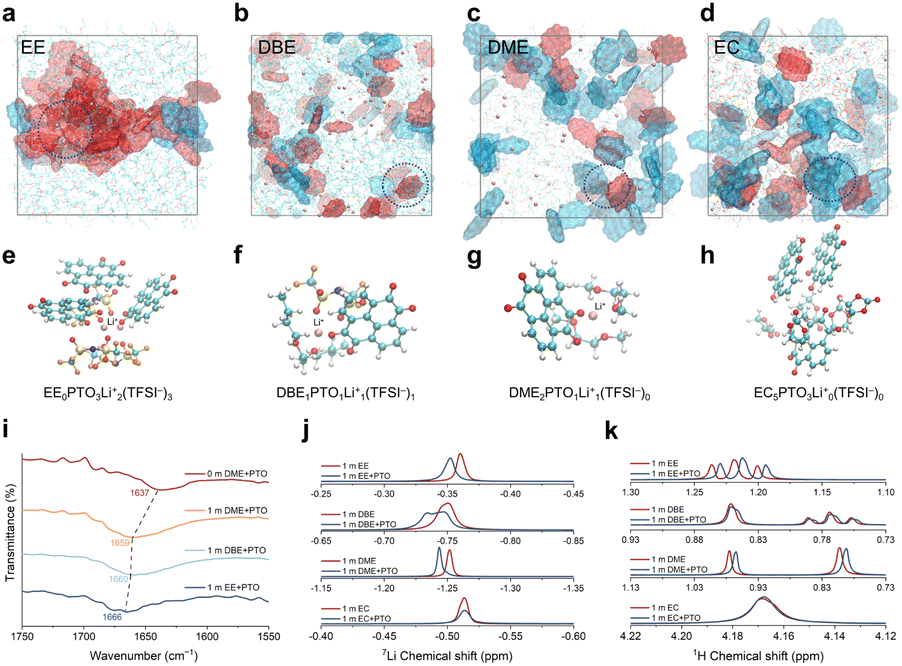 | ||
| Fig. 4 MD snapshots for PTO dissolved in (a) 1 m LiTFSI/EE, (b) 1 m LiTFSI/DBE, (c) 1 m LiTFSI/DME and (d) 1 m LiTFSI/EC electrolytes.here The red molecules represent PTO that participates in the formation of solvation structures, while the blue molecules represent free PTO. (e–h) Primary solvation structures of dissolved PTO molecules, corresponding to the dashed circles in (a–d). The cyan, white, red, blue, orange and pink balls represent C, H, O, N, F and Li, respectively. (i) IR spectra of dissolved PTO in different electrolytes. (j) 7Li NMR spectra before and after dissolving PTO in 1 m electrolytes. (k) 1H NMR spectra before and after dissolving PTO in 1 m electrolytes. | ||
Experimentally, we performed various spectral characterization studies on the electrolytes before and after the dissolution of PTO. Fig. 4i shows the IR spectra of the CO stretching mode of dissolved PTO near 1650 cm−1 (full spectra are shown in Fig. S18a–d†). Compared to DME solvent, the peak of the carbonyl group in 1 m LiTFSI/DME electrolyte exhibits a remarkable blue shift from 1637 cm−1 to 1659 cm−1. This blue shift can be attributed to the transition of PTO from the free state to the coordination state (Fig. 4i). The carbonyl group peaks in 1 m LiTFSI/DBE and 1 m LiTFSI/EE electrolytes are close to those in 1 m LiTFSI/DME, indicating a similar coordination state of PTO in DBE and EE electrolytes. The indistinguishable IR spectra of 1 m LiTFSI/EC electrolyte before and after dissolving PTO demonstrate that PTO rarely enters the Li-ion solvation shell (Fig. S18e†). Furthermore, the different states of dissolved PTO in electrolytes with varying solvent polarity are confirmed by liquid NMR. The downfield displacement of the 7Li chemical shift indicates an enhanced deshielding effect on the Li+ nucleus (Fig. 4j), which can be attributed to the participation of PTO in the Li+ solvation sheath.48 The upfield shifts in 1H NMR after dissolving PTO in 1 m LiTFSI/EE, 1 m LiTFSI/DBE and 1 m LiTFSI/DME electrolytes (Fig. 4k) suggest that PTO molecules enter the solvation shell and coordinate with Li ions.49 In contrast, the negligible chemical shifts observed in both 7Li and 1H NMR for 1 m LiTFSI/EC electrolyte after dissolving PTO indicate that the addition of PTO has a minor impact on the Li-ion solvation structures, with most of the dissolved PTO molecules remaining in the free state. The specific solvation structures of the electrolytes with dissolved PTO were also confirmed by Raman spectra (Fig. S19†), which is in complete agreement with the results of the above analysis.
2.4 Electrochemical performance
To evaluate the stability of organic electrode materials during the charge and discharge processes, we performed in situ UV-vis experiments for Li‖PTO batteries using 1 m LiTFSI/EE, 1 m LiTFSI/DME and 1 m LiTFSI/DBE electrolytes. As shown in Fig. 5a–c, the absorption maxima at around 260 nm indicate that the PTO electrode undergoes severe, moderate and slight dissolution in 1 m LiTFSI/EE, 1 m LiTFSI/DME and 1 m LiTFSI/DBE electrolytes, respectively, which is consistent with the solvation free energy calculations (Fig. 2a). The discrepancy in the dissolution behaviours of PTO can also be visualized from the color differences in the in situ test cuvettes (panels inside Fig. 5a–c). Note that the free state of PTO in electrolytes (e.g., 1 m DME/LiTFSI) exhibits an orange solution. The purple color shown in the cuvettes can be attributed to the side reaction between the dissolved PTO and the lithium metal anode (Fig. S20†). Furthermore, to rule out the possibility of lithiated PTO dissolution in the electrolytes, we evaluated the solubility of the PTO cathode in 1 m LiTFSI/DME at 3.0 V (pristine PTO), 2.4 V (partially lithiated PTO) and 1.5 V (fully lithiated PTO), respectively. The UV-vis spectra demonstrate that the solubility of lithiated PTO is negligible, indicating that the dissolution of pristine PTO is a key factor affecting the performance of Li–PTO batteries (Fig. S21†).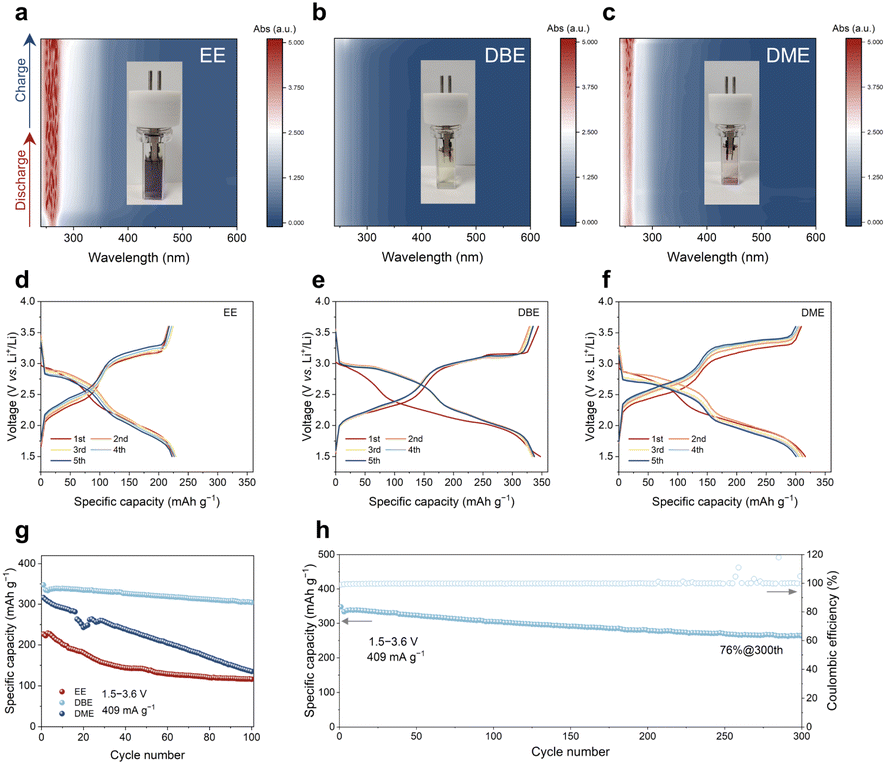 | ||
| Fig. 5 In situ UV-vis spectra of Li‖PTO batteries using (a) 1 m LiTFSI/EE, (b) 1 m LiTFSI/DBE and (c) 1 m LiTFSI/DME electrolytes. Discharge and charge profiles of PTO at different cycles with (d) 1 m LiTFSI/EE, (e) 1 m LiTFSI/DBE and (f) 1 m LiTFSI/DME electrolytes. (g) Cycling performance of PTO in different electrolytes at 1C during first 100 cycles. (h) Cycling performance of PTO in 1 m LiTFSI/DBE electrolyte at 1C during 300 cycles. | ||
Next, we compared the cycling performances of Li–PTO batteries using 1 m LiTFSI/EE, LiTFSI/DBE and LiTFSI/DME electrolytes, respectively. The linear sweep voltammetry (LSV) curves in Fig. S22† indicate that all electrolytes fulfill the requirements for Li–PTO battery operation, as their anodic decomposition potential exceed 4.5 V. The discharge and charge profiles for the first five cycles are shown in Fig. 5d–f, where all curves exhibit clear and reversible two discharge/charge plateaus. The initial discharge capacities of PTO in 1 m LiTFSI/DME and 1 m LiTFSI/DBE electrolytes are above 320 mA h g−1 at 1C (1C = 409 mA g−1). However, the PTO electrode with 1 m LiTFSI/EE electrolyte exhibits a much lower initial capacity of 227 mA h g−1. Note that there are notable differences in the capacity voltage curves of PTO in different electrolytes. As shown in Fig. S23,† the charge/discharge voltage plateaus of PTO in 1 m LiTFSI/DBE electrolyte are significantly lower/higher than those of PTO in 1 m LiTFSI/EE and 1 m LiTFSI/DME electrolytes. This difference may be related to the differences in solid–liquid redox reactions and solid–solid redox reactions. The Li–PTO batteries using 1 m LiTFSI/EE and LiTFSI/DME electrolytes experience significant capacity degradation during cycling (Fig. 5g), primarily due to the severe dissolution of PTO. The gradual loss of active mass from the cathode into the electrolyte and onto the lithium metal anode leads to ‘shuttle reactions’, fast capacity decay and fluctuations in coulombic efficiency (Fig. S24†). In contrast, PTO exhibits superior reversibility of electrochemical redox reactions in 1 m LiTFSI/DBE electrolyte, achieving a high initial capacity of 350 mA h g−1 and a good capacity retention of 76% after 300 cycles at 1C (Fig. 5h). This performance is at a leading level compared to previously reported Li–organic batteries (Table S1†). Notably, to further improve the performance of Li–PTO batteries, the single-solvent electrolyte used in this study can be extended to a multi-solvent electrolyte by adding co-solvents or additives.
2.5 The unified dissolution picture of organic electrode materials
The mechanisms described above can be integrated to build a unified picture of the dissolution of organic electrode materials in electrolytes. In this framework, the solvent dielectric constant, ε, is shown to be the critical parameter governing the dissolution process. Driven by the competition between the interactions of organic electrodes with ion-solvation structures and free solvents, the dissolution can be classified into three cases (Fig. 6): | ||
| Fig. 6 Schematic diagram for the dissolution mechanisms of organic electrode materials. | ||
(i) Non-polar solvents (e.g., EE): in this scenario, the increase in the proportion of ion-solvation structures (primarily Li+-anion clusters) outweighs the decrease in the free-solvent component as the electrolyte concentration increases. This leads to an ion-solvation-dominated dissolution mechanism, where the addition of lithium salts significantly promotes the dissolution of organic electrode materials.
(ii) Weakly polar solvents (e.g., DME): here, the increase in the proportion of ion-solvation structures primarily offsets the decrease in the free solvent component, resulting in a weak dependence of solubility on electrolyte concentration.
(iii) Strongly polar solvents (e.g., EC): in this case, the increase in the free-solvent component surpasses the decrease in the component of ion-solvation structures, leading to a free-solvent-dominated dissolution mechanism. Consequently, the dissolution of organic electrodes is effectively inhibited in high-concentration electrolytes.
It is worth noting that this unified dissolution framework can be extended to electrolytes with other lithium salts (e.g., LiPF6, Fig. S25†) and to other organic cathodes (e.g., 1,4-benzoquinone (BQ), 9,10-anthraquinone (AQ) and 1,4,5,8-naphthalenetetracarboxylic dianhydride (NTCDA), Fig. S26†). Moreover, the proposed model clarifies the ongoing debate about the impact of electrolyte concentration on the dissolution of organic electrode materials, providing invaluable insights into the dissolution mechanisms of organic electrodes for Li–organic batteries.
3 Conclusions
In summary, we have proposed a unified model that elucidates the dissolution mechanism of organic electrode in various electrolytes. By combining the theoretical calculations and experimental spectroscopic characterization, we find that the competition of the interactions of organic electrodes with free solvents and ion-solvation structures in electrolytes predominantly governs the dissolution of organic cathodes. We further demonstrate that this competitive dissolution mechanism can be tuned through solvent polarity. Our study clarifies the controversy regarding whether dissolution is suppressed or enhanced in high-concentration electrolytes and explains the anomalously high solubility of organic electrodes in electrolytes with non-polar solvents. Based on the insight gained from the unified model, we propose that balancing the effects of ion-solvation structures and free solvents can be an efficient strategy to inhibit the dissolution of organic electrodes. This is beneficial for the targeted regulation of the dissolution of organic electrodes to promote further development of organic batteries.Data availability
Data are available upon request from the authors.Author contributions
S. Z. and W. X. designed the project. S. Z. and Y. Z. performed all the experiments including characterization and electrochemical tests. S. Z. and W. X. conducted theoretical calculations. S. X., Q. Z., Y. L., K. Z. and Z. Y. carried out the analyses. J. C. supervised the project. All authors discussed the results and contributed to the preparation of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFB2402200), the National Natural Science Foundation of China (22393900, 92372001, 22121005, 22203047 and 22020102002), and the Haihe Lab of ITAI (Multi-scale and Efficient Computational Platform for Advanced Functional Materials, 22HHXCJC00007).Notes and references
- J. Xiao, F. Shi, T. Glossmann, C. Burnett and Z. Liu, Nat. Energy, 2023, 8, 329–339 CrossRef.
- S. Wang, L. Wang, S. David, T. Liu, C. Zhan and K. Amine, Chem. Soc. Rev., 2024, 53, 3561–3578 RSC.
- X. Zhou, F. Hong, S. Wang, T. Zhao, J. Peng, B. Zhang, W. Fan, W. Xing, M. Zuo, P. Zhang, Y. Zhou, G. Lv, Y. Zhong, W. Hua and W. Xiang, eScience, 2024, 4, 100276 CrossRef.
- Q. Wang, Z. Yao, J. Wang, H. Guo, C. Li, D. Zhou, X. Bai, H. Li, B. Li, M. Wagemaker and C. Zhao, Nature, 2024, 629, 341–347 CrossRef CAS PubMed.
- W. Huang, T. Liu, L. Yu, J. Wang, T. Zhou, J. Liu, T. Li, R. Amine, X. Xiao, M. Ge, L. Ma, S. N. Ehrlich, M. V. Holt, J. Wen and K. Amine, Science, 2024, 384, 912–919 CrossRef CAS PubMed.
- G. Ji, J. Wang, Z. Liang, K. Jia, J. Ma, Z. Zhuang, G. Zhou and H. Cheng, Nat. Commun., 2023, 14, 584 CrossRef CAS PubMed.
- T. Cui, X. Li and Y. Fu, eScience, 2024, 4, 100245 CrossRef.
- Y. Lu and J. Chen, Nat. Rev. Chem, 2020, 4, 127–142 CrossRef CAS PubMed.
- J. Kim, Y. Kim, J. Yoo, G. Kwon, Y. Ko and K. Kang, Nat. Rev. Mater., 2023, 8, 54–70 CrossRef.
- P. Poizot, J. Gaubicher, S. Renault, L. Dubois, Y. Liang and Y. Yao, Chem. Rev., 2020, 120, 6490–6557 CrossRef CAS PubMed.
- J. Wang, A. E. Lakraychi, X. Liu, L. Sieuw, C. Morari, P. Poizot and A. Vlad, Nat. Mater., 2021, 20, 665–673 CrossRef CAS PubMed.
- J. Wang, H. Liu, C. Du, B. Liu, H. Guan, Y. Liu, S. Guan, Z. Sun and H. Yao, eScience, 2024, 4, 100224 CrossRef.
- Y. Lu, Y. Ni and J. Chen, Acc. Chem. Res., 2024, 57, 375–385 CrossRef CAS PubMed.
- Y. Chen and C. Wang, Acc. Chem. Res., 2020, 53, 2636–2647 CrossRef CAS PubMed.
- J. Geng, Y. Ni, Z. Zhu, Q. Wu, S. Gao, W. Hua, S. Indris, J. Chen and F. Li, J. Am. Chem. Soc., 2023, 145, 1564–1571 CrossRef CAS PubMed.
- S. Bai, B. Kim, C. Kim, O. Tamwattana, H. Park, J. Kim, D. Lee and K. Kang, Nat. Nanotechnol., 2021, 16, 77–84 CrossRef CAS PubMed.
- Z. Li, Q. Jia, Y. Chen, K. Fan, C. Zhang, G. Zhang, M. Xu, M. Mao, J. Ma, W. Hu and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207221 CrossRef CAS PubMed.
- X. Xu, S. Ren, H. Wu, H. Li, C. Ye, K. Davey and S.-Z. Qiao, J. Am. Chem. Soc., 2024, 146, 1619–1626 CrossRef CAS PubMed.
- Y. Chen, J. Li, Q. Zhu, K. Fan, Y. Cao, G. Zhang, C. Zhang, Y. Gao, J. Zou, T. Zhai and C. Wang, Angew. Chem., Int. Ed., 2022, 61, e202116289 CrossRef CAS PubMed.
- H. Peng, J. Xiao, Z. Wu, L. Zhang, Y. Geng, W. Xin, J. Li, Z. Yan, K. Zhang and Z. Zhu, CCS Chem., 2023, 5, 1789–1801 CrossRef CAS.
- Z. Xiong, L. Gu, Y. Liu, H. Wang, L. Shi, X. Wu, L. Liu and Z. Chen, CCS Chem., 2024, 6, 2835–2844 CrossRef CAS.
- Y. Lu, Z. Yang, Q. Zhang, W. Xie and J. Chen, J. Am. Chem. Soc., 2023, 146, 1100–1108 CrossRef PubMed.
- M. Li, R. P. Hicks, Z. Chen, C. Luo, J. Guo, C. Wang and Y. Xu, Chem. Rev., 2023, 123, 1712–1773 CrossRef CAS PubMed.
- H. Dong, O. Tutusaus, Y. Liang, Y. Zhang, Z. Lebens-Higgins, W. Yang, R. Mohtadi and Y. Yao, Nat. Energy, 2020, 5, 1043–1050 CrossRef CAS.
- M. Lee, J. Hong, J. Lopez, Y. Sun, D. Feng, K. Lim, W. C. Chueh, M. F. Toney, Y. Cui and Z. Bao, Nat. Energy, 2017, 2, 861–868 CrossRef CAS.
- T. Cai, Y. Han, Q. Lan, F. Wang, J. Chu, H. Zhan and Z. Song, Energy Storage Mater., 2020, 31, 318–327 CrossRef.
- L. Liu, Z. Shadike, N. Wang, Y. Chen, X. Cai, E. Hu and J. Zhang, eScience, 2024, 4, 100268 CrossRef.
- H. W. Lee, Y. Kim, J. E. Kim, J. Y. Kim, J. Y. Jang, J. Choi and W. J. Kwak, Adv. Energy Mater., 2024, 14, 2303033 CrossRef CAS.
- V. W. h. Lau, I. Moudrakovski, J. Yang, J. Zhang and Y. M. Kang, Angew. Chem., Int. Ed., 2020, 132, 4052–4063 CrossRef.
- W. Zhang, H. Sun, P. Hu, W. Huang and Q. Zhang, EcoMat, 2021, 3, e12128 CrossRef CAS.
- L. Sieuw, A. Jouhara, É. Quarez, C. Auger, J.-F. Gohy, P. Poizot and A. Vlad, Chem. Sci., 2019, 10, 418–426 RSC.
- E. Stefanis and C. Panayiotou, Int. J. Thermophys., 2008, 29, 568–585 CrossRef CAS.
- P. Xiao, X. Yun, Y. Chen, X. Guo, P. Gao, G. Zhou and C. Zheng, Chem. Soc. Rev., 2023, 52, 5255–5316 RSC.
- N. Yao, X. Chen, X. Shen, R. Zhang, Z. H. Fu, X. X. Ma, X. Q. Zhang, B. Q. Li and Q. Zhang, Angew. Chem., Int. Ed., 2021, 133, 21643–21648 CrossRef.
- Z. Yu, P. E. Rudnicki, Z. Zhang, Z. Huang, H. Celik, S. T. Oyakhire, Y. Chen, X. Kong, S. C. Kim, X. Xiao, H. Wang, Y. Zheng, G. A. Kamat, M. S. Kim, S. F. Bent, J. Qin, Y. Cui and Z. Bao, Nat. Energy, 2022, 7, 94–106 CrossRef CAS.
- Y. Chen, Z. Yu, P. Rudnicki, H. Gong, Z. Huang, S. C. Kim, J.-C. Lai, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2021, 143, 18703–18713 CrossRef CAS PubMed.
- H. Fu, H. Chen, M. Blazhynska, E. Goulard Coderc de Lacam, F. Szczepaniak, A. Pavlova, X. Shao, J. C. Gumbart, F. Dehez, B. Roux, W. Cai and C. Chipot, Nat. Protoc., 2022, 17, 1114–1141 CrossRef CAS PubMed.
- L. F. Song and K. M. Merz Jr, J. Chem. Inf. Model., 2020, 60, 5308–5318 CrossRef CAS PubMed.
- S. Grimme, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 211–228 CAS.
- R. A. Pratiwi and A. B. D. Nandiyanto, Indonesian J. Educ. Res. Technol., 2022, 2, 1–20 CrossRef.
- K. Chen, X. Shen, L. Luo, H. Chen, R. Cao, X. Feng, W. Chen, Y. Fang and Y. Cao, Angew. Chem., Int. Ed., 2023, 62, e202312373 CrossRef CAS PubMed.
- J. Wang, P. Apostol, D. Rambabu, X. Guo, X. Liu, K. Robeyns, M. Du, Y. Zhang, S. Pal, R. Markowski, F. Lucaccioni, A. E. Lakraychi, C. Morari, J.-F. Gohy, D. Gupta and A. Vlad, Sci. Adv., 2023, 9, eadg6079 CrossRef CAS PubMed.
- Y. Huang, C. Fang, W. Zhang, Q. Liu and Y. Huang, Chem. Commun., 2019, 55, 608–611 RSC.
- C. Guo, K. Zhang, Q. Zhao, L. Pei and J. Chen, Chem. Commun., 2015, 51, 10244–10247 RSC.
- J. Holoubek, H. Liu, Z. Wu, Y. Yin, X. Xing, G. Cai, S. Yu, H. Zhou, T. A. Pascal, Z. Chen and P. Liu, Nat. Energy, 2021, 6, 303–313 CrossRef CAS PubMed.
- L.-Q. Wu, Z. Li, Z.-Y. Fan, K. Li, J. Li, D. Huang, A. Li, Y. Yang, W. Xie and Q. Zhao, J. Am. Chem. Soc., 2024, 146, 5964–5976 CrossRef CAS PubMed.
- N. Yao, X. Chen, Z.-H. Fu and Q. Zhang, Chem. Rev., 2022, 122, 10970–11021 CrossRef CAS PubMed.
- Y. Chen, Z. Ma, Y. Wang, P. Kumar, F. Zhao, T. Cai, Z. Cao, L. Cavallo, H. Cheng, Q. Li and J. Ming, Energy Environ. Sci., 2024, 17, 5613–5626 RSC.
- Q. Li, Z. Cao, W. Wahyudi, G. Liu, G.-T. Park, L. Cavallo, T. D. Anthopoulos, L. Wang, Y.-K. Sun, H. N. Alshareef and J. Ming, ACS Energy Lett., 2020, 6, 69–78 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4sc07932a |
| This journal is © The Royal Society of Chemistry 2025 |