
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotomediated core modification of organic photoredox catalysts in radical addition: mechanism and applications†
Yajun
Zhang
a,
Dandan
Jiang
b,
Zheng
Fang
 ac,
Ning
Zhu
*a,
Naixian
Sun
a,
Wei
He
a,
Chengkou
Liu
a,
Lili
Zhao
*b and
Kai
Guo
*ac
ac,
Ning
Zhu
*a,
Naixian
Sun
a,
Wei
He
a,
Chengkou
Liu
a,
Lili
Zhao
*b and
Kai
Guo
*ac
aCollege of Biotechnology and Pharmaceutical Engineering, Nanjing Tech University, 30 Puzhu Rd S., Nanjing 211816, China. E-mail: ningzhu@njtech.edu.cn; guok@njtech.edu.cn; Fax: +86 2558139901; Tel: +86 25581399301
bInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Nanjing Tech University, 30 Puzhu Rd S., Nanjing 211816, China. E-mail: ias_llzhao@njtech.edu.cn
cState Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Tech University, 30 Puzhu Rd S., Nanjing 211816, China
First published on 10th June 2021
Abstract
Dihydrophenazines and their analogues have been widely used as strong reducing photoredox catalysts in radical chemistry, such as organocatalyzed atom transfer radical polymerization (O-ATRP). However, when dihydrophenazines were employed as organic photoredox catalysts (OPCs) to mediate O-ATRP, the initiator efficiency was nonquantitative due to cross-coupling between dihydrophenazines and radical species. Here, a new kind of core modification for dihydrophenazines, phenoxazines and phenothiazines was developed through this cross-coupling process. Mechanistic studies suggested that the radical species would be more likely to couple with OPC' radical cations rather than the ground-state OPC. Core modification of OPCs could stabilize the radical ions in an oxidative quenching catalytic cycle. Significantly, core modifications of OPCs could lower the energy of light required for photoexcitation. Compared with their noncore-modified counterparts, all the core-modified dihydrophenazines and phenoxazines exhibited efficient performance in controlling O-ATRP for the synthesis of poly(methyl methacrylate) with higher initiator efficiencies under the irradiation of simulated sunlight.
1. Introduction
The revival of radical chemistry in organic synthesis during the past decade has initiated a resurgence in interest in photoredox catalysis. The employment of photoredox catalysis has been demonstrated to be a promising strategy in the discovery and optimization of new synthetic methodologies.1–6 Ruthenium and iridium polypyridyl complexes, which were firstly demonstrated as photoredox catalysts and stood at the forefront of transition metal chromophores, were both capable of engaging in driving chemical transformations through the generation of reactive open-shell species via photoexcitation.5,7–10 Despite the success of ruthenium and iridium-containing photoredox catalysts, there were still some limitations in using these transition metal complexes. For example, when ruthenium and iridium-containing photoredox catalysts were used in polymer synthesis,3,11,12 they suffered from purification challenges for polymer products. Organocatalyzed methods are thus highly desirable for circumventing the challenge of metal contamination.13–16 Moreover, the use of organic photoredox catalysts (OPCs) provides access to new synthetic methodologies that are difficult to realize by other means.17One of the important applications of organic photoredox catalysis is organocatalyzed atom transfer radical polymerization (O-ATRP), a powerful tool for the synthesis of polymers with targeted molecular weights (Mw) and low molecular weight dispersities (Đ).18 Hawker15 and Matyjaszewski groups19 first demonstrated that phenothiazines were strong reducing OPCs for the polymerization of methyl methacrylate (MMA). Further developments were recently reported by the Miyake group,20–25 wherein dihydrophenazines and phenoxazines were effective OPCs for O-ATRP under visible-light irradiation. Since the seminal reports of these OPCs, noncore-modified dihydrophenazines, phenoxazines and phenothiazines were further demonstrated as successful OPCs for O-ATRP (Table S3†). The proposed O-ATRP mechanism mediated by these strong reducing OPCs followed an oxidative quenching pathway (Fig. 1A).26 After effective light absorption, the photoexcited OPCs (1PC* or 3PC*) directly reduce an alkyl bromide through an outer sphere electron transfer to give the active radical for polymerization propagation, as well as the radical cation (2PC˙+). The deactivation of the propagating radical requires the 2PC˙+ species to regenerate the ground state PC and a dormant polymer.
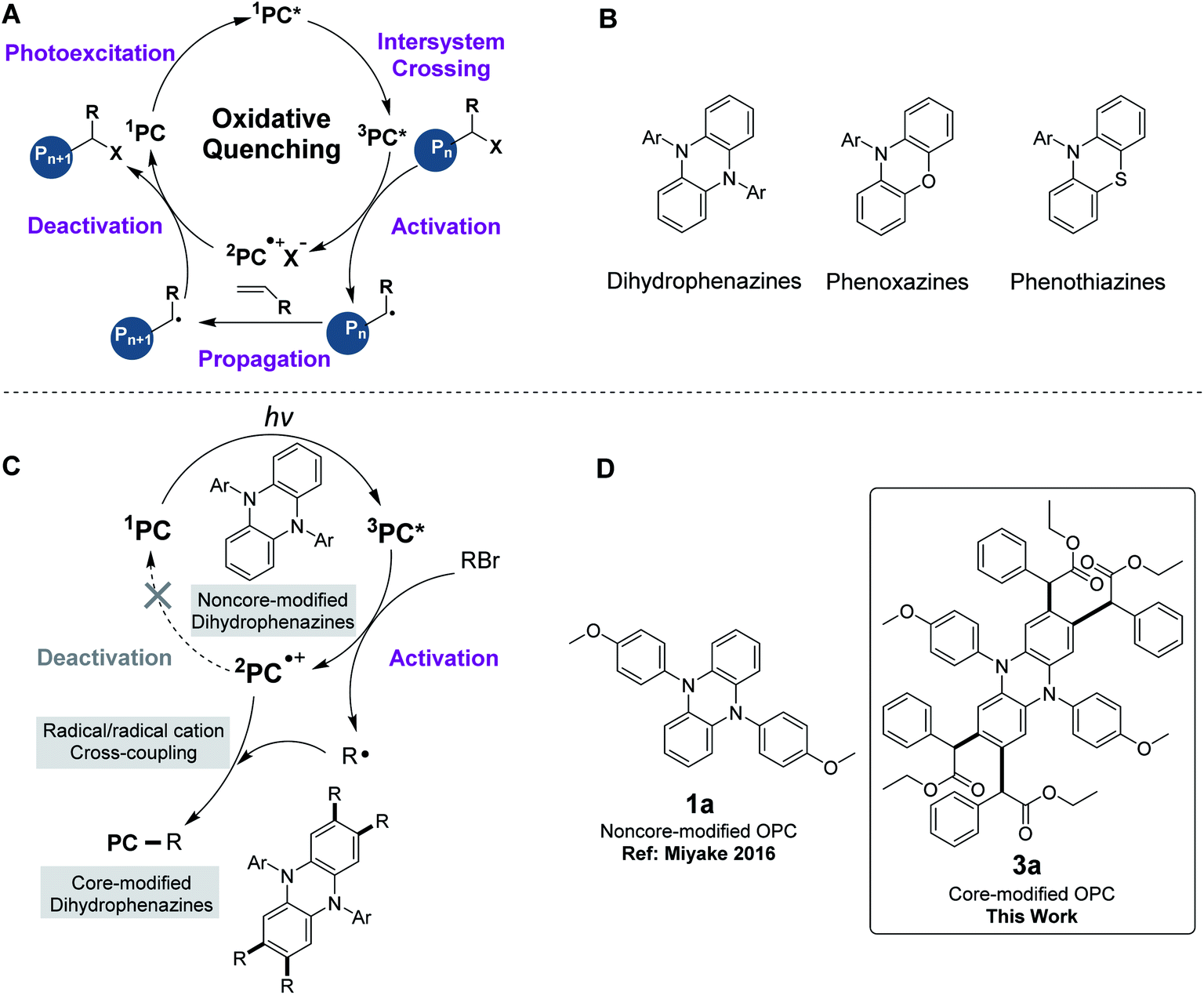 | ||
| Fig. 1 Core modification of OPCs in O-ATRP and core-modified OPCs developed in this work. (A) Proposed mechanism of O-ATRP. (B) Organic photoredox catalysts. (C) Proposed mechanism for core modification of dihydrophenazines. (D) The structures of noncore-modified OPC 1a (Miyake 2016) and core-modified OPC 3a (this work). | ||
However, when 5,10-diaryl-5,10-dihydrophenazines were used as OPCs in O-ATRP, the initiator efficiencies only reached ∼60–80%.20,21 We hypothesized that the cross-coupling between dihydrophenazines and radical species might contribute to the premature radical termination, which resulted in low initiator efficiencies in O-ATRP. To prove this hypothesis, we employed 5,10-diaryl-5,10-dihydrophenazines (1a to 1c) as OPCs for the polymerization of MMA under simulated sunlight at room temperature. To our surprise, the corresponding tetrasubstituted core-modified OPCs (3a, 3b and 3c) were detected and all noncore-modified OPCs were consumed during the early period of O-ATRP (Table S2,† entries 1–3). Noncore-modified dihydrophenazines could couple with the radical species at the 2-, 3-, 7- and 8-positions of the phenazine core to produce tetrasubstituted OPCs, which could still be applied as catalysts in the O-ATRP of MMA.
This undesirable core modification of OPCs in O-ATRP might cause the changes of the photophysical and electrochemical properties of the OPCs (i.e., introducing of the alkyl group into the core of OPCs), the feed ratio of polymerization (e.g., consuming part of the initiator) and proton concentration in solution (i.e., releasing HBr). Furthermore, when noncore-modified OPCs were used in the modification of polymers,27–30 they might be grafted onto macroinitiators, thus affecting the performance of the resultant products and also limiting the application of these OPCs. Very recently, a similar observation of two initiator-derived alkyl radicals with the addition of the phenazine catalyst was reported by the Miyake group.31 However, during our work in core modification of phenazines, we found that disubstituted core-modified phenazine may still be involved in this undesirable side reaction when employed as an organic photoredox catalyst to mediate ATRP. Therefore, it is particularly meaningful to explore the detailed reaction mechanism of the core modification of OPCs in the whole catalysis and develop a series of tetrasubstituted core-modified OPCs.
Herein, a new method of core modification for dihydrophenazines through a radical/radical cation cross-coupling process was developed and a series of tetrasubstituted core-modified OPCs were prepared. Phenoxazines and phenothiazines, similar to dihydrophenazines, were also investigated, which could react with alkyl halides to produce a core-modified product. Core modification of OPCs could stabilize radical ions in an oxidative quenching catalytic cycle. Compared with noncore-modified counterparts, all the corresponding core-modified dihydrophenazines and phenoxazines could be applied as effective catalysts for O-ATRP to produce polymers with higher initiator efficiency.
2. Results and discussion
2.1. Core modification of dihydrophenazines
 | ||
| Fig. 2 Substrate scope of core modification of dihydrophenazines. Reaction conditions: a reaction mixture of 1 (100 mg, 1.0 equiv.) and 2 (10 equiv.) in 1,4-dioxane (10 mL) was irradiated with a 300 W simulative sunshine bulb at room temperature under N2 for 5 h; isolated yields are shown. aStructure of 3a obtained by X-ray crystallography; H atoms are omitted for clarity. Color code, C: black, O: red, and N: dark blue (CCDC 2043491†). | ||
 | ||
| Fig. 3 Proposed mechanisms for the reaction between 1a and 2a. The proposed mechanism was simplified as a monosubstituted core modification of 1a. Pathway A: the monosubstituted core-modified product IV was formed via a radical/radical cation cross-coupling process. Pathway B: the monosubstituted core-modified product IV was formed via cross-coupling between 1a and radical generated by 2a. | ||
As shown in Fig. 4A, Cu/Me6-TREN, which was usually used for the activation of alkyl halides,38 was selected as a catalyst system to activate 2a to generate the corresponding radical species I in darkness. However, no desired product was detected, implying that the generated radical species I cannot react with the substrate 1a. This observation indicated that cross-coupling between 1a and radical species I was difficult, which suggested that the pathway B might be less feasible. This is further supported by density functional theory (DFT) calculations at the BP86+D3(BJ)/def2-TZVPP (CPCM, SOL = DMAC)//BP86+D3(BJ)/def2-SVP (CPCM, SOL = DMAC) level of theory. As detailed in Fig. 4B, the reaction starts with the excitation of the photoredox catalyst 1a by sunlight giving the triplet excited-state complex 31a* and is endergonic by 44.5 kcal mol−1, which is comparable to previously reported values.19,39 With the addition of the substrate 2a, the intermediate 1a˙+Br− and the radical species I are generated, which are 7.2 kcal mol−1 less stable than the initial reactants. After crossing a small barrier of 6.2 kcal mol−1 (i.e., 1a˙+Br− + I → TS1), the carbon–carbon coupling step is completed to generate the slightly more stable intermediate IM1, which is 4.7 kcal mol−1 more stable than the initial reactants. Subsequently, HBr could be easily released from IM1 to give the monosubstituted core-modified product IV. The whole process is exoergic by 5.2 kcal mol−1, implying a thermodynamically feasible reaction course. The following three equivalent substrate 2a addition steps, which will finally lead to the final product 3a, proceed in a similar reaction course. The whole reaction, from 1a and 2a to a tetrasubstituted product 3a, is exoergic by 30.4 kcal mol−1, which can provide thermodynamic driving force for the reaction to proceed. As shown by the comparison in Fig. 4B, the barrier for the carbon–carbon coupling step via the transition state TS1′ (i.e., pathway B) is 10.1 kcal mol−1 higher than that of TS1 (i.e., pathway A), implying that the radical species I is more likely to couple with 1a˙+Br−. Hence, the radical/radical cation cross-coupling process via pathway A is kinetically more favorable.
 | ||
| Fig. 4 Mechanistic investigations. (A) Control experiments. (B) Computed Gibbs energy profile (ΔG in kcal mol−1) and electronic energy [ΔE in brackets] at the BP86+D3(BJ)/def2-TZVPP (CPCM, SOL = DMAC)//BP86+D3(BJ)/def2-SVP (CPCM, SOL = DMAC) of the reaction. The proposed mechanism is simplified as a monosubstituted core modification of 1a. | ||
2.2. Core modification of phenoxazines and phenothiazines
Noncore-modified phenoxazines and phenothiazines, possessing a conformation similar to that of dihydrophenazines, have been widely used as sufficiently strong reducing OPCs in O-ATRP.15,19,24,28,29,40–43 We thus wondered whether the radial/radial cation cross-coupling process could also be applied in the core modification of phenoxazines and phenothiazines at the 3- and 7-positions of the core of the corresponding OPCs. Simplified computational mechanistic studies revealed that these noncore-modified OPCs can couple with radical species through the radical/radical cation cross-coupling process with reasonable barriers (e.g., <21.1 kcal mol−1), as shown by the black pathways in Fig. 5B, which was then confirmed by experimental synthesis of 6a and 7a (Fig. 5C).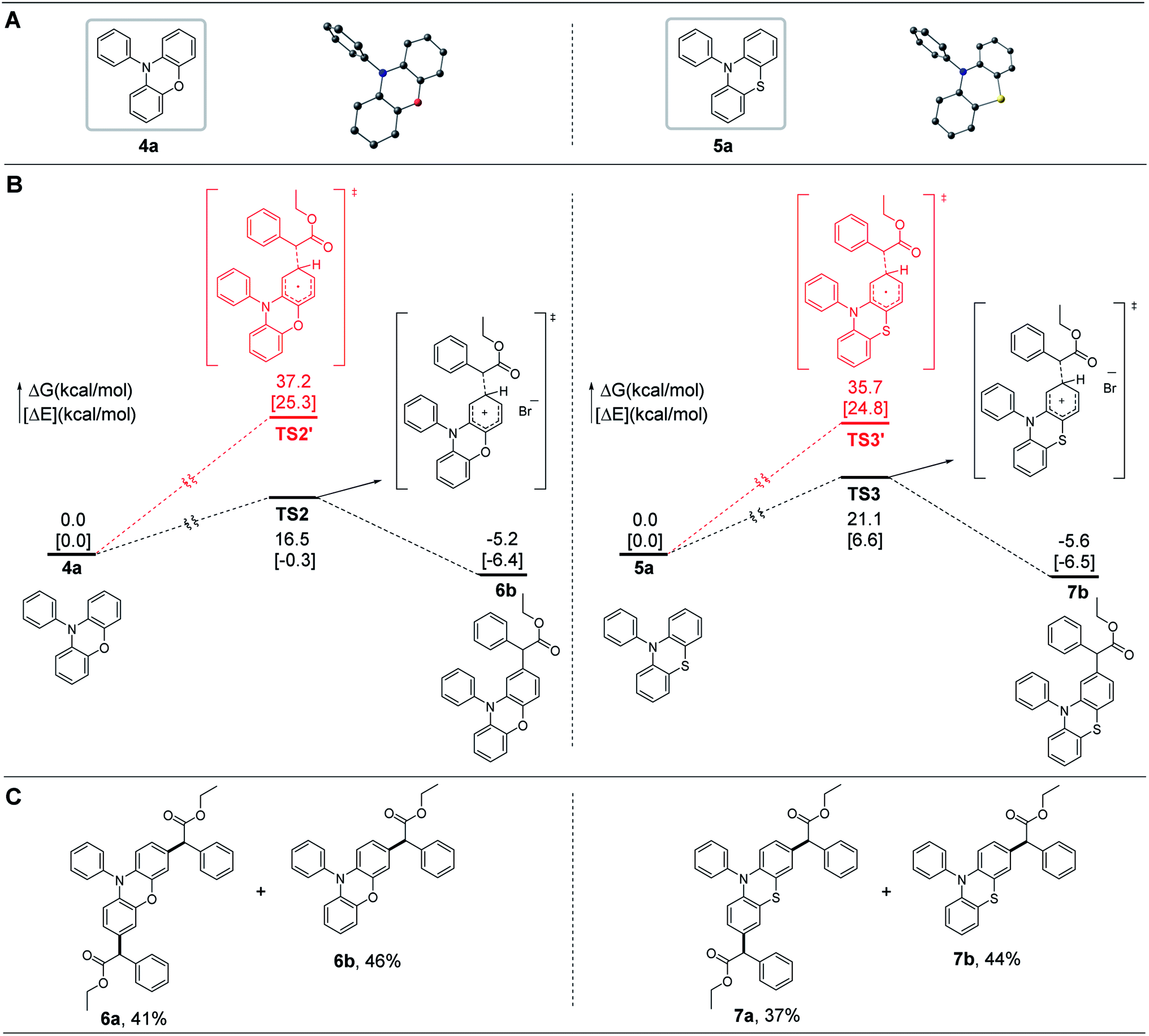 | ||
| Fig. 5 Core modification of phenoxazines and phenothiazines. (A) Molecular structures of 4a and 5a. Hydrogen atoms are omitted for clarity (color code, C: black, O: red, N: blue, and S: yellow). (B) Simplified computed Gibbs energy profile (ΔG in kcal mol−1) and electronic energy [ΔE in brackets] at the BP86+D3(BJ)/def2-TZVPP (CPCM, SOL = DMAC)//BP86+D3(BJ)/def2-SVP (CPCM, SOL = DMAC) of the reaction. (C) Core modifications of 4a and 5a. Reaction conditions: a reaction mixture of a noncore-modified OPC (4a or 5a) (100 mg, 1.0 equiv.) and 2a (3.0 equiv.) in 1,4-dioxane (10 mL) was irradiated with a 300 W simulative sunshine bulb at room temperature under N2 for 5 h; isolated yields are shown. | ||
2.3. Photophysical and electrochemical properties of OPCs
Core-modified OPCs 3, 6 and 7 all exhibit a red shift in the maximum wavelength of absorption (λmax,abs) and a significantly higher molar extinction coefficient (ε) compared to the corresponding noncore-modified OPCs (Tables 1 and S4†). Core modification of OPCs can improve the efficiency of photoexcitation with the absorption profile tailing into the lower-energy light regime. It has been reported that OPCs absorb visible light instead of ultraviolet (UV) light and can reduce the incidence of side reactions.5,23,44,45 So, it is meaningful to gain insights into the effects of these core modifications on the photon absorption of OPCs.46| OPC | λ max (nm) | ε max (M−1 cm−1) | λ em,max (nm) | E S1,exp (eV) | E 1/2(2PC˙+/1PC)c (V vs. SCE) | E 0*(2PC˙+/1PC*)d (V vs. SCE) |
|---|---|---|---|---|---|---|
| a λ max, maximum absorption wavelength; εmax, molar absorptivity at λmax. b λ em,max, maximum emission wavelength; ES1,exp, lowest singlet excited state energy determined from λem,max. c Measurements were performed in a three-compartment electrochemical cell with Ag/AgCl (submerged in 3 M KCl solution) as the reference electrode and NBu4PF6 in DMAc (0.10 M) as the electrolyte solution. d Singlet excited state reduction potentials were calculated as E0*(2PC˙+/1PC*) = E1/2(2PC˙+/1PC) − ES1,exp. More data are provided in Table S4. | ||||||
| 1a | 373 | 5600 | 467 | 2.66 | 0.29 | −2.37 |
| 3a | 383 | 8900 | 490 | 2.53 | 0.33 | −2.20 |
| 3f | 383 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
489 | 2.54 | 0.36 | −2.18 |
| 3h | 378 | 6800 | 478 | 2.59 | 0.36 | −2.23 |
| 3i | 379 | 7500 | 478 | 2.59 | 0.36 | −2.23 |
| 3j | 380 | 7700 | 477 | 2.60 | 0.36 | −2.24 |
| 4a | 324 | 7900 | 394 | 3.15 | 0.85 | −2.30 |
| 6a | 332 | 10900 |
401 | 3.09 | 0.82 | −2.27 |
| 5a | 319 | 3600 | 445 | 2.79 | 0.83 | −1.96 |
| 7a | 325 | 4300 | 455 | 2.73 | 0.83 | −1.90 |
TD-DFT calculations at the uM06/6-311+G** (CPCM, SOL = H2O)//uM06/6-31G** level of theory were performed to investigate the orbitals involved in the photoexcitation of OPCs at their corresponding λmax,abs. As shown in Fig. 6, the highest occupied molecular orbitals (HOMOs) of 1a and core-modified chromophore 3a are all localized on the phenazine core with similar orbital energies (−5.06 eV and −5.31 eV for 1a and 3a, respectively), suggesting that core modifications have no obvious interference on the energy of the πHOMO. However, the energies and shapes of the unoccupied molecular orbitals are significantly changed by core modifications. For noncore-modified OPC 1a, the energy of πLUMO+1 (LUMO, lowest unoccupied molecular orbital) involved in photoexcitation is −0.88 eV. UV-vis spectra show that the λmax,abs of PC 1a is 373 nm, which agrees with the computationally predicted result (from πHOMO into πLUMO+1 with a 70% contribution). When four alkyl substituents (equipped with phenyl or carbonyl groups) were installed onto the core of 1a, the λmax,abs red shifts from 373 to 383 nm (3a). Photonic energy at this corresponding λmax,abs is consistent with excitation from πHOMO into πLUMO+4, πLUMO+11, and πLUMO+13, with contributions of 61%, 11%, and 30%, respectively. To our surprise, the high-lying orbitals of 3a indicate that core substituents can also be involved in photon absorption. A similar trend is observed in Table 1 and Fig. S9–S11† (3h, 3i, 3j). These observations indicated that the installed alkyl groups (equipped with phenyl or carbonyl groups) play a beneficial role in the absorption of photons, permitting photoexcitation for using lower-energy visible light.
 | ||
| Fig. 6 TD-DFT calculations of orbitals and UV-Vis spectra of 1a and 3a. (Top) TD-DFT calculations and computationally predicted percentage contribution of orbitals involved in the photoexcitation of 1a and 3a at their corresponding λmax,abs. (Bottom) UV-Vis spectra of 1a and 3a acquired in DMAc. | ||
According to a modified Marcus model,18 a high reducing excited state is an essential parameter to promote the activation step in O-ATRP. However, in the design of OPCs, an inherent challenge is lowering the energy of photoexcitation without the loss of reducing power of the excited state.23 Here, the installation of alkyl groups (equipped with phenyl or carbonyl groups) onto the core of chromophore motifs was developed as a new strategy for the preparation of strong reducing OPCs. Interestingly, a series of visible light absorbing OPCs with high reducing singlet excited state reduction potentials (−2.17 V to −2.34 V versus SCE) were developed through the core modification of dihydrophenazines (Tables 1 and S4†). Subsequent to the activation process, the ground state OPC is oxidized to a radical cation (2PC˙+). In order to complete the catalytic cycle of ATRP, 2PC˙+ must have the capacity to oxidize the propagating radical (approximately −0.8 V vs. SCE for the alkyl bromides commonly employed in ATRP) and regenerate 1PC sufficiently.20 As shown in Tables 1 and S4,† core modification of dihydrophenazines has no obvious interference on E1/2, and the 2PC˙+ species of core-modified OPCs all possess appropriate E1/2(2PC˙+/1PC) (E1/2(2PC˙+/1PC) > −0.8 V vs. SCE).
Briefly, these core modifications of OPCs can lower the energy of light needed for photoexcitation but with little effect on the reducing power of the excited state and the oxidizing power of the radical cation. Similar trends are observed in the core modification of phenoxazines and phenothiazines (Tables 1 and S4†).
2.4. Applications of core-modified OPCs in O-ATRP
O-ATRP is a promising variant of traditional ATRP mediated by transition-metal catalysts, for this polymerization method to overcome the challenge of metal contamination.15,20,24,47–49 Noncore-modified dihydrophenazines, phenoxazines and phenothiazines were widely used as OPCs in O-ATRP (Table S3†). However, when these chromophores were used as catalysts in the polymerization of olefines, these OPCs could couple with radical species generated by initiators through a radical/radical cation cross-coupling process (vide supra). So, core modification of OPCs could stabilize the radical ions in the catalytic cycle of O-ATRP.Therefore, it is particularly meaningful to develop a family of core-modified OPCs. The core-modified OPCs (3, 6a, and 7a) were applied as strong reducing catalysts in O-ATRP of MMA. Compared with their noncore-modified counterparts, the core-modified dihydrophenazines and phenoxazines exhibited efficient performance in controlling the polymerization with higher initiator efficiencies, whereas the core-modified phenothiazines did not (Tables 2, S6 and S7†). 3b, 3d–3i, and 3l provided the best performance in controlling the O-ATRP of MMA with low Đ (Đ ≤ 1.12). Notably, when the disubstituted OPC 3n was used in O-ATRP, the tetrasubstituted compound S-2 was detected in the reaction (Fig. S4†), which highlights the importance of core modification for OPCs once again.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| OPC | Time (h) | Convb (%) | M w (kDa) | M n (kDa) | Đ (Mw/Mn) | M n,theo (kDa) | I*f (%) |
|
a All polymerizations were conducted using ethyl α-bromophenylacetate (EBP) as the initiator in a ratio of 1000:10:1 of [MMA]:[EBP]:[PC] with DMAc as a solvent.
b Calculated from 1H NMR results.
c Detected by SEC.
d All calculated by using Mw/Mn.
e
M
n,theo calculated by using [MMA]/[EBP] × MW of monomer × yield + MW of initiator.
f
I* = Mn,theo/Mn,exp.
g The polymerizations were conducted under a 40 W 365 nm LED. More data are provided in Table S6.
|
|||||||
| 1a | 6 | 74.3 | 11.2 | 9.13 | 1.23 | 7.71 | 84 |
| 1b | 6 | 70.0 | 10.5 | 8.51 | 1.23 | 7.38 | 87 |
| 1c | 6 | 64.5 | 15.4 | 13.5 | 1.15 | 8.81 | 65 |
| 1d | 6 | 56.0 | 19.3 | 18.0 | 1.07 | 14.5 | 81 |
| 3a | 6 | 88.2 | 16.6 | 13.5 | 1.16 | 11.6 | 86 |
| 3b | 6 | 85.1 | 13.1 | 12.0 | 1.09 | 13.5 | 113 |
| 3c | 6 | 84.9 | 12.9 | 11.3 | 1.14 | 15.6 | 138 |
| 3d | 6 | 54.3 | 13.6 | 12.8 | 1.06 | 10.7 | 84 |
| 3f | 6 | 73.6 | 16.1 | 14.7 | 1.09 | 14.7 | 100 |
| 3g | 6 | 77.7 | 16.3 | 15.0 | 1.09 | 13.9 | 93 |
| 3h | 6 | 71.6 | 16.1 | 14.5 | 1.11 | 12.8 | 88 |
| 3i | 6 | 97.6 | 16.5 | 14.7 | 1.12 | 12.6 | 86 |
| 3l | 6 | 86.5 | 18.1 | 16.6 | 1.09 | 14.3 | 86 |
| 4a | 10 | 75.7 | 15.7 | 13.6 | 1.15 | 13.0 | 96 |
| 6a | 10 | 67.0 | 16.7 | 14.3 | 1.17 | 14.0 | 98 |
3. Conclusions
In summary, photomediated core modification of dihydrophenazines, phenoxazines and phenothiazines was developed through a radical/radical cation cross-coupling process. A series of core-modified OPCs were produced in moderate to good yields. Experimental and computational mechanistic studies revealed that the radical species generated by alkyl halides were more likely to couple with the OPCs' radical cation rather than ground-state OPCs. Core modification of OPCs could stabilize the radical ions in an oxidative quenching catalytic cycle. The photophysical and electrochemical properties of these prepared core-modified OPCs were further investigated. Core modifications of OPCs can lower the energy of light needed for photoexcitation with little effect on the reducing power of the excited state and the oxidizing power of the radical cation. These core-modified chromophores were strong reducing OPCs for O-ATRP. Except for phenothiazines, core-modified dihydrophenazines and phenoxazines enabled improved initiator efficiencies in the O-ATRP of MMA. The polymerizations mediated by these core-modified OPCs were conducted in a well-controlled fashion and polymers with low Đ were obtained. We believe that our work in core modification of OPCs would broaden the application of these OPCs in O-ATRP/other radical addition reactions and provide guidance for the development of more reasonable catalyst design strategies.Data availability
All experimental and computational data has been included in the article and supporting information. No more data associated with this article will be provided.Author contributions
Y. Z. performed the experiments and wrote the manuscript; D. J. conducted the computational work; Z. F., N. S., W. H. and C. L. participated in the experiment; Y. Z., N. Z., L. Z. and K. G. analyzed the data; N. Z., L. Z. and K. G. conceived the idea and directed the project; all authors made contributions to the revision of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by the National Key Research and Development Program of China (2016YFB0301501), the National Science Foundation of China (21776130, 21878145, and 21973044), the Jiangsu Synergetic Innovation Center for Advanced Bio-Manufacture (X1821 and X1802), the Postgraduate Research & Practice Innovation Program of Jiangsu Province, and Nanjing Tech University (39837123 and 39837132). We also appreciated the high performance center of Nanjing Tech University for supporting the computational resources.Notes and references
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 Search PubMed.
- M. Chen, M. Zhong and J. A. Johnson, Chem. Rev., 2016, 116, 10167–10211 Search PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 Search PubMed.
- Q. Michaudel, V. Kottisch and B. P. Fors, Angew. Chem., Int. Ed., 2017, 56, 9670–9679 Search PubMed.
- C. K. Prier, D. A. Rankic and D. W. MacMillan, Chem. Rev., 2013, 113, 5322–5363 Search PubMed.
- D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 Search PubMed.
- D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, Tetrahedron Lett., 1978, 14, 1255 Search PubMed.
- T. J. Bergen, D. M. Hedstrand and W. H. Kruizinga, J. Org. Chem., 1979, 44, 4953–4962 Search PubMed.
- J. M. Narayanam and C. R. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 Search PubMed.
- J. J. Devery, J. J. Douglas, J. D. Nguyen, K. P. Cole, R. A. Flowers and C. R. J. Stephenson, Chem. Sci., 2015, 6, 537–541 Search PubMed.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 Search PubMed.
- N. Zivic, M. Bouzrati-Zerelli, A. Kermagoret, F. Dumur, J.-P. Fouassier, D. Gigmes and J. Lalevée, ChemCatChem, 2016, 8, 1617–1631 Search PubMed.
- K. Matyjaszewski and N. V. Tsarevsky, J. Am. Chem. Soc., 2014, 136, 6513–6533 Search PubMed.
- S. Shanmugam and C. Boyer, J. Am. Chem. Soc., 2015, 137, 9988–9999 Search PubMed.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 Search PubMed.
- A. P. Vogt and B. S. Sumerlin, Soft Matter, 2009, 5, 2347–2351 Search PubMed.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 Search PubMed.
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, J. Lee, P. C. Nandajan, R. Wannemacher, B. Milián-Medina, L. Lüer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794–804 Search PubMed.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 Search PubMed.
- J. C. Theriot, C.-H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 Search PubMed.
- J. P. Cole, C. R. Federico, C. H. Lim and G. M. Miyake, Macromolecules, 2019, 52, 747–754 Search PubMed.
- C. H. Lim, M. D. Ryan, B. G. McCarthy, J. C. Theriot, S. M. Sartor, N. H. Damrauer, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2017, 139, 348–355 Search PubMed.
- B. G. McCarthy, R. M. Pearson, C. H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 5088–5101 Search PubMed.
- R. M. Pearson, C. H. Lim, B. G. McCarthy, C. B. Musgrave and G. M. Miyake, J. Am. Chem. Soc., 2016, 138, 11399–11407 Search PubMed.
- S. M. Sartor, B. G. McCarthy, R. M. Pearson, G. M. Miyake and N. H. Damrauer, J. Am. Chem. Soc., 2018, 140, 4778–4781 Search PubMed.
- B. L. Buss, C. H. Lim and G. M. Miyake, Angew. Chem., Int. Ed., 2020, 59, 3209–3217 Search PubMed.
- E. H. Discekici, A. H. St Amant, S. N. Nguyen, I. H. Lee, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2018, 140, 5009–5013 Search PubMed.
- B. Narupai, Z. A. Page, N. J. Treat, A. J. McGrath, C. W. Pester, E. H. Discekici, N. D. Dolinski, G. F. Meyers, J. Read de Alaniz and C. J. Hawker, Angew. Chem., Int. Ed., 2018, 57, 13433–13438 Search PubMed.
- G. S. Park, J. Back, E. M. Choi, E. Lee and K.-s. Son, Eur. Polym. J., 2019, 117, 347–352 Search PubMed.
- N. You, C. Zhang, Y. Liang, Q. Zhang, P. Fu, M. Liu, Q. Zhao, Z. Cui and X. Pang, Sci. Rep., 2019, 9, 1869 Search PubMed.
- B. McCarthy, S. Sartor, J. Cole, N. Damrauer and G. M. Miyake, Macromolecules, 2020, 53, 9208–9219 Search PubMed.
- B. Huang, Y. Zhao, C. Yang, Y. Gao and W. Xia, Org. Lett., 2017, 19, 3799–3802 Search PubMed.
- Y. J. Mao, B. X. Wang, Q. Z. Wu, K. Zhou, S. J. Lou and D. Q. Xu, Chem. Commun., 2019, 55, 2019–2022 Search PubMed.
- G. Tu, C. Yuan, Y. Li, J. Zhang and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 15597–15601 Search PubMed.
- X. G. Wang, Y. Li, L. L. Zhang, B. S. Zhang, Q. Wang, J. W. Ma and Y. M. Liang, Chem. Commun., 2018, 54, 9541–9544 Search PubMed.
- C. Yuan, L. Zhu, C. Chen, X. Chen, Y. Yang, Y. Lan and Y. Zhao, Nat. Commun., 2018, 9, 1189 Search PubMed.
- C. Yuan, L. Zhu, R. Zeng, Y. Lan and Y. Zhao, Angew. Chem., Int. Ed., 2018, 57, 1277–1281 Search PubMed.
- D. Konkolewicz, Y. Wang, M. Zhong, P. Krys, A. A. Isse, A. Gennaro and K. Matyjaszewski, Macromolecules, 2013, 46, 8749–8772 Search PubMed.
- X. Zhao, Y. Liu, R. Zhu, C. Liu and D. Zhang, Inorg. Chem., 2019, 58, 12669–12677 Search PubMed.
- X. Hu, Y. Zhang, G. Cui, N. Zhu and K. Guo, Macromol. Rapid Commun., 2017, 38, 1700399 Search PubMed.
- G. Ramakers, G. Wackers, V. Trouillet, A. Welle, P. Wagner and T. Junkers, Macromolecules, 2019, 52, 2304–2313 Search PubMed.
- J. Wang, L. Yuan, Z. Wang, M. A. Rahman, Y. Huang, T. Zhu, R. Wang, J. Cheng, C. Wang, F. Chu and C. Tang, Macromolecules, 2016, 49, 7709–7717 Search PubMed.
- G. Zeng, M. Liu, C. Heng, Q. Huang, L. Mao, H. Huang, J. Hui, F. Deng, X. Zhang and Y. Wei, Appl. Surf. Sci., 2017, 399, 499–505 Search PubMed.
- E. Frick, A. Anastasaki, D. M. Haddleton and C. Barner-Kowollik, J. Am. Chem. Soc., 2015, 137, 6889–6896 Search PubMed.
- T. G. Ribelli, D. Konkolewicz, S. Bernhard and K. Matyjaszewski, J. Am. Chem. Soc., 2014, 136, 13303–13312 Search PubMed.
- M. V. Bobo, A. M. Arcidiacono, P. J. Ayare, J. C. Reed, M. R. Helton, T. Ngo, K. Hanson and A. K. Vannucci, ChemPhotoChem, 2021, 5, 51–57 Search PubMed.
- G. M. Miyake and J. C. Theriot, Macromolecules, 2014, 47, 8255–8261 Search PubMed.
- M. Kato, M. Kamigaito, M. Sawamoto and T. Higashimura, Macromolecules, 1995, 28, 1721–1723 Search PubMed.
- J.-S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2043491. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc02258j |
| This journal is © The Royal Society of Chemistry 2021 |