
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccessing lanthanide-based, in situ illuminated optical turn-on probes by modulation of the antenna triplet state energy†
Alexia G.
Cosby
 a,
Joshua J.
Woods
b,
Patrick
Nawrocki
c,
Thomas J.
Sørensen
c,
Justin J.
Wilson
b and
Eszter
Boros
*a
a,
Joshua J.
Woods
b,
Patrick
Nawrocki
c,
Thomas J.
Sørensen
c,
Justin J.
Wilson
b and
Eszter
Boros
*a
aDepartment of Chemistry, Stony Brook University, Stony Brook, New York, 11794, USA. E-mail: eszter.boros@stonybrook.edu
bDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York, 14853, USA
cNano-Science Center & Department of Chemistry, University of Copenhagen, Universitetsparken 5, 2100, København Ø, Denmark
First published on 14th June 2021
Abstract
Luminescent lanthanides possess ideal properties for biological imaging, including long luminescent lifetimes and emission within the optical window. Here, we report a novel approach to responsive luminescent Tb(III) probes that involves direct modulation of the antenna excited triplet state energy. If the triplet energy lies too close to the 5D4 Tb(III) excited state (20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 cm−1), energy transfer to 5D4 competes with back energy transfer processes and limits lanthanide-based emission. To validate this approach, a series of pyridyl-functionalized, macrocyclic lanthanide complexes were designed, and the corresponding lowest energy triplet states were calculated using density functional theory (DFT). Subsequently, three novel constructs L3 (nitro-pyridyl), L4 (amino-pyridyl) and L5 (fluoro-pyridyl) were synthesized. Photophysical characterization of the corresponding Gd(III) complexes revealed antenna triplet energies between 25800 and 30400 cm−1 and a 500-fold increase in quantum yield upon conversion of Tb(L3) to Tb(L4) using the biologically relevant analyte H2S. The corresponding turn-on reaction can be monitored using conventional, small-animal optical imaging equipment in presence of a Cherenkov radiation emitting isotope as an in situ excitation source, demonstrating that antenna triplet state energy modulation represents a viable approach to biocompatible, Tb-based optical turn-on probes.
500 cm−1), energy transfer to 5D4 competes with back energy transfer processes and limits lanthanide-based emission. To validate this approach, a series of pyridyl-functionalized, macrocyclic lanthanide complexes were designed, and the corresponding lowest energy triplet states were calculated using density functional theory (DFT). Subsequently, three novel constructs L3 (nitro-pyridyl), L4 (amino-pyridyl) and L5 (fluoro-pyridyl) were synthesized. Photophysical characterization of the corresponding Gd(III) complexes revealed antenna triplet energies between 25800 and 30400 cm−1 and a 500-fold increase in quantum yield upon conversion of Tb(L3) to Tb(L4) using the biologically relevant analyte H2S. The corresponding turn-on reaction can be monitored using conventional, small-animal optical imaging equipment in presence of a Cherenkov radiation emitting isotope as an in situ excitation source, demonstrating that antenna triplet state energy modulation represents a viable approach to biocompatible, Tb-based optical turn-on probes.
Introduction
The optical probe mediated detection of changes in receptor, enzyme or analyte expression can reveal disease-related biological activity such as aberrant enzymatic activity,1–4 modification of intracellular pH,5–7 production of reactive oxygen species,8 or drastic changes in the extra- and intracellular concentration of analytes such as metal cations,9–12 or inorganic salts.13 Ideally, such optical responsive probes produce a ratiometric response, but in lieu of this, a measurable turn-on effect is preferred over turn-off response, as the absence of an optical signal can be quenched by factors other than the lack of analyte. Organic chromophores have been extensively explored and established as tools for probing intra- and extracellular biochemistry. The luminescence emission from discrete lanthanide complexes provides an attractive alternative to organic chromophores for biological imaging applications.14 Lanthanide luminescence produces narrow emission bands within the optical window with diminished photobleaching, large pseudo-Stokes shifts, and long luminescent lifetimes that enable time-resolved imaging applications. However, the need of short-wave UV excitation of sensitizing antennae outside the biological optical window, especially for Tb(III), has limited the use of lanthanide-based sensing probes to in cellulo experiments. In our previous work, we have successfully demonstrated that Cherenkov radiation (CR) of positron-emissive radioisotopes can serve as an in situ excitation source and thus opens opportunities for lanthanide-based in vivo optical imaging.15,16 Motivated by these results, we hypothesized that the in situ excitation of Tb(III)-based turn-on probes in presence of a CR emissive isotope could provide means to detect the aberrant levels of biologically relevant analytes in vivo. Based on our prior work, we had determined, that CR-mediated excitation of Tb(III)-complexes low μM concentrations required Tb-based quantum yields (ΦTb) of at least 0.1 or above. We therefore proposed that a toggle between ΦTb <0.01 (“off”) and ΦTb >0.05 (“on”) could produce a detectable Tb(III) turn-on signal in the presence of a CR emissive isotope.Several strategies have previously been pursued to devise responsive, luminescent lanthanide complexes. The efficient excitation of lanthanide excited states relies on energy transfer from an organic chromophore within the immediate coordination environment of the lanthanide ions.17–20 Hence, the modification of the efficiency of this energy transfer process provides a convenient handle for turn-on probes. One approach includes the reduction of oscillatory quenching upon coordination of an analyte of interest to the first coordination sphere, effectively replacing a water molecule (Fig. 1A). This has been explored for the sensing of F−, where corresponding Eu(III) emission increases 9-fold upon binding of F−, demonstrating fluoride sensing at concentrations of as low as 20 μM.21 Inner-sphere water displacement has also been employed to detect CN−, HCO3−, and ATP.22–24 A second approach explores the caging of the organic antenna chromophore upon enzyme activity. Borbas and coworkers have demonstrated the feasibility of this approach by enzyme triggered assembly of the lanthanide-sensitizing antenna (coumarin). The formation of the coumarin antenna sensitizes Tb(III) or Eu(III) 5- to 10-fold more efficiently as compared to the benzoyl-precursor, with “turn-on” probe quantum yields of 0.010 and 0.016 for Eu(III) and Tb(III) respectively.25 A third approach involves the photoelectron transfer (PeT) mediated quenching of antenna emission (Fig. 1B). A Tb(III)-probe developed by Nagano and coworkers employed a photoelectron transfer quencher directly coordinated to the aniline antenna moiety, resulting in intraligand quenching of the antenna excited triplet state (T1).26 Upon reaction with N-acetyltransferase, the PeT quenched antenna was converted into an optically active probe producing a 10-fold increase in emission as determined by time-resolved microscopy.
 | ||
| Fig. 1 Schematic and accompanying Jablonksi diagrams of previous approaches (A and B) and our approach (C) to lanthanide-based turn-on probes for analyte or enzyme sensing. (A) Lanthanide emission is quenched through O–H oscillators until an analyte replaces the coordination water molecule.21 (B) Antenna chromophore possesses a photoelectron (PeT) quencher, limiting excitation into the excited singlet state (S1) and subsequent energy cascade to lanthanide excited state. Enzymatic cleavage releases the PeT quencher.26 (C) Our approach is the direct modulation of the antenna triplet excited state (T1), where interaction with analyte or enzyme would increase T1 to enable efficient energy transfer to the lanthanide excited state. | ||
In addition to the presence of oscillatory quenchers, caged antennae or PeT quenchers, the relative quantum yield of lanthanide complexes relies on the efficient energy transfer from the dye to the electronic energy levels of the lanthanide ion, which requires rates of energy transfer from the excited states of the antenna to the lanthanide center that can compete with the rates of the other active pathways of deactivation in the antenna.27 The importance of the relative energies of T1 and the 5D0/1 levels for Eu(III), and the 5D4 for Tb(III) has been discussed and demonstrated extensively by Latva, Raymond, Parker and others.27–30
While many literature examples have demonstrated elimination of a luminescence quencher as described above, no attempts have yet been made to modify the antenna triplet state directly and irreversibly. Parker and coworkers observed an increase in the excited triplet state energy (21300 cm−1 to 22000 cm−1) upon protonation of phenathridine-antenna Eu(III)/Tb(III) complexes (see ESI Fig. S2†).7 The ΦTb increased 27-fold to 0.025 at pH 6.5–7, although the ΦEu only increased (3-fold) upon protonation at pH 1.5, which is outside of the physiological pH range.
A significant drawback of each one of these approaches (Fig. 1 & S1†) is the low quantum yield (ΦLn) of the corresponding turn-on probes resulting in limited brightness, rendering them not applicable for bioimaging applications beyond time-resolved microscopy (for a summary of primary turn-on examples see Table S1†). Furthermore, the low ΦLn of these compounds, even in the on-state, would not produce signal in presence of a CR emitter.
Here, we propose that the triplet energy level can be adjusted by direct and irreversible functional group transformation of the antenna, providing unprecedented access to activatable lanthanide probes with comparatively high quantum yields in their corresponding turn-on state. As we demonstrate, these activatable lanthanide probes are ideal candidates for biological optical imaging with Cherenkov-radiation, owing to the high energy wavelengths (∼200–400 nm) required for excitation.
Here, we designed a library of lanthanide complexes baring antennae with varying T1 energies relative to the lanthanide excited state, allowing for subsequent and irreversible chemical modification of the antenna in a single step (i.e. reduction) to produce antennae capable of efficiently sensitizing Tb(III). Guided by density functional theory calculations of the ground state energy of antenna singlet and triplet states, we selected Tb(L3) (“off”), Tb(L4) and Tb(L5) (both “on”). Following chemical synthesis and characterization, the photophysical properties of each complex were measured, including relative T1 energy levels, quantum yields and turn-on response to a biologically relevant analyte (H2S) in presence of a CR emitter as an in situ excitation source.
Results and discussion
Molecular design and DFT
We designed and synthesized a series of terbium(III) complexes which utilize a macrocyclic chelator based on cyclen, and a pyridyl antenna which incorporates varied substituents at the para-position (Fig. 2). The chelator forms kinetically inert and highly stable Ln(III) complexes, with the coordinating pyridyl substituent forming an 8-coordinate complex.31 We proposed that tuning of the triplet state energy can be achieved by single-step chemical modification of the antenna substituent. | ||
| Fig. 2 Structures of previously investigated complexes [Tb(L1)]− and [Tb(L2)]−, leading to the design and of Tb(L3)–Tb(L5) featured in this study. | ||
According to Latva's empirical principle, the lowest energy antenna triplet excited state should be at least 1800 cm−1 above the photoluminescent lanthanide excited state.27 If the change in energy between these states is too low, back energy transfer processes become more probable, leading to severely diminished lanthanide-based radiative de-excitation and increased ligand-based phosphorescence/fluorescence. Modulation of triplet energy of pyridyl and picolinate-based antennas can be induced by the incorporation of electron-withdrawing substituents such as nitro, cyano or carbonyl groups which are expected to lower the relative ligand triplet energy. This has been observed previously by Faulkner and Sherry, who reported a nitro-substituted antennae with decreased lanthanide luminescence.32,33 In contrast, electron-donating functional groups such as amines, effectively increase the ligand triplet state energy.29
In our previous study, we observed a 1.4-fold increase in ΦTb with a picolinate antenna incorporating an amine in the para position [Tb(L2)]−, when compared to the corresponding picolinate antenna [Tb(L1)]− (Fig. 2).16 We attributed this increase to the 1330 cm−1 blue-shift of the triplet excited state energy for [Tb(L2)]− relative to [Tb(L1)]−, which decreases the likelihood of a back energy transfer to the antenna. The high quantum yield of [Tb(L2)]− motivated the design of an antenna that possessed a poor triplet energy match and could be transformed into the para-NH2-picolinate for a particularly efficient luminescence turn-on. Inspired by work from others on nitro-to-amine reduction mediated sensing with organic fluorophores, we proposed a nitro-moiety could operate as the “off” state.34–38 Synthetic challenges to furnish the nitro-picolinate precursor (ethyl 6-(bromomethyl)-4-nitro-2-pyridinecarboxylate) derivative of [Tb(L2)]− in sufficient quantities led to the design of Tb(L3), a more readily accessible coordination complex. In addition to the electron-donating amine of Tb(L4), we hypothesized that a fluoro-substituted complex Tb(L5) could also provide a sufficiently blue-shifted T1 relative to the nitro-moiety.
To predict the relative T1 energies of the Tb(III) complexes in the presence of the antenna bearing different substituents, we carried out density functional calculations (DFT) at the TPSSh/LCRECP/6-31G(d,p) level of theory. The complexes were optimized in the ground state singlet state (GS) in gas phase and the T1 energies were estimated using the adiabatic-transition approach, where the energy difference between T1 and GS corresponds to the optimized geometry of each state. This method gives the lowest energy difference between the S0 and T1 states, corresponding to the 0–0 emission energy. In contrast, the commonly used vertical approach, where T1 and the S0 share the same geometry, gives a larger energy gap, since the calculated T1 state exists in a vibrationally excited state.39–41 Solvation effects were included in the calculations using the solvent model based on density (SMD) method and the previously optimized polarizable continuum model (PCM) radius was used for Tb(III) in all solvation calculations without scaling (α = 1.0).42 Spin density plots of the optimized T1 state for all of the complexes confirmed that the spin density was located on the antenna arm of the ligand (Fig. 3A). In agreement with our hypothesis, the calculated T1 energy of Tb(L3) is significantly lower than that of Tb(L4) and Tb(L5) (Table 1).
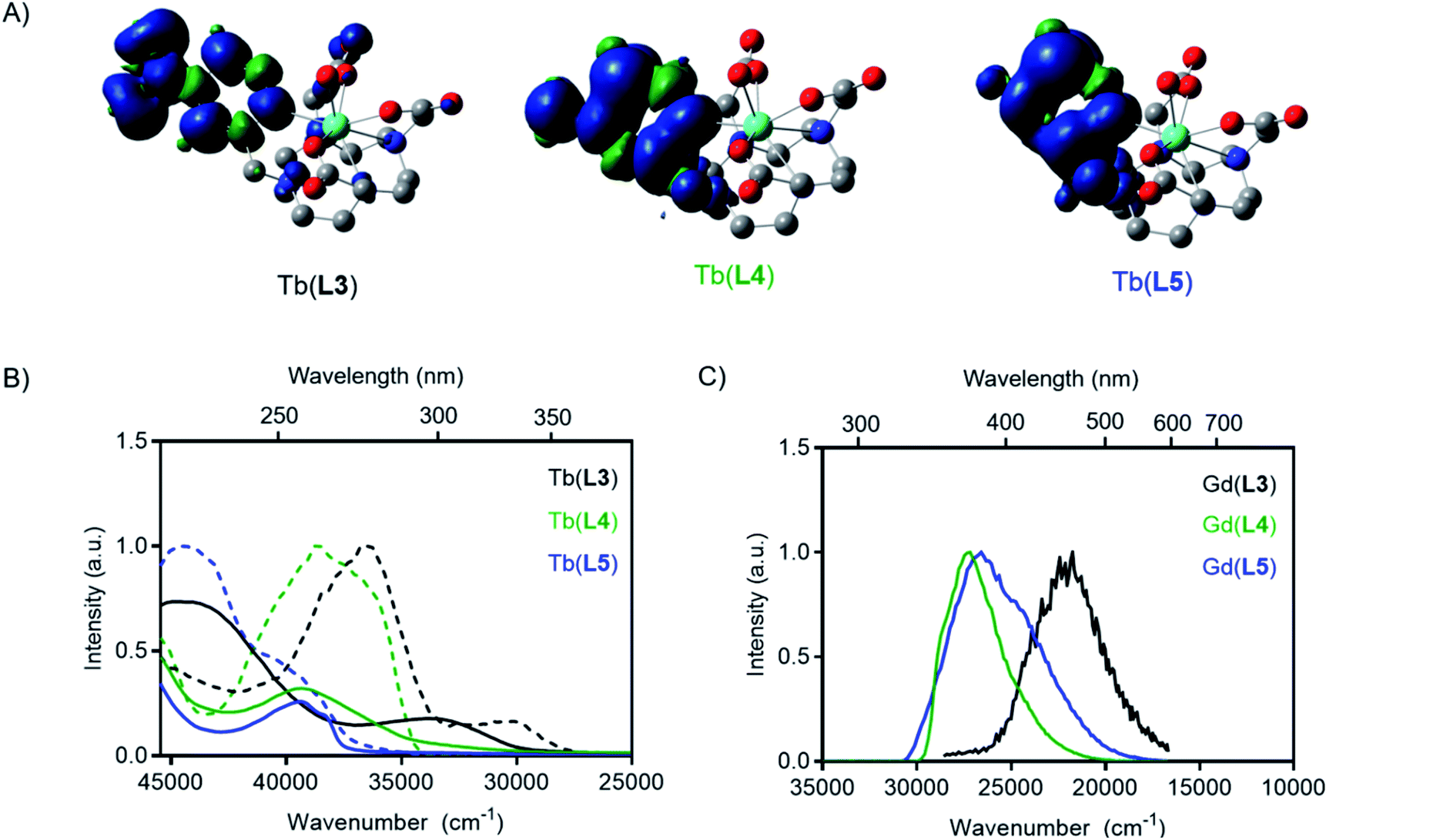 | ||
| Fig. 3 (A) Spin-density plots of optimized T1 state of each Tb(III) complex. (B) Absorbance (solid) and excitation (dashed) spectra of Tb(III) complexes. (C) Phosphorescence spectra of Gd(III) complexes. All measurements in PBS buffer at RT (absorption, excitation) or 77 K (phosphorescence). | ||
| Compound | Experimentalb S1 (cm−1) | Experimentalb T1 (cm−1) | Calculated T1 (cm−1) | Φ Tb | q | x-fold ΦTb increasee |
|---|---|---|---|---|---|---|
| a All experimental values recorded in PBS buffer. b Measured on Gd(III) complex. c Estimated relative error 10–15%. d Hydration state (±20%) values calculated according to literature methods. e Relative to Tb(L3). | ||||||
| Tb(L3) | 33333 |
25773 |
17307 |
0.0002 | 1.3 | — |
| Tb(L4) | 38462 |
29586 |
26625 |
0.10 | 1.1 | 500 |
| Tb(L5) | 39370 |
30395 |
27063 |
0.093 | 0.9 | 465 |
Synthesis
After affirming our original hypothesis with DFT, we embarked on the chemical synthesis of the L3, L4 and L5 ligand systems. L3 was synthesized by alkylating tri-tert-butyl protected macrocycle (tBu-DO3A, tri-tert-butyl 1,4,7,10-tetraazacyclododecane-1,4,7-triacetate) with a para-nitropyridyl chromophore [2-methyl(sulfonyloxymethyl)-4-nitropyridine], which was synthesized in three steps from commercially available 2-methylpyridine N-oxide to yield tert-butyl protected intermediate S1 (for a synthesis scheme, see ESI Fig. S1†). Subsequent deprotection under acidic conditions produced L3. To afford the electron-donating amine substituent, compound S1 was reduced by Pd-catalyzed hydrogenation and purified using semi-preparative HPLC to yield S2, followed by deprotection under acidic conditions to provide ligand L4. The fluorinated substituent L5 was synthesized by alkylation of 2-(bromomethyl)-4-fluoropyridine (synthesized in one step from 4-fluoro-2-methylpyridine) with tBu-DO3A, followed by deprotection of precursor S3 under acidic conditions. Tb(III) complexes were formed in water by mixing the Tb(III) with ligand under acidic conditions followed by adjustment of pH to 7 and removal free Tb(III) by reverse phase chromatography.Photophysical measurements
Photophysical measurements of each Tb(III) complex were carried out to determine how substituents on the ligand pyridine influenced T1 and overall quantum yield. For each complex, the spectrochemical properties were measured in PBS (pH = 7.0) at room temperature, including absorption, emission spectra, and corresponding quantum yields. Luminescent lifetime values of all complexes were determined in PBS and D2O to determine inner-sphere hydration (q).43 A summary of the results is provided in Table 1. A 5200 cm−1 blue-shift was observed in the experimental absorbance maxima from Tb(L3) to Tb(L4), indicating an increase in the relative excited singlet state energies (Fig. 3). Similarly, a blue-shift of the absorbance maximum was observed for the modification of Tb(L3) to complex Tb(L5) (6000 cm−1).Time-gated phosphorescence spectra were recorded at 77 K for Tb(III) and Gd(III) complexes of L3, L4 and L5 ligands. The energy of the excited T1 states were determined as the onset of the phosphorescence emission peak from the Gd(III) complexes (see ESI†). As the lanthanide-based excited state of Gd(III) is too high in energy (32200 cm−1) to be excited by the ligands studied, the result is pure ligand-based emission. The absorbance and excitation spectra of Tb(III) complexes, as well as the phosphorescence emission from the T1 state obtained from the Gd(III) complexes are shown in Fig. 3.
The phosphorescence excitation spectra of the Gd(III) complexes are in good agreement with the steady-state absorption measurements. Excitation measurements of the Tb(III) complexes however revealed that two absorbing species are simultaneously present in all samples (See Fig. 3 and in ESI, Fig. S9 to S12†). The excitation spectrum obtained when monitoring Tb(III)-based emission result in a blue shifted excitation spectrum compared to the excitation spectrum obtained when monitoring ligand-based emission. The two excitation spectra appear to agree with the neutral and the protonated forms of the pyridyl groups that exist in an equilibrium at the PBS moderated pH under the cryogenic, 77 K measurement conditions. At room temperature, the protonated species is not readily detected, leading to the conclusion that it does not play a significant role in the modulation of emission properties or the de-excitation pathway of M(L4) complexes in subsequent experiments.
The absorption and excitation spectra of L3 have many components, but a low intensity peak at 330 nm resembling the charge transfer band was observed, and the T1 state was readily accessed by excitation through the charge transfer band. Like the absorption maxima, the triplet excited states also resulted in a blue shift of Gd(L4) and Gd(L5) relative to Gd(L3). The experimental triplet state energy of L3 was only 5700 cm−1 greater than the 5D4 level for Tb(III), while the experimental triplet energy of L4 and L5 are 9500 and 9900 cm−1 greater respectively. The variation in T1 indicates competing back energy transfer pathways for L3 reduce overall Tb(III) sensitization. A further explanation for decreased Tb(III) emission of L3 could be that the energy cascade from the lowest-lying excited T1 state populates the 5D3 state and is subsequently quenched by oxygen before populating the 5D4 excited state.44 The DFT-calculated and experimentally determined T1 values follow the same trend, where reduction or conversion of L3 to L4 or L5 increases the triplet state energy level.
Quantum yield measurements were carried out as described previously, using [Tb(L1)]− (ΦLn = 0.47) as the reference compound.16 Quantum yield was 0.0002 for Tb(L3), which corresponds well to diminished efficiency of transfer from the corresponding T1 state. Substitution of the electron-withdrawing nitro-substituent (L3) with an amine (L4), increased the quantum yield of Tb(L4) 500-fold, to 0.10. Similarly, replacement of the nitro-group with a fluoro-group improves the quantum yield 465-fold to 0.09 for Tb(L5), also in agreement with a significant blue shift of T1. Albeit trends predicted by our DFT calculations persist, we note a deviation of 3000–8000 cm−1. This indicates that instead of the existence of a T1 energy level energy transfer threshold that correlates with the 5D4 excited state, rather the rate of back energy transfer appears to control the efficiency of the lanthanide based de-excitation.
Hydrogen sulfide sensing
With evidence of the 500-fold quantum yield increase from Tb(III) complexes of L3 and L4, reduction of Tb(L3) we sought to chemically transform Tb(L3) to Tb(L4) using a biologically relevant analyte. An analyte of biological importance, hydrogen sulfide has gained interest in the past decade in the context of non-invasive molecular imaging, as it plays important roles in apoptosis, insulin signaling, blood pressure regulation, and neuromodulation.45,46 Previous H2S sensitive optical probes rely on the presence of an accessible nitroaromatic component which acts as a PeT quencher, but in the presence of aqueous HS− is reduced to an amine.47,48 Motivated by the reactivity of previous nitroaromatic-containing metal complexes, we assessed Na2S as an in situ H2S source to reduce Tb(L3) without significant degradation of the corresponding complex (Fig. 4A). Indeed, in the presence of 100 equivalents of Na2S, full conversion of Tb(L3) to Tb(L4) was obtained. The direct functional group transformation was followed by HPLC and further affirmed by mass spectrometry (ESI Fig. S21†). Of note, the reaction produced no detectable side products, which bodes well for selective sensing applications. The conversion reaction was also monitored in situ by measurement of increasing fluorescence emission produced by Tb(L4) in the presence of 100 equivalents of Na2S (Fig. 4B). Plotting of relative intensity of the emission peak at 548 nm, corresponding to the diagnostic 5D4–7F5 transition reveals a 31-fold increase over 5 hours, which is comparable to other H2S-sensing systems.48–50 Selectivity of Tb(L3) was tested against a series of biologically relevant anions and reducing agents such as NaOAc, NaI, NaHCO3, Cys. A 31-fold increase was only obtained in the presence of Na2S (Fig. 4D), while 100 equivalents of other analytes produced no detectable luminescence increase. | ||
| Fig. 4 (A) Reaction scheme of Tb(L3) (10 μM) reacting with 100 equiv. Na2S (1 mM) to form Tb(L4) in situ. (B) Emission spectra over time upon reduction of 10 nmol (10 μM) Tb(L3) (523 nm peak = scattering). (C) Fold-increase of emission over 5.5 hours. (D) Relative fold-change of emission of 10 nmol (10 μM) Tb(L3) in response to NaOAc, NaI, NaHCO3, NaCl, Na2SO4, Cys, and Na2S (100 equiv.) in PBS (dashed line = 1; error bars represent standard deviation, n = 3). | ||
Optical imaging with CRET
To validate that our T1 energy mediated probe turn-on produces an appropriate optical signal that can be readily detected using conventional, small animal imaging equipment, we conducted experiments in the presence of the Cherenkov emissive radioisotope 18F (βavg+ = 0.25 MeV, 97%, t1/2 = 109 min). As we have previously shown, Cherenkov radiation (CR) provides a convenient in situ excitation source for luminescent Tb(III) complexes.16 CR-imaging was carried out with complexes Tb(L3), Tb(L4), and Tb(L5), imaging phantoms containing 1–150 nmol probe (as measured by ICP-OES). Based on our previous CRET studies, Tb(III) complexes with ΦTb of 0.10 possess a limit of detection of 5 nmol (25 μM), which corresponds to 1–2 orders of magnitude greater sensitivity than clinically administered, T1-based MRI contrast agents in phantoms and small animal imaging experiments.51,52Here, we prepared samples containing complex and 10 μCi (10 μL) of 18F. This amount of activity compares well to quantities used for targeted positron emission tomography (PET) imaging in small animals. Tube phantoms containing complex and 18F were subsequently imaged on a small animal optical imager (top down view), with blocked excitation and open emission from 500 to 875 nm (Fig. 5A). The images acquired affirm results obtained using conventional fluorescence measurements: the “off” complex Tb(L3) provides no significant radiance output relative to 18F in PBS (lower left, Fig. 5A). Region of interest (ROI) analysis of radiance indicates that turn-on complexes Tb(L4) and Tb(L5) demonstrate 220- and 190-fold radiance increase relative to Tb(L3) at concentrations above 25 nmol (125 μM), and a clearly discernable optical signal with only 5 nmol of complex (Fig. 5B) in accordance with our previous studies and estimates for corresponding limits of detection.
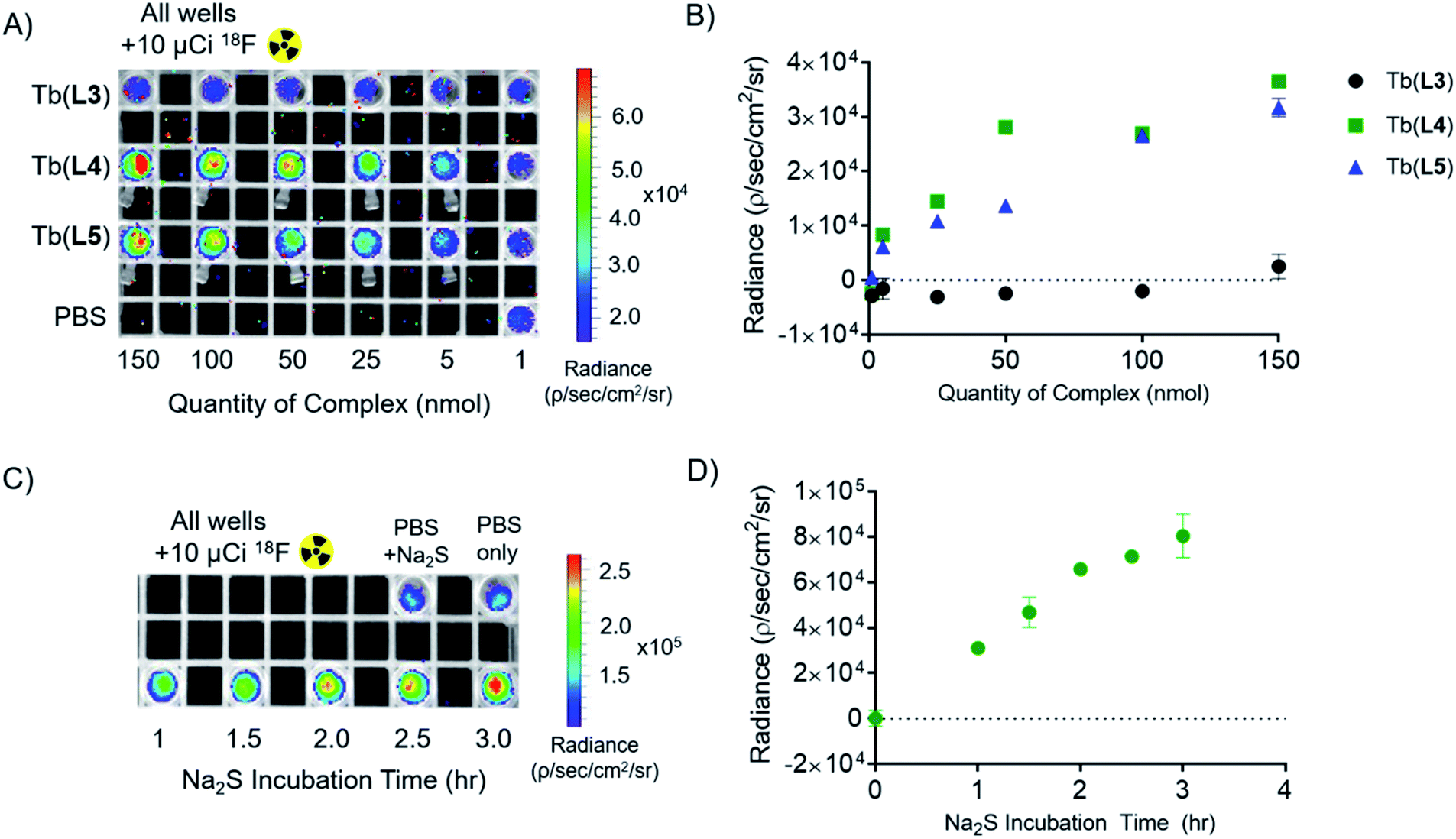 | ||
| Fig. 5 Cherenkov-radiation energy transfer (CRET) imaging experiments and quantitation of detected radiance. (A) Plate image of all complexes in 200 μL of PBS buffer doped with 10 μCi Na18F per well. (B) Quantified radiance output of part A, buffer-only CR sample subtracted. (C) Plate image of 10 nmol Tb(L3) with 100 equivalents Na2S over 3.5 hour incubation period. (D) Quantified radiance output of part C, buffer + Na2S CR sample subtracted. Error bars represent standard deviation from ROI analysis (n = 3). | ||
In order to observe probe turn-on in presence of a CR source, 100 equivalents of Na2S (1000 nmol, 100 μL) were added to Tb(L3) (10 nmol, 100 μL). 10 μCi of 18F was added to each well and samples were imaged with blocked excitation and open emission (Fig. 5C) at different lengths of Na2S incubation (1.0, 1.5, 2.0, 2,5, 3.0, and 3.5 hours) following Na2S addition. A 480-fold emission turn-on is observed from the in situ formation of Tb(L4) at 3.5 hours, quantified as radiance increase (Fig. 5D) relative to 18F in PBS. By utilizing Cherenkov radiation, this Na2S-reduction experiment demonstrates the first example of an analyte sensing platform with broad applicability and facile detection of lanthanide turn-on in absence of a short-wave laser excitation source or time resolution.
Conclusions
Herein, we report a new approach to generate lanthanide-based optical imaging turn-on probes through the direct chemical modification of the antenna triplet state and relative rates of back energy transfer (BET). We validated our rational design by estimating respective S1 and T1 states prior to synthesis and photophysical complex characterization of three pyridine-functionalized, aza-macrocycle-based Tb(III) complexes. Experimentally determined S1 and T1 states affirmed computationally predicted trends, but showed a 3000–8000 cm−1 deviation of the predicted energy levels which is consistent with previous literature findings, and therefore directly guides subsequent ligand design strategies.53 The water-soluble Tb(III) complex series illustrates how conversion of the nitro-pyridine functionalized complex Tb(L3) to the corresponding amino-pyridine Tb(L4) or fluoro-pyridine Tb(L5) functionalized macrocyclic complexes results in a blue-shift of corresponding S1 and T1 states.The increased energy gap between the antenna T1 state and the 5D4 Tb(III) excited state results in decreased BET efficiency for Tb(L4) and Tb(L5) complexes, which produces 500- and 465-fold increases in relative lanthanide-based quantum yield ΦTb. This is the largest increase in quantum yield reported for discrete, biocompatible lanthanide complexes (Table S1†). As a proof-of-concept, we demonstrate that turn-on of Tb(L4) emission by reductive, chemical transformation from Tb(L3) with Na2S, an in situ source of hydrogen sulfide, is feasible. We employed in situ excitation using Cherenkov radiation to detect as little as 5 nmol of Tb(L4) and Tb(L5) with conventional, small animal optical imaging equipment. The turn-on probe design employed here obviates the need for large probe quantities otherwise incompatible with biological applications or time-resolved deconvolution typical for luminescent lanthanide-based probes to identify the desired luminescence turn-on. Furthermore, we also note that the probe concentration required to produce a detectable optical signal with Tb(L4) correlates closely with probe concentrations recently used to validate organic turn-on sensors in vitro and in vivo.54,55
While we successfully employed H2S as a proof-of-concept turn-on analyte, we note that quantities employed here are at the upper limit of biologically encountered analyte concentration range. Future studies will focus on enhancing reactivity of Tb(III) complexes, broadening the scope of analytes that elicit a detectable turn-on response, and direct in vivo validation of our CR-mediated sensing approach. Second generation probes with enhanced sensitivity will be especially suitable for analyte-based, preclinical imaging in small animals or intrasurgical detection requiring limited depth penetration. We also note that Tb(L5) is suitable for the synthesis of a self-illuminating CR probe by incorporation of radioactive 18F to displace the NO2 functional group.
Experimental section
All starting materials were purchased from Acros Organics, Alfa Aesar, Sigma Aldrich, or TCI America and used without further purification. NMR spectra (1H, 13C, 19F) were collected on a 700 MHz AdvanceIII Bruker, 500 MHz or 400 MHz Bruker instrument at 25 °C and processed using TopSpin 4.0.6. Chemical shifts are reported as parts per million (ppm). Mass spectrometry: low-resolution electrospray ionization (ESI) mass spectrometry and high-resolution ESI mass spectrometry were carried out at the Stony Brook University Institute for Chemical Biology and Drug Discovery (ICB&DD) Mass Spectrometry Facility with an Agilent LC/MSD and Agilent LC-UV-TOC spectrometers respectively. Preparative HPLC was carried out using a Shimadzu HPLC-20AR equipped with a Binary Gradient, pump, UV-Vis detector, and manual injector. Method A (preparative purification method, on Phenomenex Luna 10 μm C18 column [250 mm × 21.2 mm, 100 Å, AXIA packed]): A = 0.1% TFA in water, B = 0.1% TFA in MeCN. Gradient: 0–1 min: 5% B, 1–5 min: 5–35% B, 5–23.5 min: 35–75% B, 23.5–24 min: 75–95% B, 24–26 min: 95% B, 27–30.1 min 95–5% B. Method B preparative purification method, (preparative purification method, on Phenomenex Luna 10 μm C18 column [250 mm × 10 mm, 100 Å, AXIA packed]): A = 10−2 M ammonium formate in water, B = 10% 10 mM ammonium formate in water, 90% MeCN. Gradient: 0–1 min: 5% B, 1–5 min: 5–35% B, 5–23.5 min: 35–75% B, 23.5–24 min: 75–95% B, 24–26 min: 95% B, 27–30.1 min 95–5% B. Analytical HPLC analysis was carried out using a Shimadzu HPLC-20AR equipped with a binary gradient, pump, UV-Vis detector, and autoinjector on a Phenomenex Luna 5 μm C18 column (150 mm × 3 mm, 100 Å, AXIA packed). Method C (analysis of Ln(III)-complexes): A = 0.1% TFA in water, B = 0.1% TFA in MeCN with a flow rate of 0.8 mL min−1, UV detection at 220 and 254 nm. Gradient: 0–5 min: 95% A. 5–24 min: 5–95% B gradient. CombiFlash purifications were performed on RediSep RF Gold C18 column (50G, 100 Å) using Method D (A = 0.1% TFA in water, B = 0.1% TFA in MeCN. Gradient: 0–1.4 CV: 10% B, 1.4–2.1 CV: 10–20% B, 2.1–17.1 CV: 20–50% B, 17.1–21.9 CV: 50–100%, 21.9–23 CV: 100% B, 23–24.1 CV 10% B. Flow rate: 40 mL min−1).UV-VIS spectra were collected with a NanoDrop 1C instrument (AZY1706045). Spectra were recorded from 200 to 900 nm in a quartz cuvette with 1 cm path length. Room temperature luminescence measurements were carried out on a Hitachi F-7100 FL spectrophotometer at room temperature in Dulbecco's phosphate-buffered saline (DPBS). Wavelength scans were collected by exciting at the appropriate wavelength for antenna-mediated excitation and minimization of scattering interference. Luminescence emission spectra were collected under luminescence scan mode: excited at λex (Table S6†) with emission collection from 300 to 800 nm; 1.0 nm excitation and 5.0 nm emission slit widths, chopping speed of 40 Hz, 1200 nm min−1 scan speed, 0.5 s response time, PMT voltage = 400 V. Lifetime measurements were executed using the following settings: Scan time 20 ms; chopping speed of 40 Hz; excitation wavelength specific to each complex (Table S6†) and emission wavelength of 545.0 nm; 0 second delay; excitation and emission slit widths of 10 nm each; 0.5 second response. Complexes were dissolved in DPBS buffer or D2O (samples were resuspended and lyophilized in D2O repeatedly prior to measurement). A quartz cuvette with a 1 cm pathlength was used. ICP-OES was carried out using an Agilent 5110 ICP-OES. A 10-point standard with respect to terbium was used and fits were found to be with R2 of 0.9999. Concentrations were then back-calculated to the stock sample concentration. Concentrations of each lanthanide complex were diluted based on stock concentrations determined by ICP-OES. IVIS Lumina Series II from Caliper LifeScience small animal imager was used to image phantoms. All scans were collected with blocked excitation, open emission filter (500–875 nm, average band width 20 nm), and a collection time of 5 minutes. Images were analyzed with Living Image software version 4.3.1. Regions of interest were determined in triplicate with the ROI tool for each sample. Radiance values for each complex were subtracted from the Cherenkov-only sample (Na18F in PBS). Lanthanide complexation, general procedure: Tb(OTf)3 (1.2 equiv.) or GdCl3·6H2O (1.2 equiv.) was added to a solution of ligand (1 equiv.) in 1.0 mL of water. The pH was adjusted to 7.0 with 1 M NaOH and stirred at room temperature overnight. Crude complex was loaded onto a C18 SepPak cartridge and extracted with 8 mL of 100:0 H2O:MeCN to 70:30 H2O:MeCN gradient. The fractions containing product were pooled and lyophilized, resulting in a white solid. Further purification under method B was applied as necessary.
Na2S turn-on
For all in situ Na2S experiments, quantity of Tb(L3) was pre-determined by ICP-OES. Na2S stock solutions were prepared in PBS immediately prior to addition. Fluorimetry-based experiments were carried out by diluting 10 nmol of Tb(L3) in PBS and adding 100 equivalents Na2S for a final volume of 1 mL. The reaction was monitored via luminescence emission from a conventional fluorimeter under the identical conditions listed above (λex = 260 nm). For HPLC verification of Tb(L3) in situ reduction, 10 nmol of Tb(L3) in 100 μL was activated with 100 equivalents of Na2S (100 μL). Selectivity experiments were conducted in the same manor, with 10 nmol of complex and 100 equivalents of Na2S in a total volume of 1 mL PBS. I0 was measured after 5 minutes of incubation with each respective analyte/inorganic salt (each pre-formed stock solutions in PBS); It was measured after 5 hours. Fold-increase of intensity is measured as Itvs. I0 of the Tb(III) peak at 548 nm. The Cherenkov-radiation imaging experiment was also carried out with 10 nmol of Tb(L3) in 100 μL PBS and activated with 100 equivalents of Na2S (100 μL) for time points 1.0, 1.5, 2.0, 2.5, 3.0, and 3.5 hours prior to 18F addition and imaging.Macrocyclic starting material tri-tert-butyl 1,4,7,10-tetraazacyclododecane-1,4,7-triacetate (tert-butyl DO3A) was prepared according to literature procedure.56 Bromo-4-fluoro-pyridine was also synthesized in one step from commercially available 2-methyl-4-fluoropyridine in one step according to a patented procedure.57
{4,10-Bis(carboxymethyl)-7-[(4-nitro-2-pyridyl)methyl]-1,4,7,10-tetraaza-1-cyclododecyl}acetic acid, L3
Tert-Butyl {4,10-bis(tert-butoxycarbonylmethyl)-7-[(4-nitro-2-pyridyl)methyl]-1,4,7,10-tetraaza-1-cyclododecyl}acetate, S1, (34.1 mg, 0.023 mmol) was added to a 2:1 solution of TFA and DCM and stirred overnight at room temperature. The solvent was removed in vacuo, and the product was re-dissolved in H2O. The solution was then lyophilized to yield L3 as an off-white solid (11.1 mg, 0.022 mmol, Rt (method C) = 3.0 minutes). 1H NMR (CD3OD, 500 MHz): δ 8.88 (d, 1H), 8.23 (s, 1H), 8.10 (s, 1H), 4.13 (br s, 2H), 3.76–3.57 (m, 16H), 3.23–3.21 (m, 6H). 13C NMR (CD3OD, 125 MHz): δ 173.12, 169.36, 159.54, 156.12, 152.71, 117.43, 116.77, 58.33, 55.34, 54.12, 52.25, 50.48, 49.62. ESI-MS calcd. for C20H30N6O8: 482.2125. Found: 483.2198 [M + H]+.{10-[(4-Amino-2-pyridyl)methyl]-4,7-bis(carboxymethyl)-1,4,7,10-tetraaza-1-cyclododecyl}acetic acid, L4
Tert-Butyl {10-[(4-amino-2-pyridyl)methyl]-4,7-bis(tert-butoxycarbonylmethyl)-1,4,7,10-tetraaza-1-cyclododecyl}acetate, S2, (7.1 mg, 0.011 mmol) was added to a 2:1 solution of TFA and DCM and stirred overnight at room temperature. The solvent was removed in vacuo, and the product was re-dissolved in H2O. The solution was then lyophilized to yield L4 as an off-white solid (4.7 mg, 0.010 mmol, (method C) = 1.89 minutes). 1H NMR (CD3OD, 400 MHz): 7.90 (d, 1H), 6.86 (s, 1H), 6.72 (d, 2H), 4.07 (br s, 2H), 3.96–3.66 (m, 4H), 3.26–2.93 (m, 18H). 13C NMR (CD3OD, 100 MHz): δ 177.27, 176.46, 175.85, 161.89, 149.85, 142.07, 111.23, 109.06, 57.47, 56.14, 56.13, 55.44, 54.34, 51.78. ESI-MS calcd. for C20H32N6O6: 452.2383. Found: 453.2457 [M + H]+.{4,10-Bis(carboxymethyl)-7-[(4-fluoro-2-pyridyl)methyl]-1,4,7,10-tetraaza-1-cyclododecyl}acetic acid, L5
Tert-Butyl {4,10-bis(tert-butoxycarbonylmethyl)-7-[(4-fluoro-2-pyridyl)methyl]-1,4,7,10-tetraaza-1-cyclododecyl}acetate, S3, (15.2 mg, 0.023 mmol) was added to a 2:1 solution of TFA and DCM and stirred overnight at room temperature. The solvent was removed in vacuo, and the product was re-dissolved in H2O. The solution was then lyophilized to yield L5 as an off-white solid (97%, 10.3 mg, 0.023 mmol, (method C) = 2.01 minutes). 1H NMR (CD3OD, 400 MHz): δ 8.58 (m, 1H), 7.35 (d, 1H), 7.22 (m, 1H), 4.62 (br s, 2H), 4.11 (br s, 2H), 3.69–3.49 (m, 14H), 3.22 (m, 6H). 13C NMR (CD3OD, 125 MHz): δ 173.05, 172.01, 169.91, 158.79, 153.12, 120.96, 118.64, 116.34, 58.17, 55.77, 53.82, 52.61, 51.37, 50.75, 50.17. 19F NMR: (CD3OD, 376 MHz): −100.16. (CD3OD, 376 MHz): ESI-MS calcd. for C20H30FN5O6: 455.2180. Found: 456.2256 [M + H]+.Computational details
DFT calculations were performed using the Gaussian 16 software.58 The starting geometries for the complexes were taken from the crystal structure of a similar complex.59 Geometries of the complexes were optimized in the gas phase at 298 K using the TPSSh functional.60 Light atoms (H, N, C, O, F) were treated with the 6-31G(d,p) basis set, and the Tb(III) center was assigned the large core relativistic effective core potential (LCRECP) and related (7s6p5d)/5s4p3d basis set.61) The LCRECP includes 52 electrons in the core of the metal center leaving the outermost 5s, 5p, 5d, and 6s electrons to be handled explicitly. As such, the GS calculations were performed in the pseudosinglet state and T1 calculations were performed in the pseudotriplet state. Frequency calculations were performed at the same level of theory to confirm that the optimized structures were local minima on the potential energy surface and to determine the thermodynamic properties of the complexes. Solvation energies were calculated using the SMD method62 and the thermodynamic properties in H2O were estimated by taking the thermodynamic corrections from gas-phase calculations. Optimized coordinates for the complexes in the GS and T1 states are given in the ESI.†Data availability
Raw data for imaging and spectroscopy is available as an x, y number document upon request.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support for this work was provided by the National Science Foundation (CHE 1942434, NSF CAREER, E. B.), the Novo Nordisk Foundation (TJS), Carlsbergfondet (TJS), Villum Fonden (grant #14922, TJS).Notes and references
- H. Ito, T. Terai, K. Hanaoka, T. Ueno, T. Komatsu, T. Nagano and Y. Urano, Chem. Commun., 2015, 51, 8319–8322 RSC.
- S. C. Zondlo, F. Gao and N. J. Zondlo, J. Am. Chem. Soc., 2010, 132, 5619–5621 CrossRef CAS PubMed.
- E. Pershagen and K. E. Borbas, Angew. Chem., Int. Ed., 2015, 54, 1787–1790 CrossRef CAS PubMed.
- E. Pershagen, J. Nordholm and K. E. Borbas, J. Am. Chem. Soc., 2012, 134, 9832–9835 CrossRef CAS PubMed.
- B. K. McMahon, R. Pal and D. Parker, Chem. Commun., 2013, 49, 5363–5365 RSC.
- J. E. Whitaker, R. P. Haugland and F. G. Prendergast, Anal. Biochem., 1991, 194, 330–344 CrossRef CAS PubMed.
- D. Parker, P. Kanthi Senanayake and J. A. Gareth Williams, J. Chem. Soc., Perkin Trans. 2, 1998, 2, 2129–2140 RSC.
- E. Del Giorgio and T. J. Sørensen, Molecules, 2020, 25, 1959 CrossRef CAS PubMed.
- M. L. Aulsebrook, B. Graham, M. R. Grace and K. L. Tuck, Tetrahedron, 2014, 70, 4367–4372 CrossRef CAS.
- W. Xu, Y. Zhou, D. Huang, M. Su, K. Wang and M. Hong, Inorg. Chem., 2014, 53, 6497–6499 CrossRef CAS PubMed.
- T. J. Rink, R. Y. Tsien and T. Pozzan, J. Cell Biol., 1982, 95, 189–196 CrossRef CAS PubMed.
- A. Minta, J. P. Y. Kao and R. Y. Tsien, J. Biol. Chem., 1989, 264, 8171–8178 CrossRef CAS.
- S. H. Hewitt, G. Macey, R. Mailhot, M. R. J. Elsegood, F. Duarte, A. M. Kenwright and S. J. Butler, Chem. Sci., 2020, 11, 3619–3628 RSC.
- G. Vereb, E. Jares-Erijman, P. R. Selvin and T. M. Jovin, Biophys. J., 1998, 74, 2210–2222 CrossRef CAS PubMed.
- A. G. Cosby, S. H. Ahn and E. Boros, Angew. Chem., Int. Ed., 2018, 57, 15496–15499 CrossRef CAS PubMed.
- A. G. Cosby, G. Quevedo and E. Boros, Inorg. Chem., 2019, 58, 10611–10615 CrossRef CAS PubMed.
- M. C. Heffern, L. M. Matosziuk and T. J. Meade, Chem. Rev., 2014, 114, 4496–4539 CrossRef CAS PubMed.
- L. G. Nielsen, A. K. R. Junker and T. J. Sørensen, Dalton Trans., 2018, 47, 10360–10376 RSC.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214–217 CrossRef CAS.
- B. Alpha, R. Ballardini, V. Balzani, J.-M. Lehn, S. Perathoner and N. Sabbatini, Photochem. Photobiol., 1990, 52, 299–306 CrossRef CAS.
- S. J. Butler, Chem. Commun., 2015, 51, 10879–10882 RSC.
- S. Y. Huang and V. C. Pierre, Chem. Commun., 2018, 54, 9210–9213 RSC.
- D. G. Smith, R. Pal and D. Parker, Chem.–Eur. J., 2012, 18, 11604–11613 CrossRef CAS PubMed.
- S. H. Jung, K. Y. Kim, J. H. Lee, C. J. Moon, N. S. Han, S.-J. Park, D. Kang, J. K. Song, S. S. Lee, M. Y. Choi, J. Jaworski and J. H. Jung, ACS Appl. Mater. Interfaces, 2017, 9, 722–729 CrossRef CAS PubMed.
- E. Pershagen and K. E. Borbas, Angew. Chem., Int. Ed., 2015, 54, 1787–1790 CrossRef CAS PubMed.
- T. Terai, K. Kikuchi, Y. Urano, H. Kojima and T. Nagano, Chem. Commun., 2012, 48, 2234–2236 RSC.
- M. Latva, H. Takalo, V.-M. Mukkala, C. Matachescu, J. C. Rodríguez-Ubis and J. Kankare, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- A. D'Aléo, E. G. Moore, J. Xu, L. J. Daumann and K. N. Raymond, Inorg. Chem., 2015, 54, 6807–6820 CrossRef PubMed.
- A. P. S. Samuel, J. Xu and K. N. Raymond, Inorg. Chem., 2009, 48, 687–698 CrossRef CAS PubMed.
- S. Shuvaev, M. Starck and D. Parker, Chem.–Eur. J., 2017, 23, 9974–9989 CrossRef CAS PubMed.
- T. J. Sørensen and S. Faulkner, Acc. Chem. Res., 2018, 51, 2493–2501 CrossRef PubMed.
- M. Woods, G. E. Kiefer, S. Bott, A. Castillo-Muzquiz, C. Eshelbrenner, L. Michaudet, K. McMillan, S. D. K. Mudigunda, D. Ogrin, G. Tircsó, S. Zhang, P. Zhao and A. D. Sherry, J. Am. Chem. Soc., 2004, 126, 9248–9256 CrossRef CAS PubMed.
- O. A. Blackburn, M. Tropiano, L. S. Natrajan, A. M. Kenwright and S. Faulkner, Chem. Commun., 2016, 52, 6111–6114 RSC.
- E. Llobet, J. Brunet, A. Pauly, A. Ndiaye and C. Varenne, Sensors, 2017, 17, 391 CrossRef PubMed.
- J. Katla and S. Kanvah, Photochem. Photobiol. Sci., 2018, 17, 42–50 CrossRef CAS PubMed.
- X. Huang, H. Liu, J. Zhang, B. Xiao, F. Wu, Y. Zhang, Y. Tan and Y. Jiang, New J. Chem., 2019, 43, 6848–6855 RSC.
- Y. L. Pak, J. Li, K. C. Ko, G. Kim, J. Y. Lee and J. Yoon, Anal. Chem., 2016, 88, 5476–5481 CrossRef CAS PubMed.
- Y. Takano, H. Echizen and K. Hanaoka, Antioxid. Redox Signaling, 2017, 27, 669–683 CrossRef CAS PubMed.
- L. Babetto, S. Carlotto, A. Carlotto, M. Rancan, G. Bottaro, L. Armelao and M. Casarin, Dalton Trans., 2020, 49, 14556–14563 RSC.
- C. Greco, G. Moro, L. Bertini, M. Biczysko, V. Barone and U. Cosentino, J. Chem. Theory Comput., 2014, 10, 767–777 CrossRef CAS PubMed.
- S.-y. Baek, S.-Y. Kwak, S.-T. Kim, K. Y. Hwang, H. Koo, W.-J. Son, B. Choi, S. Kim, H. Choi and M.-H. Baik, Nat. Commun., 2020, 11, 2292 CrossRef CAS PubMed.
- U. Cosentino, A. Villa, D. Pitea, G. Moro and V. Barone, J. Phys. Chem. B, 2000, 104, 8001–8007 CrossRef CAS.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. d. Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 2, 493–503 RSC.
- A. K. R. Junker and T. J. Sørensen, Dalton Trans., 2019, 48, 964–970 RSC.
- O. Kabil and R. Banerjee, J. Biol. Chem., 2010, 285, 21903–21907 CrossRef CAS PubMed.
- M. M. Gadalla and S. H. Snyder, J. Neurochem., 2010, 113, 14–26 CrossRef CAS PubMed.
- X. Wang, L. An, Q. Tian and K. Cui, RSC Adv., 2019, 9, 33578–33588 RSC.
- Z. Dai, L. Tian, B. Song, Z. Ye, X. Liu and J. Yuan, Anal. Chem., 2014, 86, 11883–118839 CrossRef CAS PubMed.
- Y. Yao, L. Delgado-Rivera, H. Samareh Afsari, L. Yin, G. R. J. Thatcher, T. W. Moore and L. W. Miller, Inorg. Chem., 2018, 57, 681–688 CrossRef CAS PubMed.
- M. Tropiano and S. Faulkner, Chem. Commun., 2014, 50, 4696–4698 RSC.
- J. Wahsner, E. M. Gale, A. Rodríguez-Rodríguez and P. Caravan, Chem. Rev., 2019, 119, 957–1057 CrossRef CAS PubMed.
- B. Driehuys, J. Nouls, A. Badea, E. Bucholz, K. Ghaghada, A. Petiet and L. W. Hedlund, ILAR J., 2008, 49, 35–53 CrossRef CAS PubMed.
- D. Jacquemin, B. Mennucci and C. Adamo, Phys. Chem. Chem. Phys., 2011, 13, 16987–16998 RSC.
- Z. Chen, X. Mu, Z. Han, S. Yang, C. Zhang, Z. Guo, Y. Bai and W. He, J. Am. Chem. Soc., 2019, 141, 17973–17977 CrossRef CAS PubMed.
- M. Y. Lucero, A. K. East, C. J. Reinhardt, A. C. Sedgwick, S. Su, M. C. Lee and J. Chan, J. Am. Chem. Soc., 2021, 143, 7196–7202 CrossRef CAS PubMed.
- K. Kikuchi, F. Sugihara, S. Mizukami, Y. Yoshioka, H. Matsushita and T. Nakamura, Angew. Chem., Int. Ed., 2014, 54, 1007–1010 Search PubMed.
- R. Blunt, A. J. Eatherton, V. Garzya, M. P. Healy, J. Myatt and R. A. Porter, WO2011012622A1, 2011.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C01, Wallingford, CT, 2016 Search PubMed.
- S. Aime, A. S. Batsanov, M. Botta, J. A. K. Howard, M. P. Lowe and D. Parker, New J. Chem., 1999, 23, 669–670 RSC.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- A. Nicklass, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1995, 102, 8942–8952 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02148f |
| This journal is © The Royal Society of Chemistry 2021 |