
Direct synthesis of CsPbX3 perovskite nanocrystal assemblies†
Chujie
Wang
a,
Sri K.
Matta
bc,
Chun Kiu
Ng
 a,
Chang
Cao
a,
Manoj
Sharma
*a,
Anthony S. R.
Chesman
d,
Salvy P.
Russo
b and
Jacek J.
Jasieniak
*a
a,
Chang
Cao
a,
Manoj
Sharma
*a,
Anthony S. R.
Chesman
d,
Salvy P.
Russo
b and
Jacek J.
Jasieniak
*a
aARC Centre of Excellence in Exciton Science, Department of Materials Science and Engineering, Monash University, Clayton, Victoria 3800, Australia. E-mail: manoj.sharma@monash.edu; jacek.Jasieniak@monash.edu
bARC Centre of Excellence in Exciton Science, School of Science, RMIT University, Melbourne 3000, Australia
cCenter for Computational Sciences, University of Tsukuba, Japan
dCSIRO Manufacturing, Ian Wark Laboratories, Research Way, Clayton, VIC 3168, Australia
First published on 12th December 2023
Abstract
Inorganic CsPbX3 (X = Cl, Br, I) perovskite nanocrystals (NCs) possess many advantageous optoelectronic properties, making them an attractive candidate for light emitting diodes, lasers, or photodetector applications. Such perovskite NCs can form extended assemblies that further modify their bandgap and emission wavelength. In this article, a facile direct synthesis of CsPbX3 NC assemblies that are 1 μm in size and are composed of 10 nm-sized NC building blocks is reported. The direct synthesis of these assemblies with a conventional hot-injection method of the NCs is achieved through the judicious selection of the solvent, ligands, and reaction stoichiometry. Only under selective reaction conditions where the surface ligand environment is tuned to enhance the hydrophobic interactions between ligand chains of neighbouring NCs is self-assembly achieved. These assemblies possess narrow and red-shifted photoluminescence compared to their isolated NC counterparts, which further expands the colour gamut that can be rendered from inorganic perovskites. This is demonstrated through simple down-converting light emitters.
Introduction
Inorganic CsPbX3 (X = Cl, Br, I) perovskite nanocrystals (PNCs) have attracted great research interest because of their high photoluminescence quantum yield (PLQY),1 narrow photoluminescence (PL) emission full-width half-maximum (FWHM),2 defect-tolerant band structure,3 and readily tunable emission wavelength across the visible spectrum.4 PNCs are typically synthesized by a colloidal method, stabilized with long-chain hydrocarbon ligands, and possess good solution processability.5 With all these advantageous properties, PNCs have demonstrated promise in applications such as light-emitting diodes (LED),6 lasers,7 photodetectors,8 and solar cells.9 To support these applications, ongoing research into PNCs is needed around structurally controlled nanoparticles and their assemblies, both in solution and in suitable host matrices. While the study of direct PNC synthesis is prominent,10 the formation of NC assemblies requires more attention.NCs assembled into ordered structures are known as supercrystals (SCs). Such assemblies can modify their optoelectronic properties,11 inducing narrower emission linewidths, coherent superfluoroscence, and increased stimulated emission.12–15 To date, the main approaches for assembling NCs into such assemblies include: (1) direct synthesis through ultrasonication and elevated precursor concentration;12 (2) slowly evaporating the solvent of previously synthesized and monodisperse PNC dispersions;13 (3) introducing polar anti-solvents or complexing agents into a PNC dispersion to drive assembly;16,17 and (4) post-synthesis ligand exchange.18 The assembly of perovskites is not limited to cube shaped NCs, with reports extending the assembly of perovskites to other morphologies, such as nanoplatelets (NPLs),19 nanorods,20 and nanosheets.21 However, with the exception of ultrasonication,12 SC assembly methods have been limited largely to two-step processes, requiring initial hot-injection synthesis of PNC dispersions and a separate, subsequent, assembly method.
The synthesis of PNC assemblies has been largely reliant on inter-molecular forces, namely hydrogen bonding, dipole–dipole interactions (i.e. van der Waals forces, VdW),22 or electrostatic attractions.23 In contrast, the role of hydrophobic interactions, which are an inherent feature of alkyl ligand-stabilized NC systems, have not been well understood in the direct synthesis of PNC-SCs.24 For gold nanoparticles, the hydrophobic interaction between the long chain hydrocarbon capping ligands on the nanoparticle surface has been reported to effectively modulate their assembly.25 The hydrophobic interaction originates from the tendency of two hydrophobes (e.g. hydrocarbon ligands) to mutually attract, resulting in the rearrangement of nearby solvent molecules, and hence increasing the system's overall entropy. The strength of the hydrophobic interaction can be an order of magnitude larger than the van der Waals force, and is dependent on two primary factors: (1) the length and shape of the hydrophobes, with long and straight hydrocarbon chains ensuring greater hydrophobicity and less steric hindrance, giving rise to stronger hydrophobic interaction; and (2) the polarity of the solvent, with more polar solvents decreasing the solubility of the hydrophobes, leading to increased hydrophobic interaction.26 Although PNCs are typically capped with long-chain hydrocarbon ligands (e.g. oleic acid, OLA, and oleylamine, OLA), their assembly into SCs by intrinsic hydrophobic interactions has not yet been reported.
Herein, a facile direct synthetic method for perovskite SCs of CsPbCl3, CsPbBr3, and CsPbI3 is reported. The SCs are up to 1 μm in size and are assembled from 10 nm-sized NC building blocks during the hot-injection synthesis. The synthesis of the SCs follows the conventional hot-injection method of the PNCs, with slight modifications to the solvent, ligands, and stoichiometry employed in the reaction. It is found that these tailored reaction conditions enhance the effective hydrophobic interactions between the PNCs to drive their assembly into SCs. The degree of this hydrophobic interaction is manipulated by modifying the ligand hydrocarbon chain structure and length, which eventually influences the SC's formation. Optical characterization of these SCs show greatly red-shifted PL emission and narrowed PL FWHM. Such CsPbX3 PNC-SCs are further shown to be readily introduced into hybrid polymer-SC matrices that can be used within colour-tunable down-converting light-emitting devices.
Methods
Materials
Cesium carbonate (Cs2CO3, 99.995% trace metal basis), lead(II) chloride (PbCl2, 99.999% trace metals basis), lead(II) bromide (PbBr2, 99.999% trace metals basis), lead(II) iodide (PbI2, 99.999% trace metal basis), dodecane (anhydrous, ≥99%), 1-octadecene (ODE, 90%, technical grade), oleylamine (OLA, 70%, technical grade), octylamine (99%), hexadecylamine (HDA, 98%), octadecylamine (97%), hydrochloric acid (ACS reagent, 37 wt% in H2O), hydroiodic acid (57 wt% in H2O, distilled, stabilized, 99.95%), polymethyl methacrylate (PMMA, MW∼350![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), and toluene (99.5%, analytical reagent), were purchased from Sigma-Aldrich. Isopropanol (IPA, 99.5%, analytical reagent), hexane (99.5% analytical reagent), and hydrobromic acid (48 wt% in H2O, ≥99.99%) were purchased from Merck. Dodecylamine (98%) was purchased from Alfa Aesar. Bis(2,4,4-trimethylpentyl) phosphinic acid (TMPPA, 85%) was purchased from Cytec. All reagents were used without further purification.
000), and toluene (99.5%, analytical reagent), were purchased from Sigma-Aldrich. Isopropanol (IPA, 99.5%, analytical reagent), hexane (99.5% analytical reagent), and hydrobromic acid (48 wt% in H2O, ≥99.99%) were purchased from Merck. Dodecylamine (98%) was purchased from Alfa Aesar. Bis(2,4,4-trimethylpentyl) phosphinic acid (TMPPA, 85%) was purchased from Cytec. All reagents were used without further purification.
Synthesis
:3 (v/v) ODE:IPA ratio. The mixture was then centrifuged for 5 min at 10000 rpm in a Beckman Coulter AllegraTM X-22R Centrifuge. The supernatant was discarded, and the CsPbX3 NC precipitate was redispersed in hexane.
000 rpm. The clear supernatant was discarded, and the CsPbX3 SC precipitate was redispersed in hexane.
Characterizations
For TEM sample preparation, 10 μl of either the NC or SC solutions were diluted in 1 ml of hexane, and the TEM sample grid (Ted Pella inc. ultrathin carbon film) was immersed into the mixture and the solvent was then evaporated.
Computational methods
Periodic Density Functional Theory (DFT) calculations were performed using the Vienna Ab initio simulation package (VASP) software platform.27,28 For all calculations (geometry optimizations and single point energy) the Perdew–Burke–Ernzerhof (GGA-PBE) exchange–correlation functional with the associated Projector-Augmented-Wave (PAW) pseudopotentials were used.29,30 The planewave cut-off was 400 eV or 550 eV in different models of the study but maintained the consistency of the cut-off within the same system analysis. As the supercell sizes used in this study were large (more than 400 atoms), Gamma point sampling was used to sample the Brillouin Zone (BZ). The convergence criteria for electronic convergence and ionic relaxation loop convergence was 10−5 eV and 10−4 eV, respectively. In the aperiodic (surface) direction a minimum vacuum space of 20 Å was used to minimize interactions with periodic images of the supercell in that direction. Grimme's scheme was also used to incorporate van der Waals's interactions.30,31 The 3D model visualisation is done using VESTA software.32Results and discussion
Synthesis of CsPbX3 supercrystals
As shown in Fig. 1a, our synthesis of CsPbX3 generally follows a modified hot-injection method developed by Kovalenko et al.,1 in which Cs2CO3 and PbX2 precursors are dissolved separately in solutions containing ligands, with the Cs-precursor subsequently being injected into the PbX2 mixture at 140 °C to yield the nanocrystalline products. These products are isolated via centrifugation with the addition of a polar anti-solvent and then redispersed in hexane. As per our previous report, bis-(2,2,4-trimethylpentyl) phosphinic acid (TMPPA, chemical formula in Fig. S1†) has been used as the acidic ligand to produce stable cubic-phase CsPbI3.33 Herein, the CsPbI3 system was also used as an example to demonstrate the synthesis of normal NCs and self-assembled SCs. When the conventional unsaturated solvent (octadecene, ODE) and amine ligand (OLA) are used with the TMPPA, monodisperse cubic-phased CsPbI3 NCs are synthesised (Fig. 1b–d). However, upon modifying the reaction stoichiometry of the Cs, Pb and I precursors, utilising saturated (aliphatic) solvent (dodecane) and an amine ligand (hexadecylamine, HDA), direct synthesis of CsPbI3 SCs could be achieved (Fig. 1e–g). The effects of these two modifications will be discussed shortly. | ||
| Fig. 1 (a) Schematic illustration of CsPbX3 NC and SC syntheses. Structure illustration, low and high magnification TEM images for (b)–(d) CsPbI3 NCs and (e–g) SCs. Insets of (c) and (f) are indexed SADP of NCs and SCs. | ||
As seen from transmission electron microscopy (TEM) images (Fig. 1g), the assemblies of CsPbI3 within the SCs are on the 0.1–1 μm scale, which is comparable to the reported CsPbBr3 SCs.12,17 Such a larger size of the CsPbI3 SCs is further confirmed through dynamic light scattering (DLS) measurements, which show significantly larger mean sizes (∼1 μm, Fig. S2†) for the SCs, compared with the typical 10–100 nm for normal NC dispersions. Additional TEM and HAADF images at different magnifications are provided in Fig. S3 and S4.† At low magnification (Fig. S3a†), isolated NCs can still be observed in the sample, however, the SCs comprise the majority of the product. When inspected at a higher magnification (Fig. S3c†), a periodic inter-connected morphology comprising of the ∼10 nm sized NC building blocks can be clearly identified. This alignment of the NC building blocks suggests that assembly occurs due to a ligand-directed process.13 SADP of these SCs proves that the individual NCs are cubic-phased, while also providing evidence of preferential orientations within the assemblies (Fig. 1f inset). Notably, when changing the halide component from iodide to bromide and chloride, the respective CsPbBr3 and CsPbCl3 SCs can also be achieved. Energy Dispersive X-ray (EDX) elemental mapping and X-ray Diffraction (XRD) patterns are included in the SI for the Cl, Br, and I SC analogues. As shown by EDX mapping, the Cs, Pb, and X elements are evenly distributed throughout the SCs (Fig. S5†). The corresponding XRD patterns have been further indexed to their perovskite phases (Fig. S6†).
A distinct feature of directly synthesised SCs is that they can be purified from the crude reaction solution via centrifugation without any anti-solvent. As a result of their size, when redispersed in hexane and stored, the SCs spontaneously precipitate, leaving a colourless supernatant. Such a stronger sedimentation effect of the SCs is in stark contrast with the OLA-capped NCs, which form stable dispersions in hexane. Considering the similar chain length and a functional binding group of HDA and OLA, we believe the SC formation to arise from the different steric and/or hydrophobic interactions provided by these ligands (vide infra). Notably, it has been previously shown that NCs capped with lecithin ligands enable concentrated dispersions without precipitation or formation of SCs.45 Lecithin ligands provide multi-dentate surface coordination and strong steric hindrance to enable these dispersion properties. Therefore, to overcome these strong repulsive steric interactions, the potential role of strong hydrophobic interactions observed for the saturated straight-chained HDA (or its ammonium salt) used in this work needs to be understood.
A critical parameter affecting the formation of SCs is reactant stoichiometry. Experiments were conducted in which the concentration of the Cs and I ratios were increased relative to a fixed amount of Pb precursor through the addition of Cs precursor or hydroiodic acid as an iodine source. TEMs presented in Fig. 2 illustrate the effect of tuning the reactant stoichiometry. In a conventional hot injection synthesis of NCs, a Cs:Pb:I molar ratio of 1:4:8 yields monodisperse cube-shaped NCs (Fig. 2a).1 When the Cs content is doubled, the majority of the NCs retain the same morphology; however, a small portion of hexagonal-shaped particles also form (Fig. 2b). Meanwhile, a four-fold increase in Cs content yields only hexagonal shape particles (Fig. 2c), which are ascribed to the Cs4PbI6 phase according to the indexing of the SADP (Fig. S7†). This phase has been reported to form at high Cs:Pb ratios.34 Doubling the iodide content of the original reaction stoichiometry did not change the morphology of the product (Fig. 2d). However, at this elevated iodide ratio and progressively higher Cs content, >100 nm ordered nanocrystalline assemblies formed (Fig. 2e and f).
 | ||
| Fig. 2 TEM images of CsPbI3 NCs synthesised using HDA-HI at different Cs:Pb:I molar ratios: (a) 1:4:8, (b) 1:2:4, (c) 1:1:2, (d) 1:4:16, (e) 1:2:8, and (f) 1:1:4. All images were taken immediately after synthesis. | ||
To further explain the effect of stoichiometry on SC formation, the reaction yield of the synthesis was determined. For this, the concentration of CsPbI3 NCs was first calculated using the UV-vis absorption spectra of the samples shown in Fig. 2d–f (Fig. S8†) and our previously reported extinction coefficient of CsPbI3 NCs,35 with the reaction yield being subsequently determined by considering Cs as the limiting reagent (see ESI†). The NC reaction yields across these samples were all around 70–85% (Table S1†). Moreover, the CsPbI3 concentration increased almost four-fold (1.1 × 10−6 M to 4.8 × 10−6 M) for the 1:1:4 sample as compared to the conventional 1:4:8 stoichiometry. Such a high CsPbI3 concentration is in accordance with a previous report that a high NC concentration is essential for SC formation.12
Another critical factor determining direct SC formation is that the solvent and ligand systems used must be saturated hydrocarbons. Firstly, replacing the saturated solvent dodecane with the conventional unsaturated solvent ODE under otherwise identical reaction conditions for CsPbI3 SCs synthesis results in no SCs forming (Fig. S9†). This observation is consistent with the additional iodide content provided by the hydroiodic acid being consumed through an additional reaction with the double bond of the ODE, which is in great excess.36 Secondly, to differentiate between the use of saturated and unsaturated solvent and ligand systems, syntheses were carried out using various ligand combinations. As a starting point, a comparison of both the conventional OLA and the straight chained HDA ligands was made. The OLA produced well-dispersed nanocubes without any evidence of SC formation (Fig. S10†). Similarly, if the TMPPA in the SC reactions is replaced by the conventionally-used OLA, SCs also do not form (Fig. S11†). Instead, polydisperse nanocube structures are observed, which can be attributed to the fact that OLA is binding to the NCs’ surface.37
The use of TMPPA in an otherwise conventional synthesis of NCs results in surface chemistry that is dominated by alkyl amine/ammonium salts.3331P NMR spectroscopy indicates that this holds true for SCs formed within a saturated dodecane solvent and HDA ligand system, for which the presence of phosphorous-containing species in purified solutions of CsPbCl3, CsPbBr3 and CsPbI3 SCs cannot be detected (Fig. S12†). Therefore, it can be concluded that a surface chemistry composed of straight-chained HDA and/or its ammonium derivative are an essential aspect to the formation of the SCs.
To probe this further, TEM was used to measure the inter-NC distance of NCs synthesized using HDA and OLA ligands. Again, because the TMPPA is not present at the surface, the amine and/or its ammonium species should be the dominating surface ligand contributor. As seen in Fig. 3(a and b) the inter-NC distance for the HDA sample, either in the NC or the SC form, is determined to be around 2 nm. This is consistent with the fully extended chain length of HDA (2.18 nm),38 indicating that the HDA ligands likely interdigitate between adjacent NC surfaces. Such a configuration would ensure close contact of the hydrocarbon chains of HDA, thus favouring stronger hydrophobic interactions,24 which can further support the assembly of NCs into SCs. In comparison, the distance between the OLA-capped NCs is ∼2.5 nm (Fig. 3c), which is much larger than the extended chain length of an OLA molecule (2.0 nm). This is consistent with the bent OLA possessing a higher degree of rotational freedom, which reduces the extent that it can interdigitate.39 As a result, decreased hydrophobic interactions would be expected for OLA ligand passivation. While it has been shown that OLA surface chemistry supports SC formation, it requires ultra-high NC concentrations or narrow NC size distributions to enhance their for their formation.12,13
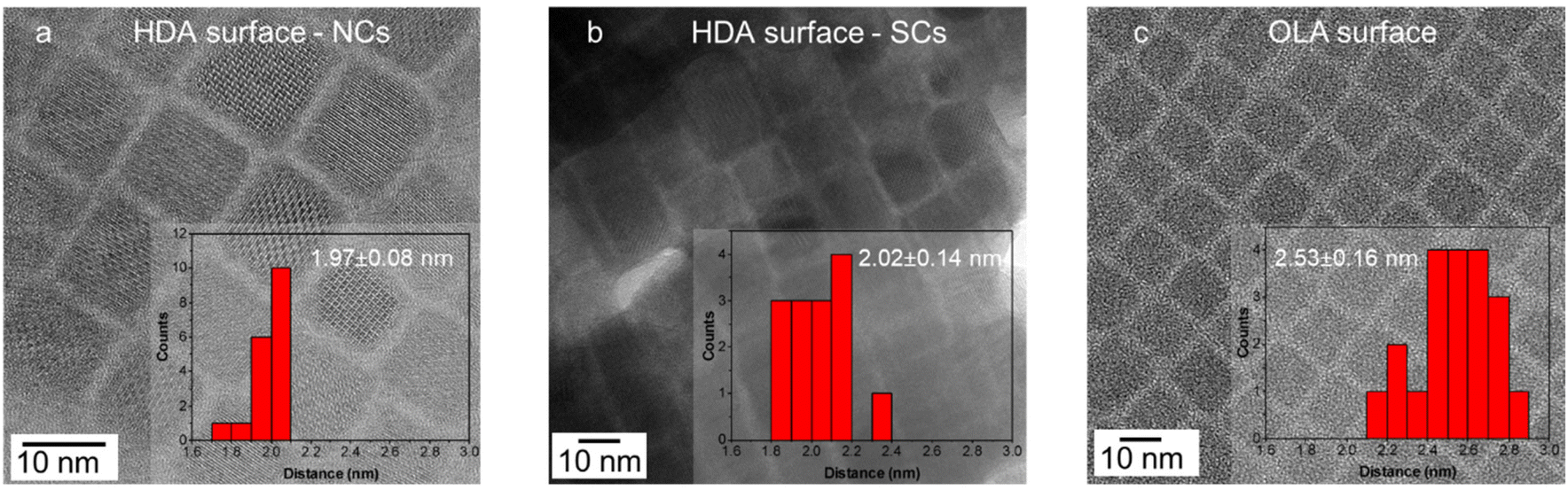 | ||
| Fig. 3 TEM images and histograms for the measurement of the inter-NC surface distance for (a) HDA-Br capped NCs, (b) HDA-Br capped SCs, (c) OLA-Br capped NCs. | ||
DFT modelling
To further support the hypothesis of ligand-interdigitation-induced SC formation, Density Functional Theory (DFT) calculations of a CsPbI3 perovskite surface with a PbI2 terminated (001) surface interacting with OLA-X and HDA-X surface ligands were performed (Fig. 4a–c). Previous studies on gold nanoparticle assemblies have indicated that the attachment angle between alkylamine ligands and the vertical c-axis of the nanoparticle is between 0° and 30°,40 whereas for the HDA ligand this angle has been experimentally and computationally determined to be 30°.41 While this provides a gauge into the bonding nature typical of such native ligands, the partial quaternization of the amine to form the ammonium salt within the synthesis of perovskites necessitates an understanding of the alkyl ammonium bromide salts.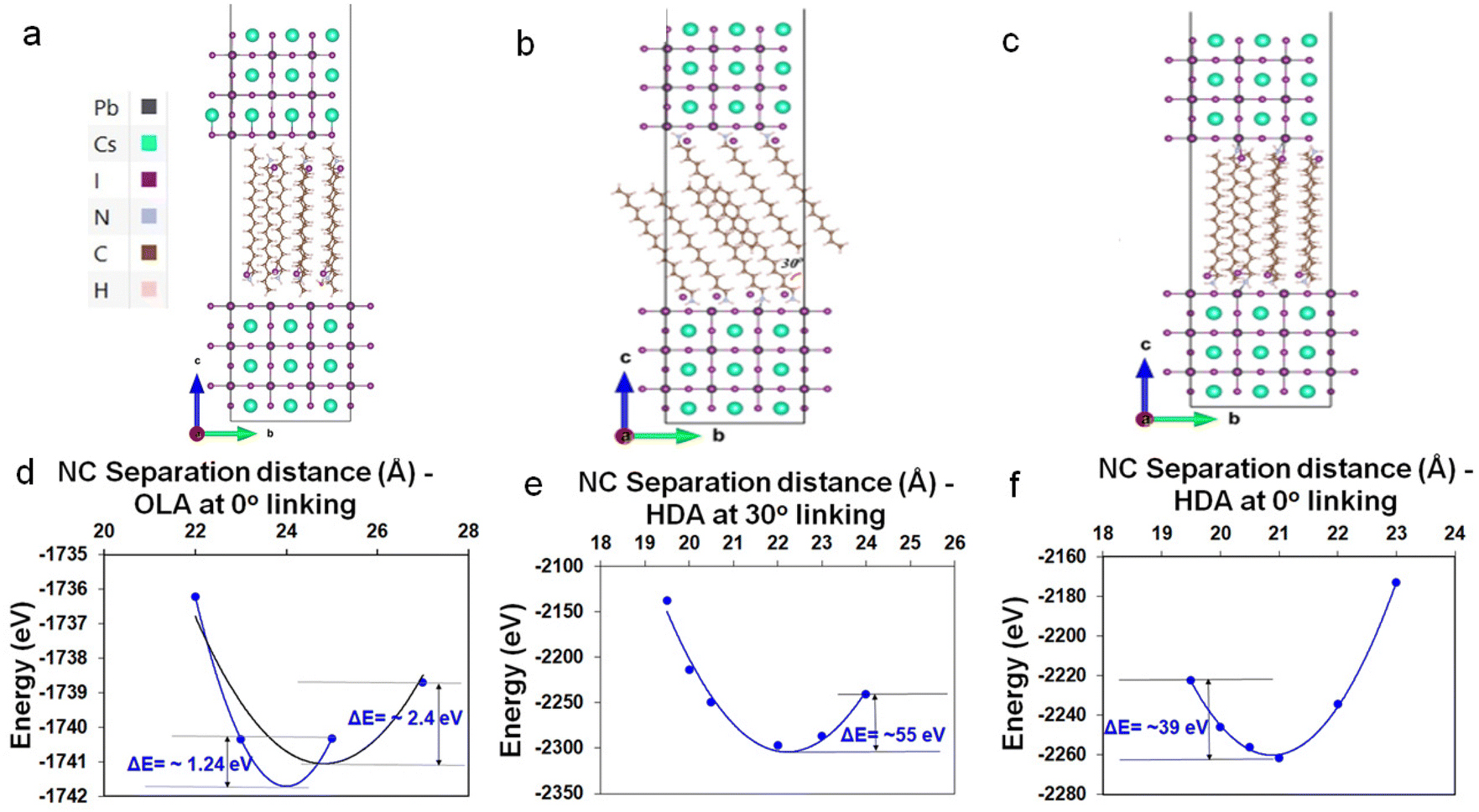 | ||
| Fig. 4 (a) 2 × 1 supercell of six-layered CsPbI3 NCs facing each other with OLA linked on PbI2 terminated (001) surfaces on both NCs at 0° angle with c-lattice direction. Similarly, (b and c) a 3 × 1 supercell with I linked at 30° and 0° with respect to c-lattice direction. The colour code for the atoms in models are: Cs (cyan), I (purple), Pb (black), N (light blue), C (brown), and H (pale purple). In the orientation given, the Pb atoms are partially obscured by the I atoms. Calculation of DFT energy (in eV) for (d) OLA and, (e and f) HDA ligands linked to nanoparticles as a function of NC separation distance (in Angstrom) at 30° and 0° with respect to c-lattice direction. The dashed lines are quadratic polynomial fits to the data as a visual guide. | ||
Our DFT calculations indicate that OLA-X has the lowest energy configuration with an attachment angle of 0°, while for the HDA-X ligand angle configurations of both 0° and 30° present the lowest energy configurations (Fig. S13†). Under these bonding conditions, the average ligand density is calculated to be 3.15 and 2.94 ligands per nm2 for the OLA-X and HDA-X ligands, respectively, which is a similar ligand density to that obtained experimentally of ∼3 ligands per nm2.35
We further explore the total energy dependence on the distance (d), between two such passivated perovskite surfaces (see Fig. 4d–f as a guide). For OLA-X, the ligands have a 0° link, which made it very difficult to obtain converged DFT energies at various separation distances. Therefore, we were only able to obtain converged results for 4 distance values; 2.2, 2.3, 2.5, and 2.7 nm (Fig. 4d). Meanwhile, for the two HDA-X ligand configurations (30° and ∼0°), we were able to obtain converged DFT energies for 6 distance values, ranging between 1.95–2.4 nm (Fig. 4e and f). We would expect the potential energy surfaces in these systems to have a complex multidimensional structure with hills, valleys, and saddle points. However, if we assume that the reaction path coordinate corresponding to the vertical separation distance between the NPs is one that would be associated with large energy variations in the ground electronic state, then the surface energy may look approximately parabolic about the atomic equilibrium positions of this coordinate. Assuming this to be the case, then under the harmonic approximation the separation distance corresponding to the local potential minima (i.e. the equilibrium separation distance) could be approximated as a distance associated with the well minimum. Therefore, in Fig. 4d–f, we have fitted the potential energy profile to a quadratic polynomial fit to approximate the equilibrium NP separation distance. This distance would be the most probable distance of separation between any two NCs in the system at ground-state if the assumptions stated above hold.
For OLA-X (Fig. 4d) we fitted the data to a parabolic fit using the first 3 data points (blue line) and all 4 data points (black line). The calculated values of the equilibrium separation distance for these fits were 2.4 (blue) and 2.48 (black). Therefore, we believe the true value lies within this range. For the HDA-X coated NCs, the local minimum in the potential energy surface is approximately 2.09 nm for the 0° attached ligand (Fig. 4f) and slightly larger at 2.22 nm for the 30° attached ligand (Fig. 4e). Both of these results are in very good agreement with the experimental results. Notably, the deeper potential energy well for the 30° attachment model versus the 0° attachment suggests that the preferred ligand bonding angle is different for HDA-X compared to the OLA-X ligands.
Effect of hydrophobic interactions
When NCs prepared using the 1:4:8 Cs:Pb:I stoichiometry with HDA surface ligands were stored under ambient conditions, they exhibited a gradual self-assembly into SCs over time. As shown in Fig. 5a and b, immediately after synthesis and purification, the sample contained well dispersed NCs, whereas after 10 days of storage at ambient condition, the UV-vis absorption spectrum (Fig. 5c) of this sample showed an increase in scattering over time, indicating the formation of SCs. Also, the PL peak of this sample red-shifted during this time, accompanied by a decrease in low wavelength emission. This is consistent with the progressive enhancement of particle electronic coupling and cascading energy transfer in the evolving SCs.3 These results suggest that the hydrophobic interaction is universal for all the NCs with the same HDA ligand environment, and it is sufficiently strong to drive SC formation even at low NC concentrations in hexane.
 | ||
| Fig. 5 TEM images and photos for the CsPbI3 crude solution synthesised with Cs:Pb:I = 1:4:8 stoichiometry that is (a) as-synthesised, (b) stored for 10 days, and (c) evolution of corresponding UV-vis absorption and PL spectra with time (inset: zoom-in PL spectra showing the redshift of the PL peaks). | ||
Phenomenologically, when NCs come into contact due to Brownian motion, ligand chains can intertwine, inducing attractive hydrophobic interactions through dispersive forces.42,43 These attractive interactions can be considered through the simplified version of Salem's equation:42
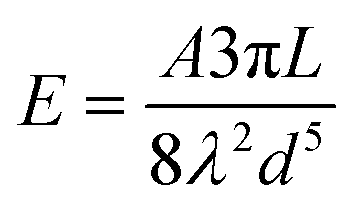 | (1) |
According to Salem's Equation, the similarity of the packing densities for both OLA and HDA in our system, indicates that the molecular overlap length is the primary factor that dictates the major differences in the attractive interactions between our NCs. From the TEM results and the accompanying DFT, nearly full intercalation for HDA-capped NCs and partial intercalation for OLA-capped NCs are observed. Based on the overlap regions, approximated as 2.03 nm (16 methylene groups) and 1.14 nm (9 methylene groups), respectively, we determine the attractive interaction of the HDA-passivated NCs to be ∼1.8 times larger than those featuring OLA. The enhanced hydrophobic interaction of the HDA-passivated NCs is evidently large enough to overcome the repulsive colloidal interactions existing at room temperature within the hexane solvent. In contrast, the bent hydrocarbon tail of the unsaturated OLA ligands weakens the chain-to-chain interaction to induce dominant inter-particle steric interactions, which result in stable colloidal dispersions. Notably, the attractive interactions for OLA-passivated NCs can be enhanced through increased concentrations and/or the addition of polar solvents, to selectively drive aggregation or self-assembly.15,16,26,45
Optical properties
Having examined the SC formation, the optical properties of the SCs are now considered. When SCs are formed, the electronic wavefunctions of the constituent NCs can overlap with each other for sufficiently small inter-particle distances, leading to enhanced electronic coupling.46 Meanwhile, the dense NC packing in SCs also leads to an increased local dielectric constant, which strengthens the screening of the coulombic interactions. These effects can reduce the exciton binding energy, slow PL recombination and induce PL red-shifting.46–48The UV-vis absorption and PL spectra of drop-cast CsPbCl3, CsPbBr3 and CsPbI3 SC films are compared with the corresponding NC films in Fig. 6a–c. All the SC samples demonstrate a greater degree of scattering in their absorption spectra, a red shifted PL, and a narrower PL FWHM. Specifically, compared with their NC analogues, a PL red shift of 36 meV, 41 meV, and 59 meV, and a reduced PL FWHM of 12 meV, 13 meV, and 21 meV are observed for the Cl, Br and I SCs, respectively. To further probe these samples, PL, PLQY and TRPL measurements are conducted on the CsPbBr3 NCs and SCs dispersed in solution and as drop cast films (Fig. S14 and S15†). The PL spectra of the SCs in solution and film form show a redshift compared to the NC analogues, with the redshift being smaller in solution (∼24 meV) as compared to films (∼51 meV). Meanwhile, the PLQYs for the samples in solution (film) were respectively determined to be 83% (35%) and 51% (32%) (Fig. S14†). Finally, TRPL measurements show that the average photoluminescence lifetime for the SCs (∼3.5 ns) remains similar compared to the NCs (∼3.9 ns) when measured in solution, while increasing in film form from 7.4 ns to 12.8 ns, respectively, due to an enhancement in the long lifetime component (Fig. S15 and Tables S2–S3†). While these collective results suggest that strong electronic coupling within the samples is unlikely (significant lifetime reductions expected), without further lower temperature and power-dependent measurements, it is not possible to distinguish between dielectric effects, photon propagation effects (self-absorption) and/or surface impurities.12,46,48,49
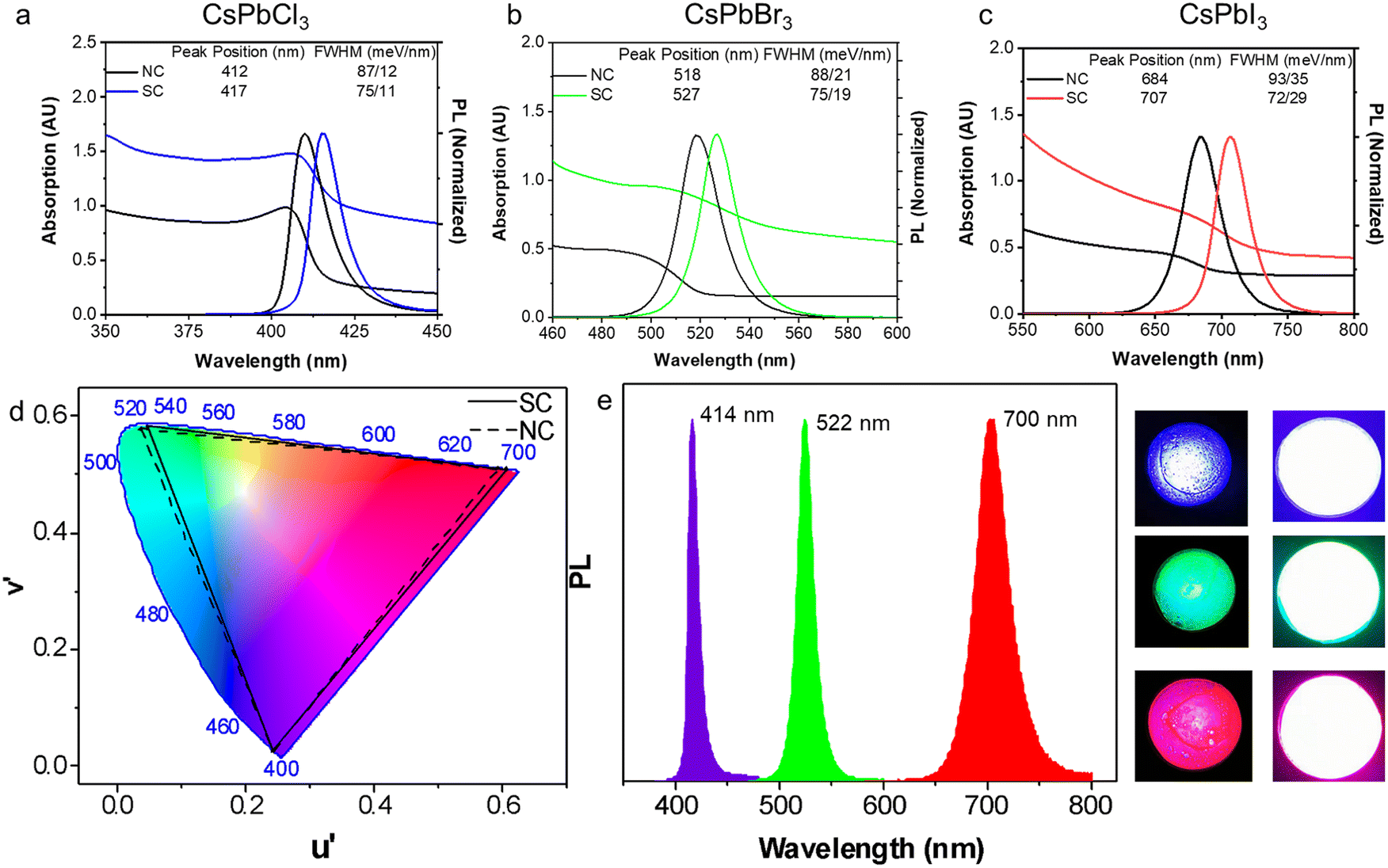 | ||
| Fig. 6 UV-vis absorption and PL spectra for NC and SC films of (a) CsPbCl3, (b) CsPbBr3 and (c) CsPbI3. (d) The emission of the NCs and SCs plotted on the colour gamut spectrum (CIE 1976); the PL is transformed into the UV through the chromaticity application in Origin software. (e) The PL spectra of the down-conversion LED devices fabricated from the SC-PMMA composites (left) and photos of the LEDs (right). | ||
A comparison of the emission wavelengths of the NCs and SCs on the colour gamut spectrum shows the red-shifted PL of the SCs slightly extends the range of emission colour beyond what can be rendered from conventional inorganic PNCs (Fig. 6d). For that reason, they are potential candidates for light emitting components and LEDs. As a preliminary proof of concept, SC-PMMA composite films were fabricated as per previous reports,1 and used as down-converters for commercially available UV LED torches (Fig. 6e). The peak PL wavelength for these SC-composite LEDs is at 414 nm, 522 nm and 700 nm for the Cl, Br and I PNC samples, respectively. While detailed studies are needed to fully quantify and understand the origins of the red shifted PL of the SCs as compared to NCs and control the assembly of SCs in solution and film forms, these results do showcase the facile nature of such SC materials as filters for achieving wide colour-gamut tunable LED emission profiles.
Conclusions
In conclusion, we have reported the facile direct synthesis of CsPbX3 perovskite supercrystals of around 1 μm in size that consist of 10 nm NC building blocks. The synthesis follows the well developed hot injection method, albeit with the conventional unsaturated solvent and ligands replaced with saturated analogues, and the stoichiometry of the precursors tuned to increase the concentration of the NCs. It is experimentally shown and verified through Density Functional Theory that smaller inter-particle distances are observed between NCs containing saturated vs. unsaturated alkyl amine ligands of similar chain length, with only the former yielding SC formation. These factors suggest that enhanced hydrophobic interactions within the saturated ligand system arising from greater interchain overlap provides the dominant driving force for NC assembly. The SCs demonstrate red-shifted absorption and PL, as well as narrowed PL linewidth compared to the discrete NC analogues. These were conducive to achieving expanded colour-gamut down-conversion prototype light-emitting devices. Overall, the facile synthesis of SCs presented here opens a new strategy to manipulate the assembly of PNCs into ordered structures through engineering of the reaction conditions and resulting surface chemistry.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Australian Research Council funded Center of Excellence in Exciton Science (CE170100026). We thank the staff in Monash Center for Electron Microscopy, Monash X-ray platform, and CSIRO NMR facility for their support. This work was also supported by computational resources provided by the Australian Government through the National Computational Infrastructure National Facility and the Pawsey Supercomputer Centre S. K. M. acknowledges the computing system resources’ support from the University of Tsukuba, Japan through the International Postdoctoral Fellowship of Japan Society for the Promotion of Science (JSPS)'s KAKENHI (Grant No. JP22F32733), during part of the computational work and finalization of this study. S. K. M. also acknowledges the FUJITSU Supercomputer PRIMEHPC FX1000 and FUJITSU Server PRIMERGY GX2570 (Wisteria/BDEC-01) facilities at the Information Technology Center, The University of Tokyo. We acknowledge Elliott Ayers for help during the editing stages of the manuscripts.References
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS PubMed.
- A. Swarnkar, R. Chulliyil, V. K. Ravi, M. Irfanullah, A. Chowdhury and A. Nag, Angew. Chem, 2015, 127, 15644–15648 CrossRef.
- J. Kang and L. W. Wang, J. Phys. Chem. Lett., 2017, 8, 489–493 CrossRef CAS PubMed.
- G. Nedelcu, L. Protesescu, S. Yakunin, M. I. Bodnarchuk, M. J. Grotevent and M. V. Kovalenko, Nano Lett., 2015, 15, 5635–5640 CrossRef CAS PubMed.
- Q. A. Akkerman, M. Gandini, F. Di Stasio, P. Rastogi, F. Palazon, G. Bertoni, J. M. Ball, M. Prato, A. Petrozza and L. Manna, Nat. Energy, 2017, 2, 16194 CrossRef CAS.
- J. Song, J. Li, X. Li, L. Xu, Y. Dong and H. Zeng, Adv. Mater., 2015, 27, 7162–7167 CrossRef CAS PubMed.
- Y. Wang, X. Li, J. Song, L. Xiao, H. Zeng and H. Sun, Adv. Mater., 2015, 27, 7101–7108 CrossRef CAS PubMed.
- Z. Dai, Q. Ou, C. Wang, G. Si, B. Shabbir, C. Zheng, Z. Wang, Y. Zhang, Y. Huang, Y. Dong, J. J. Jasieniak, B. Su and Q. Bao, J. Mater. Chem. C, 2019, 7, 5954–5961 RSC.
- A Swarnkar, A. R. Marshall, E. M. Sanehira, B. D. Chernomordik, D. T. Moore, J. A. Christians, T. Chakrabarti and J. M. Luther, Science, 2016, 354, 92–95 CrossRef CAS PubMed.
- C. K. Ng, C. Wang and J. J. Jasieniak, Langmuir, 2019, 35, 11609–11628 CrossRef CAS PubMed.
- Z. Nie, A. Petukhova and E. Kumacheva, Nat. Nanotechnol., 2010, 5, 15–25 CrossRef CAS PubMed.
- Y. Tong, E. P. Yao, A. Manzi, E. Bladt, K. Wang, M. Döblinger, S. Bals, P. Müller-Buschbaum, A. S. Urban, L. Polavarapu and J. Feldmann, Adv. Mater., 2018, 30, 1801117 CrossRef PubMed.
- G. Rainò, M. A. Becker, M. I. Bodnarchuk, R. F. Mahrt, M. V. Kovalenko and T. Stöferle, Nature, 2018, 563, 671–675 CrossRef PubMed.
- D. Lapkin, C. Kirsch, J. Hiller, D. Andrienko, D. Assalauova, K. Braun, J. Carnis, Y. Y. Kim, M. Mandal, A. Maier, A. J. Meixner, N. Mukharamova, M. Scheele, F. Schreiber, M. Sprung, J. Wahl, S. Westendorf, I. A. Zaluzhnyy and I. A. Vartanyants, Nat. Commun., 2022, 13, 1–10 Search PubMed.
- Z. Liu, X. Qin, Q. Chen, T. Jiang, Q. Chen and X. Liu, Adv. Mater., 2023, 35, 2209279 CrossRef CAS PubMed.
- J. S. Van Der Burgt, J. J. Geuchies, B. Van Der Meer, H. Vanrompay, D. Zanaga, Y. Zhang, W. Albrecht, A. V. Petukhov, L. Filion, S. Bals, I. Swart and D. Vanmaekelbergh, J. Phys. Chem. C, 2018, 122, 15706–15712 CrossRef CAS PubMed.
- K. H. Wang, J. N. Yang, Q. K. Ni, H. B. Yao and S. H. Yu, Langmuir, 2018, 34, 595–602 CrossRef CAS PubMed.
- Y. Nagaoka, K. Hills-Kimball, R. Tan, R. Li, Z. Wang and O. Chen, Adv. Mater., 2017, 29, 1606666 CrossRef PubMed.
- S. K. Mehetor, H. Ghosh and N. Pradhan, J. Phys. Chem. Lett., 2019, 10, 1300–1305 CrossRef CAS PubMed.
- S. K. Mehetor, H. Ghosh and N. Pradhan, ACS Energy Lett., 2019, 4, 1437–1442 CrossRef CAS.
- Y. Liu, M. Siron, D. Lu, J. Yang, R. dos Reis, F. Cui, M. Gao, M. Lai, J. Lin, Q. Kong, T. Lei, J. Kang, J. Jin, J. Ciston and P. Yang, J. Am. Chem. Soc., 2019, 141, 13028–13032 CrossRef CAS PubMed.
- J. Liu, K. Song, Y. Shin, X. Liu, J. Chen, K. X. Yao, J. Pan, C. Yang, J. Yin, L. J. Xu, H. Yang, A. M. El-Zohry, B. Xin, S. Mitra, M. N. Hedhili, I. S. Roqan, O. F. Mohammed, Y. Han and O. M. Bakr, Chem. Mater., 2019, 31, 6642–6649 CrossRef CAS.
- M. A. Boles, M. Engel and D. V. Talapin, Chem. Rev., 2016, 116, 11220–11289 CrossRef CAS PubMed.
- H. A. Scheraga, J. Biomol. Struct. Dyn., 1998, 16, 447–460 CrossRef CAS PubMed.
- A. Sánchez-Iglesias, M. Grzelczak, T. Altantzis, B. Goris, J. Pérez-Juste, S. Bals, G. Van Tendeloo, S. H. Donaldson, B. F. Chmelka, J. N. Israelachvili and L. M. Liz-Marzán, ACS Nano, 2012, 6, 11059–11065 CrossRef PubMed.
- P. Atkins and J. de Paula, Physical Chemistry for the Life Sciences, 2nd Edition, W. H. Freeman, 2011 Search PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- C. Wang, A. S. R. Chesman and J. J. Jasieniak, Chem. Commun., 2017, 53, 232–235 RSC.
- W. Zhai, J. Lin, Q. Li, K. Zheng, Y. Huang, Y. Yao, X. He, L. Li, C. Yu, C. Liu, Y. Fang, Z. Liu and C. Tang, Chem. Mater., 2018, 30, 3714–3721 CrossRef CAS.
- C. Wang, A. S. R. Chesman, W. Yin, L. Frazer, A. M. Funston and J. J. Jasieniak, J. Chem. Phys., 2019, 151, 121105 CrossRef PubMed.
- D. Landini and F. Rolla, J. Org. Chem., 1980, 45, 3527–3529 CrossRef CAS.
- A. Pan, B. He, X. Fan, Z. Liu, J. J. Urban, A. P. Alivisatos, L. He and Y. Liu, ACS Nano, 2016, 10, 7943–7954 CrossRef CAS PubMed.
- D. Gidalevitz, Z. Huang and S. A. Rice, Biophys. J., 1999, 76, 2797–2802 CrossRef CAS PubMed.
- Z. Wang, X. D. Wen, R. Hoffmann, J. S. Son, R. Li, C.-C. Fang, D.-M. Smilgies and T. Hyeon, PNAS, 2010, 107(40), 17119–17124 CrossRef CAS PubMed.
- L. Ramin and A. Jabbarzadeh, Langmuir, 2011, 27, 9748–9759 CrossRef CAS PubMed.
- E. De La Llave, R. Clarenc, D. J. Schiffrin and F. J. Williams, J. Phys. Chem. C, 2014, 118, 468–475 CrossRef CAS.
- L. Salem, J. Chem. Phys., 1962, 37, 2100–2113 CrossRef CAS.
- A. Antanovich, A. Prudnikau, A. Matsukovich, A. Achtstein and M. Artemyev, J. Phys. Chem. C, 2016, 120, 5764–5775 CrossRef CAS.
- J. N. Israelachvili, van der Waals Forces between Particles and Surfaces, Elsevier, 2011 Search PubMed.
- F. Krieg, Q. K. Ong, M. Burian, G. Rainò, D. Naumenko, H. Amenitsch, A. Süess, M. J. Grotevent, F. Krumeich, M. I. Bodnarchuk, I. Shorubalko, F. Stellacci and M. V. Kovalenko, J. Am. Chem. Soc., 2019, 141, 19839–19849 CrossRef CAS PubMed.
- D. Baranov, S. Toso, M. Imran and L. Manna, J. Phys. Chem. Lett., 2019, 10, 655–660 CrossRef CAS PubMed.
- A. Chernikov, T. C. Berkelbach, H. M. Hill, A. Rigosi, Y. Li, O. B. Aslan, D. R. Reichman, M. S. Hybertsen and T. F. Heinz, Phys. Rev. Lett., 2014, 113, 1–5 CrossRef PubMed.
- H. Huang, M. W. Feil, S. Fuchs, T. Debnath, A. F. Richter, Y. Tong, L. Wu, Y. Wang, M. Döblinger and B. Nickel, Chem. Mater., 2020, 32, 8877–8884 CrossRef CAS.
- N. M. Villegas, J. C. Hernandez, J. C. John and M. Sheldon, Nanoscale, 2023, 15, 9728–9737 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of computational methods, supporting figures of DLS, TEM, XRD, SADP, UV-vis absorption, NMR, etc. See DOI: https://doi.org/10.1039/d3nr04285e |
| This journal is © The Royal Society of Chemistry 2024 |