
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe photophysical properties of naphthalene bridged disilanes†
Vipin B. Kumarab,
Cassandra L. Flemingc,
Sai Shruthi Muralid,
Paul A. Humebd,
Nathaniel J. L. K. Davis bd,
Tilo Söhnelab and
Erin M. Leitao*ab
bd,
Tilo Söhnelab and
Erin M. Leitao*ab
aSchool of Chemical Sciences, The University of Auckland, Private Bag, 92019, Auckland, 1142, New Zealand. E-mail: erin.leitao@auckland.ac.nz
bThe MacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
cCentre for Biomedical and Chemical Sciences, School of Science, Auckland University of Technology, Private Bag 92006, Auckland 1142, New Zealand
dSchool of Chemical and Physical Sciences, Victoria University of Wellington, PO Box 6006140, New Zealand
First published on 16th June 2021
Abstract
Structural isomers of naphthalene-bridged disilanes were prepared via catalytic intramolecular dehydrocoupling of disilyl precursors using Wilkinson's catalyst. Interestingly, it was observed that interchanging the side groups on the silicon atoms altered the photophysical properties of the bridged disilanes. Herein, we report the first example of naphthalene bridged disilanes forming excimers in non-polar solvents. Cyclic voltammetry experiments and DFT calculations were performed to analyse the band gaps of the compounds and σ–π mixing in the bridged disilanes.
Introduction
The unique sigma electron delocalisation (σ-conjugation) present across catenated Si atoms grants polysilanes useful properties, such as absorption in the UV region and semiconducting capability, when compared to the analogous carbon-based polymers.1–3 This feature is attributed to the fact that silicon has a lower ionization energy and a higher electron affinity compared to carbon, resulting in a lower singlet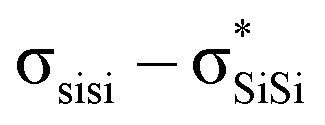 excitation energy in Si–Si bonds compared with the
excitation energy in Si–Si bonds compared with the 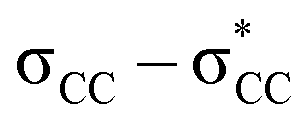 excitation energy in C–C bonds.4 Polysilanes typically absorb in the range 250–350 nm and the absorption is greatly influenced by the side groups attached to the silicon atoms. Previous reports have shown that the presence of aromatic groups on the silicon atoms cause a red shift of 25–35 nm in the UV absorption spectra and enhance σ–π mixing thereby increasing the conjugation across the system.5 Organosilanes have a number of applications in both medicinal chemistry6–8 and as energy materials.9 The photoluminescent properties of silicon based materials have provided a new platform for polysilanes to act as chemo sensors for detecting explosive materials such as 2,4,6-trinitrotoluene (TNT) and picric acid.10–12 Being photoactive in nature, long chain organosilanes have also been tested for photoconductivity and photocurrent generation.13–15
excitation energy in C–C bonds.4 Polysilanes typically absorb in the range 250–350 nm and the absorption is greatly influenced by the side groups attached to the silicon atoms. Previous reports have shown that the presence of aromatic groups on the silicon atoms cause a red shift of 25–35 nm in the UV absorption spectra and enhance σ–π mixing thereby increasing the conjugation across the system.5 Organosilanes have a number of applications in both medicinal chemistry6–8 and as energy materials.9 The photoluminescent properties of silicon based materials have provided a new platform for polysilanes to act as chemo sensors for detecting explosive materials such as 2,4,6-trinitrotoluene (TNT) and picric acid.10–12 Being photoactive in nature, long chain organosilanes have also been tested for photoconductivity and photocurrent generation.13–15
Despite having the potential to be used in various applications, the Si–Si sigma bonds cleave upon excessive exposure to UV radiation, therefore limiting the applications of these materials.5 For example, successive blue shifts were observed in the UV spectra of oligosilanes over time indicating photodecomposition.16 Sakurai et al. proposed a radical mechanism for the photodecomposition of the Si–Si bonds as they successfully isolated the silyl radicals, as detected by electron paramagnetic resonance spectroscopy, for an aryldisilane in cyclohexane.17
We propose that reinforcing the Si–Si bond in a disilane, using a tether in the form of a covalent bridge, is a potential way to support the weak Si–Si bonds. In the presence of a bridge, upon UV irradiation, the silyl radicals formed during photodecomposition will remain in close proximity to each other, therefore creating a greater chance for the two silyl radicals to recombine, reforming the covalent Si–Si bond. The bridge could be either alkyl or aromatic. Since aromatic groups attached to the silicon atoms leads to σ–π mixing, attaching aromatic substituents will presumably enhance the semi-conducting properties of the material owing to increased conjugation. Evidence for this was observed in studies conducted by Klausen et al.18 wherein the conductivity of the disilanes containing sulfur tethers were measured by a scanning tunnelling microscope (STM) break junction technique.
In our quest to prepare oligosilanes that are robust, with increased conjugation that can ultimately be used for optoelectronics, substituted naphthalene bridged disilanes seemed to be the best candidates. We have previously reported19 that naphthalene disilanes increase the overall conjugation owing to the σ–π mixing and also benefit the geometry of the resulting five membered ring system as the Si–Si bond length matches with that of the 1,8-substitution of naphthalene. In this study, we investigated the effects of modifying the substitution around the silicon atoms in the naphthalene bridged disilanes on their photophysical properties in organic solvents of varying polarity. The three target bridged disilanes (1b, 2b and 3b, Fig. 1), two of which are structural isomers of each other, were prepared.
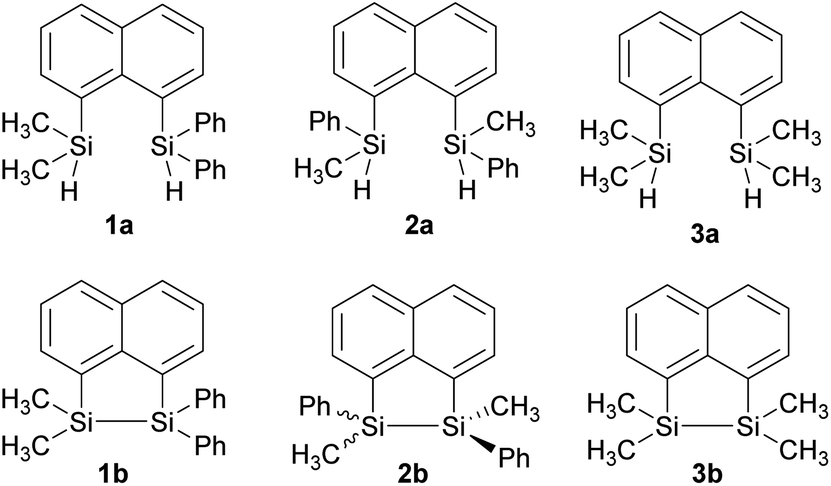 | ||
| Fig. 1 Substituted disilyl naphthalene precursors (1a, 2a and 3a) and naphthalene bridged disilanes (1b, 2b, and 3b). | ||
Results and discussion
The target naphthalene bridged disilane precursors 1a, 2a and 3a (Scheme 1) were synthesised using previously reported methodologies (Fig. S1–S7†).19–21 A two-step lithiation process to synthesize 1a was used in order to introduce the two different silanes. 1,8-Dibromonapthalene was monolithiated with n-butyllithium and further reacted with a chlorodimethylsilane (ClSiMe2H) to form 1. After purification, 1 was further lithiated and reacted with chlorodiphenylsilane (ClSiPh2H) to produce 1a as a white solid. The disilyl naphthalene derivatives (2a and 3a) were prepared from 1,8-dibromonapthalene, which was reacted with n-butyllithium to form 1,8-dilithiated naphthalene in situ. Followed by a slight excess of the respective chlorosilanes (e.g. PhMeSiClH or Me2SiClH) the formation of the required chiral and tetramethyl substituted disilyl naphthalene derivatives, 2a and 3a respectively, were obtained.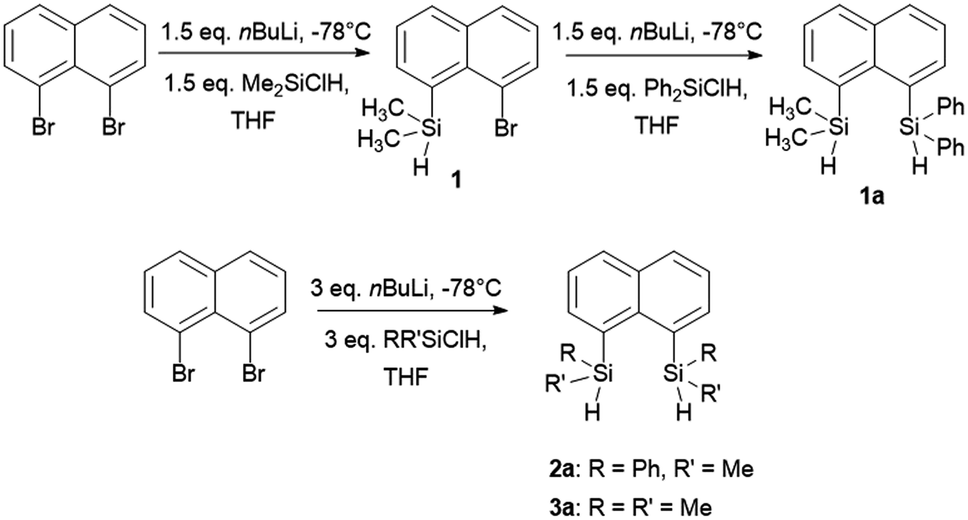 | ||
| Scheme 1 Synthesis of disilyl precursors 1a, 2a and 3a. | ||
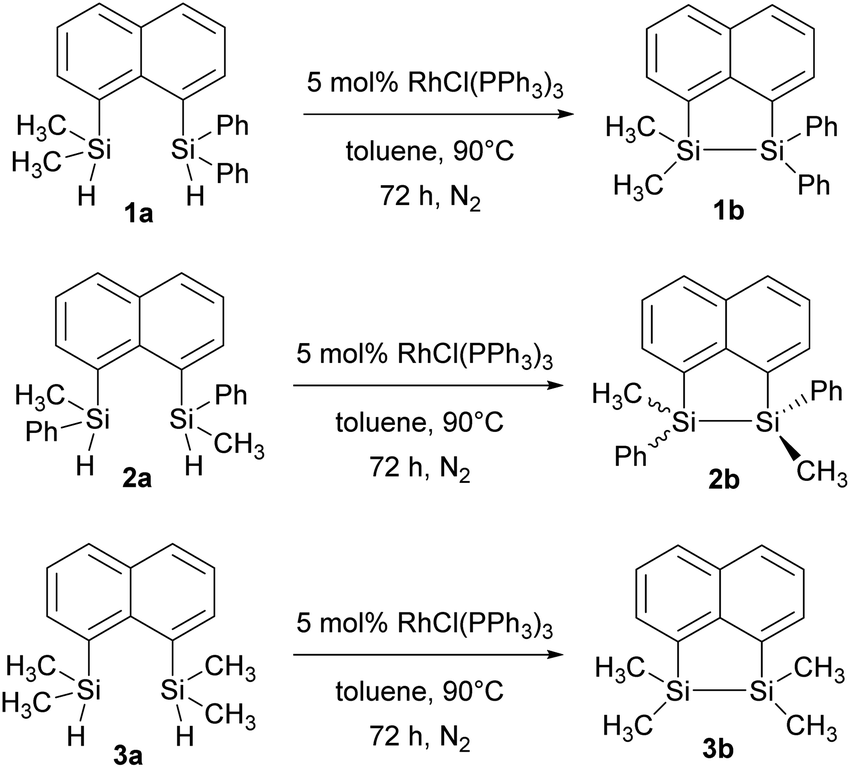
The disilyl precursors (1a, 2a and 3a) were subjected to intramolecular dehydrocoupling using 5 mol% of RhCl(PPh3)3 in toluene to produce the asymmetric naphthalene bridged disilane 1b (Fig. S8–S10†) and the symmetric naphthalene bridged disilane 2b and 3b (Scheme 2). Interestingly, it was observed that 1b and 3b existed as a crystalline solid, whereas, the chiral 2b was obtained as a waxy substance. Product 1b was dissolved in pentane and stored at −20 °C to grow single crystals (Fig. 2). However, repeated attempts to crystallise 2b, such as freezing at −20 °C and −40 °C in pentane for 24 h and 2 h, respectively, did not yield any crystalline solids, leaving the waxy substance after removal of pentane.
 | ||
| Scheme 2 Intramolecular dehydrocoupling of 1a, 2a and 3a to produce the bridged disilanes 1b, 2b and 3b. | ||
 | ||
| Fig. 2 Molecular structures of 1a (left) and 1b (right, only one of the two crystallographically independent molecules is shown) with thermal ellipsoids shown at 50% probability level. | ||
The five membered ring in 1b was observed to be slightly distorted from planarity. The C13–Si1–Si2 bond angle was 93.02° compared with the C21–Si2–Si1 bond angle of 91.86° indicating the effect of steric bulk on the two silicon centres (Table S1†). The bond angles around the silicon centre bearing the bulkier phenyl rings were narrower compared with the silicon centre containing methyl groups. The Si1–Si2 bond distance was 2.33 Å which was similar to the 3b derivative.19 NMR spectroscopy and GC-MS analysis of 2b (Fig. S11–S15†) confirmed that the product obtained was a mixture of two isomers. The cis and trans isomers of the product 2b could not be separated by silica gel column chromatography or high-pressure flash chromatography as the mixture showed no separation on TLC despite varying the solvent polarities.
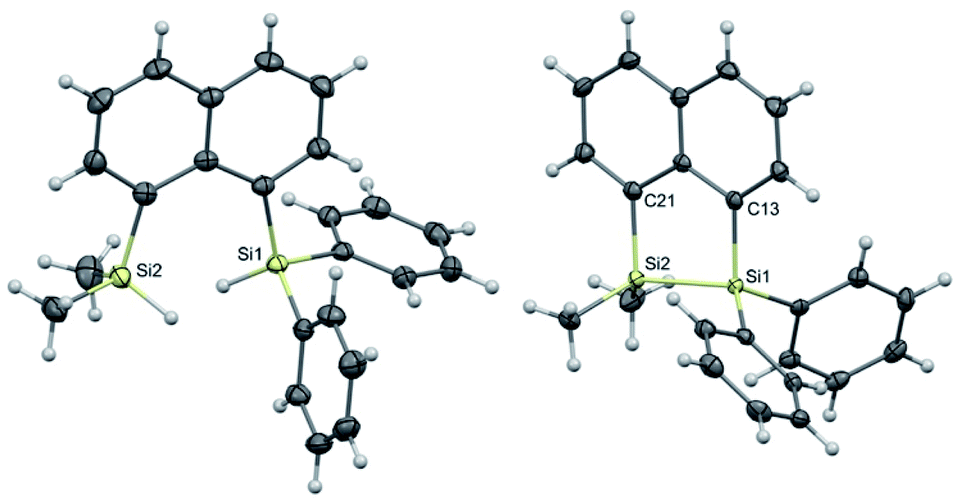
For the studies of the photophysical properties, naphthalene was selected as the parent compound. Absorption and emission spectra of naphthalene and the three-bridged disilanes (1b, 2b and 3b) were obtained in organic solvent of varying polarity (Table 1 and Fig. S17–S20†). In comparison to naphthalene, the absorption spectra of the bridged disilanes 1b, 2b and 3b displayed red-shifted absorption, thereby indicating the extended conjugation in the bridged systems. The absorption maxima of the naphthalene and compounds 1b–3b showed very little dependency on solvent polarity. The emission profiles were recorded in the same set of solvents (Fig. 3) at an excitation wavelength of 305 nm for 1b, 2b and 3b and 286 nm for naphthalene. In THF and acetonitrile, the emission spectra of the bridged silanes (1b, 2b and 3b) exhibited vibronic fine structure (maxima at ca. 335 and 345 nm), displaying a close resemblance to that of naphthalene. However, upon moving to non-polar solvents (cyclohexane and toluene), an additional broad emission band at longer wavelengths (>400 nm) is observed, suggesting the formation of excimers (excited state dimers, Fig. 3). The presence of this additional emission band in non-polar solvents was not observed for naphthalene.
| Compound | Solvent | λabsb (nm) | εmax (M−1 cm−1) | λemc (nm) | ΦFd |
|---|---|---|---|---|---|
| a nd = not determined.b Wavelength of the absorption maximum.c Wavelength of the emission maximum; for naphthalene, excitation wavelength was 286 nm. For compounds 1b–3b, excitation wavelength was 305 nm.d Fluorescence quantum yield for compounds 1b–3b in different solvent, excitation wavelength range 285–295 nm.e Emission band ascribed to the formation of excimers.f Emission band ascribed to the formation of excimers. Naphthalene was not measured in toluene due to the strong absorption of toluene in the UV region in which naphthalene absorbs. | |||||
| Naphthalene | Cyclohexane | 276 | 5976 | 324 | nd |
| Toluene | nd | nd | nd | nd | |
| THF | 276 | 4499 | 323 | nd | |
| ACN | 274 | nd | 322 | nd | |
| 1b | Cyclohexane | 292 | 4230 | 407 | 0.17 |
| Toluene | 293 | nd | 427 | 0.17 | |
| THF | 293 | 9017 | 335 | 0.007 | |
| ACN | 292 | nd | 336 | 0.05 | |
| 2b | Cyclohexane | 292 | 6797 | 335, 413e | 0.19 |
| Toluene | 293 | nd | 335, 427f | 0.15 | |
| THF | 292 | 7097 | 336 | 0.09 | |
| ACN | 292 | nd | 335 | 0.49 | |
| 3b | Cyclohexane | 290 | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 635 635 |
376 | 0.20 |
| Toluene | 289 | nd | 405 | 0.32 | |
| THF | 291 | 8345 | 335 | 0.008 | |
| ACN | 290 | nd | 335 | 0.13 | |
 | ||
| Fig. 3 Absorption (dashed) and emission (solid) profiles of (i) naphthalene, (ii) 1b, (iii) 2b, and (iv) 3b in cyclohexane (black), toluene (red), THF (blue), acetonitrile (green). Absorption spectrum is that recorded in THF. | ||
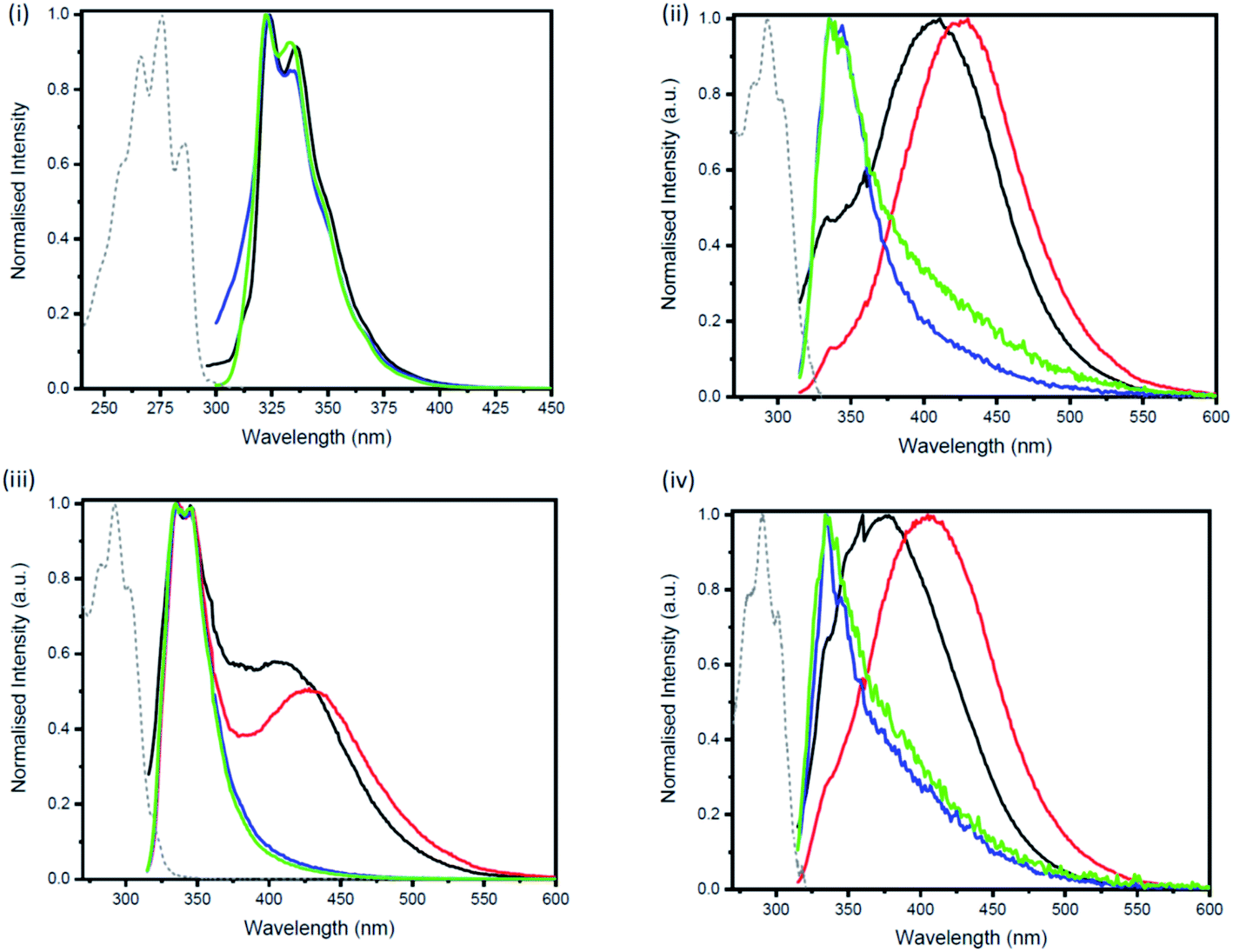
Excimers form between identical molecules, when a monomer in the excited state interacts with another monomer in the ground state, which subsequently relaxes by dissociation and involves emission of photons.22 Concentration studies were conducted in cyclohexane (Fig. 4) and THF (Fig. S21–S24†) to provide further evidence for the presence of excimers in non-polar solutions. As excimers are favoured at higher concentrations, it was anticipated that the broad band observed at >400 nm in cyclohexane would decrease upon dilution. As illustrated in Fig. 4(ii), the emission spectrum of compound 1b in cyclohexane is highly dependent on concentration. Upon dilution of a 2.54 × 10−4 M solution of 1b, the broad emission band at 407 nm significantly decreases. The same trend was also observed for compounds 2b and 3b (Fig. 4(iii and iv)). In contrast, the emission spectrum of naphthalene in cyclohexane at varying concentrations remained unchanged (Fig. 4(i)).
 | ||
| Fig. 4 Concentration dependent emission of (i) naphthalene upon excitation at 286 nm (1.76 × 10−4 M (black) → 2.33 × 10−5 M (blue)), (ii) 1b upon excitation at 305 nm (2.54 × 10−4 M (black) → 3.10 × 10−6 M (blue)), (iii) 2b upon excitation at 305 nm (1.36 × 10−4 M (black) → 1.61 × 10−6 M (blue)) and (iv) 3b upon excitation at 305 nm (3.20 × 10−5 M (black) → 1.20 × 10−6 M (blue)) in cyclohexane. | ||
Measurements of the photoluminescence quantum yields of the naphthalene bridged disilanes (1b, 2b and 3b) corroborated these results (Table 1). In THF, the three derivatives exhibit weak to moderate fluorescence (compounds 1b and 3b φF <0.01; compound 2b φF = 0.09). However, in non-polar solvents (cyclohexane and toluene) all three compounds show an increase in fluorescence quantum yields (φF = 0.15–0.32) due to the formation of excited state dimers. Interestingly, for 2b, a high fluorescence quantum yield in acetonitrile (φF = 0.49) is also observed.
Several literature examples of excimer formation in molecules containing a naphthalene moiety have been reported.22–25 Pandeeswer et al. and Boer et al. observed broad peaks in the visible regions for naphthalene diimides in polar as well as non-polar solvents.22,25 Similarly, L'Her et al. observed broad peaks in the visible regions for imidazolium-naphthalene salts in DCM.23 However, to the best of our knowledge, this is the first example where excimers are observed through intermolecular interactions between naphthalene in bridged disilane compounds. It is also important to note that this phenomenon was not observed in the disilyl naphthalene precursors, prior to the formation of the Si–Si bond. This was evident from the detailed photophysical study conducted by Maeda et al. on various silyl naphthalene compounds, similar to 1a–3a in non-polar solvents.26 As such, it appears that the Si–Si bond plays a key role in the formation of excimers in the compounds 1b–3b. The work of Karatsu et al. also highlighted the significance the presence of the Si–Si bond has on excimer formation.27–30 In their investigation, the photophysical properties of a series of permethyloligosilanes of varying length ([Me2Si]n, n = 1–4, 6), containing naphthalene or anthracene groups on the terminal silicon atoms, were explored. The two aromatic groups on the ends of the oligosilane chains have the ability interact via π-stacking, forming an intramolecular sandwich. In cases where the number of silicon atoms in the chain was greater than 1, intramolecular excimer formation was observed in cyclohexane. Interestingly, when the oligosilane chain was tetramethyl disilane (n = 2), the strongest excimer fluorescence was observed,27 indicating the disilane tether provides the optimal distance between the two end groups (i.e. naphthalene or anthracene). Furthermore, unlike in our study, excimer formation occurred for the shorter chain oligomers (n = 2, 3 for naphthalene and n = 2–4 for anthracene) in polar solvent (e.g. acetonitrile) as well.
The electrochemical behaviour of the bridged disilanes (1b, 2b, 3b) was determined using cyclic voltammetry (CV) in dry acetonitrile under inert atmosphere with ferrocene as an internal standard to calibrate the potentials (Fig. S25–S27†). The electrochemical oxidation (Eox) and reduction (Ered) potentials were used to calculate the HOMO–LUMO band gaps of the three bridged disilanes relative to ferrocene/ferrocenium (4.8 eV below the vacuum level).31,32 The band gaps for 1b and 2b were found to be similar (3.62 eV and 3.67 eV, respectively) and lower than the calculated value for compound 3b (3.85 eV) (Table S2†). This was expected due to the extra π-conjugation afforded by the phenyl substituents in compounds 1b and 2b.
DFT calculations were performed using Gaussian 09 software package,33 for 1b, the two isomers of 2b (2b-cis and 2b-trans, Fig. 5), 3b and naphthalene to calculate their band gaps in the gas phase and in solution phases using B3LYP/6-31++G**. The band gaps of 1b and 2b isomers varied form 4.01 eV to 4.07 eV and were lower in value than 3b and naphthalene (Table 2) similar to the trend observed by the CV experiments. The lower relative band gaps for compounds 1b and 2b can be attributed to the presence of phenyl substituents on each silicon atom, increasing the overall conjugation. The diagrams depicting the HOMO of the same set of compounds (Fig. 6) indicated that the naphthalene also extended the overall conjugation of the system. This extended conjugation, a result of the σ–π mixing responsible for the reduction in the band gaps, is well established in the literature.34 Another way to lower the band gap is to increase the number of Si atoms in the chain.16 It was observed that the electronic contribution from the naphthalene rings was significantly more substantial than the phenyl rings attached to the Si centres, and that all the aromatic rings contributed to the σ–π mixing therefore explaining the difference in the band gap values between the various bridged silanes (1b and 2b) and 3b. Similar results were obtained for the other naphthalene based bridged systems recently reported by our group.19
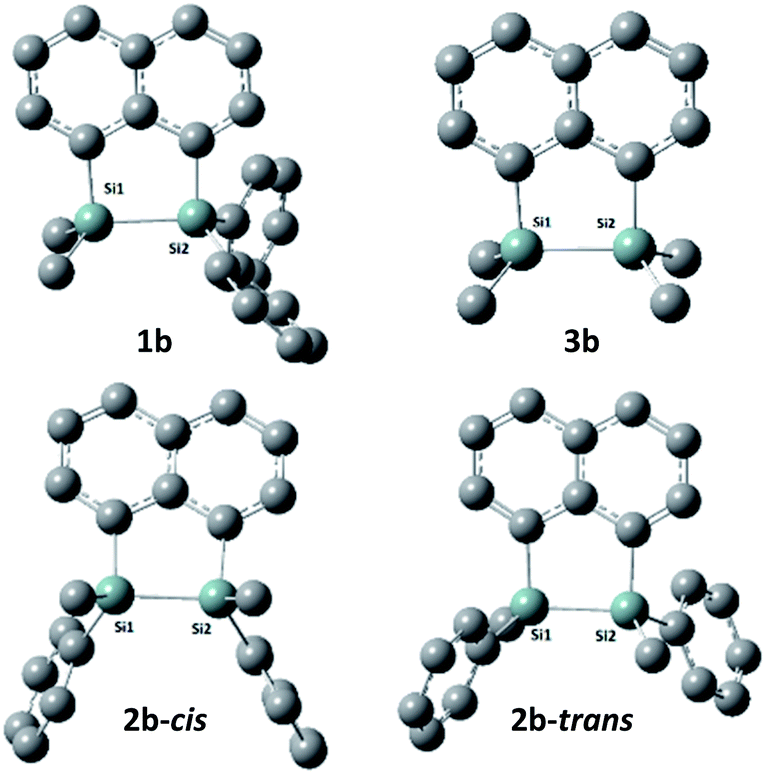 | ||
| Fig. 5 Optimized geometries of 1b, 2b(cis/trans) and 3b. (Hydrogen atoms are omitted for clarity). | ||
| Compound | Gas phase | THF | Cyclohexane | |||
|---|---|---|---|---|---|---|
| Band gap (eV) | λmax (nm) | Band gap (eV) | λmax (nm) | Band gap (eV) | λmax (nm) | |
| 1b | 4.06 | 305.0 | 4.02 | 308.0 | 4.02 | 308.0 |
| 2b-cis | 4.07 | 304.2 | 4.05 | 306.0 | 4.05 | 306.0 |
| 2b-trans | 4.04 | 306.4 | 4.01 | 308.5 | 4.02 | 308.0 |
| 3b | 4.14 | 299.0 | 4.10 | 302.0 | 4.10 | 302.0 |
| Naphthalene | 4.32 | 286.5 | 4.29 | 289.0 | 4.28 | 289.4 |
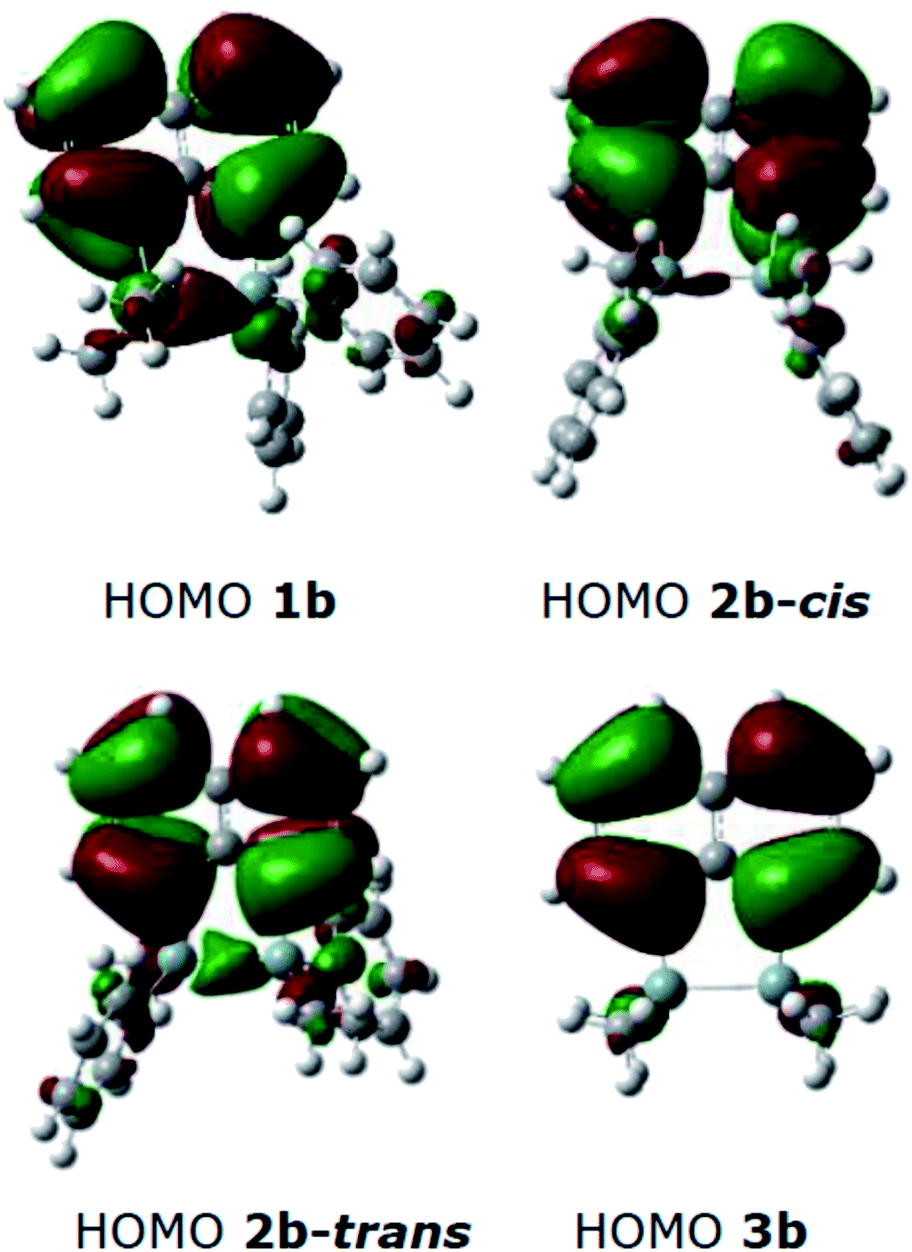 | ||
| Fig. 6 HOMO diagrams from the optimised structures of 1b, 2b(cis/trans) and 3b. | ||
Conclusions
A series of naphthalene bridged disilanes (1b, 2b, 3b) containing different arrangements of methyl and phenyl substituents at silicon were synthesised, characterised and the photophysical properties were investigated. Experimental results and DFT calculations suggested that the Si–Si bonds as well as the presence of aromatic groups, particularly naphthalene, attached to the Si atoms contributed to the lowering of the band gaps. For example, λmax was higher for 1b and 2b than 3b. Furthermore, interchanging the substitution on the silicon atoms in the disilane derivatives resulted in significant changes in the photophysical properties that were observed. The naphthalene disilane derivatives 1b (asymmetric with SiMe2 and SiPh2 substitution) and 3b (no Ph substituents) both absorbed in the UV region and showed weak emission in the near-UV region, similar to naphthalene. In contrast, the symmetric, naphthalene disilane mixture 2b(cis/trans) showed an intense emission peak in polar solvents. On switching to non-polar solvents such as cyclohexane and toluene, broadening in the visible region, corresponding to the presence of excimers, was observed in the emission profile for all three disilane molecules. As expected, concentration studies performed showed that a decrease in the concentration led to a decrease in the broad band at ca. 430 nm. The photoluminescence observed for 2b presents opportunities to use this compound in applications such as light emitting devices or as a photocurrent generator in solar cells. Further derivatising these molecules as well as studying their potential in applications is ongoing in our laboratory.Experimental
General information
All the experiments and manipulations were performed under nitrogen atmosphere in an MBraun Unilab 1200/780 glovebox or using conventional Schlenk techniques. All glassware was oven-dried overnight in a 135 °C oven. 1,8-Dibromonaphthalene and Wilkinson's catalyst were purchased from AK Scientific, chlorodimethylsilane and chlorodiphenylsilane were purchased from Sigma-Aldrich, chloromethylphenylsilane was purchased from Gelest. Materials were used directly as received. Ether and toluene were obtained from solvent purification system and later dried over activated molecular sieves (3 Å).Instrumentation
1H, 13C and 29Si{1H} DEPT NMR, 1H–29Si HSQC NMR spectra were recorded on Bruker DPX-400 (400 MHz) spectrometer. Chemical shifts were reported in parts per million (ppm) relative to tetramethylsilane and are referenced to residual protium in the NMR solvent (e.g., CHCl3 = 7.26 ppm) as the internal standard. The silicon NMR resonances were determined with a DEPT pulse sequence. Data are represented as follows: chemical shift (δ), multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = quintet, sep = septet, m = multiplet), coupling constants in Hertz (Hz) and integration. High resolution mass spectrometry measurements were made on a Bruker microTOF-QII mass spectrometer, equipped with a KD Scientific syringe pump, in positive ion ESI mode. Hard ionization mass spectrometry analysis was performed on Agilent 7890A GC + 5975C EI-MS with Agilent auto-sampler. X-ray diffraction analysis of single crystals of 1a and 1b were performed on a Rigaku Oxford Diffraction XtaLAB-Synergy-S single crystal diffractometer with a PILATUS 200 K hybrid pixel array detector using Cu Kα radiation (λ = 1.54184 Å; ESI Table 1†). The data was processed with the SHELX2018/3 and Olex2 1.3 software packages.36,37 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were inserted at calculated positions and refined with a riding model or located in the difference Fourier map and refined without restrictions (silane hydrogen atoms). Mercury 2020.3.1 was used to visualize the molecular structures.38 UV-Vis spectra were recorded on a Shimadzu UV-3600 Plus spectrophotometer and the fluorescence emission spectra were recorded on a Jasco FP-8600 spectrofluorometer. Samples were recorded in a macro quartz cuvette (light path = 10 × 10 mm). Fluorescence quantum yields were determined using an Edinburgh Photonics FLS-980 spectrophotometer equipped with an integrating sphere, using a 450 W ozone free xenon arc lamp as the light source. CV experiments were performed using a BAS CGME controlled mercury stand cell. All the DFT calculations were performed using Gaussian 09.33 The geometries of all the subject molecules were optimised using the B3LYP functional and 6-311++G** basis set.39–41Preparation of 1a
1a was synthesized via a literature procedure and the NMR data was in excellent agreement with the published compound.21Preparation of 2a
To a 100 mL Schlenk flask equipped with a stir bar was added 1,8-dibromonaphthalene (2 mmol, 0.571 g) and 15 mL THF solvent. The solution was cooled to −78 °C using a dry ice-acetone bath, to which was added n-BuLi (2.0 M in cyclohexane, 3.0 equiv., 6 mmol, 3 mL) dropwise and the mixture was stirred at −78 °C for 1 hour. To the lithiated mixture was added chloromethylphenylsilane (3 equiv., 6 mmol, 0.9 mL) and was stirred at 22 °C overnight. The reaction mixture was quenched with saturated solution of NaHCO3 (5 mL) and the aqueous layer was extracted with diethyl ether (3 × 10 mL). All the organic layers were combined and dried over Na2SO4. The crude product was obtained upon removal of solvents under vacuum as a yellow gummy substance. The crude was purified over silica gel column, using 100% hexanes as the eluent, to obtain non-separable diastereomers of 2a as a blueish-yellow gum in 64.3% yield.1H NMR (400 MHz, CDCl3): δ 7.92–7.26 (m, Ph, 16H), 5.59 (p, SiH, 2H), 0.69 (dd, CH3, 6H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 144.19, 139.80, 139.68, 139.59, 137.46, 137.42, 137.05, 137.00, 135.88, 133.66, 131.10, 129.85, 129.79, 126.53, −0.08. 29Si{1H} NMR (79.5 MHz, CDCl3): δ −20.37. EI: Found for C24H24Si2: 368.19 m/z, calc'd: 368.14 m/z.
Preparation of 1b
To a 50 mL Schlenk flask was added 1a (1 mmol, 0.37 g), Wilkinson's catalyst (5 mol%, 0.05 mmol, 46 mg) and 10 mL toluene. The mixture was heated at 90 °C for 72 hours. To the mixture was added 15 mL hexanes and the mixture was filtered through a Florisil column. The colorless solution was concentrated under high vacuum to remove the solvents and obtain the crude product. The crude was re-dissolved in pentane (7 mL) and was stored at −20 °C overnight to obtain colorless crystals of 1b in 30.0% yield.1H NMR (400 MHz, CDCl3): δ 8.02–7.38 (m, Ph, 16H), 0.56 (s, CH3, 6H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 141.86, 134.8, 133.47, 133.11, 131.78, 130.89, 128.62, 128.16, 126.94, 124.94, 124.90, −3.54. 29Si{1H} NMR (79.5 MHz, CDCl3): δ −20.62. EI: found for C24H22Si2: 366.14 m/z, calc'd: 366.12 m/z.
Preparation of 2b
To a 50 mL Schlenk flask was added 2a (1.2 mmol, 0.47 g), Wilkinson's catalyst (5 mol%, 0.06 mmol, 55 mg) and 10 mL toluene. The mixture was heated at 90 °C for 72 hours. To the mixture was added 15 mL hexane and the mixture was filtered through a Florisil column. The colorless solution was dried under high vacuum to obtain the crude product. The crude was washed with cold pentane (2 × 5 mL) to obtain pure 2b as blueish-yellow gum in 24.5% yield.From the proton NMR spectrum, it was evident that the product was a mixture of cis and trans isomers in 2:1 ratio respectively. Similar results were observed by Kim et al.,42 and based on their findings, it is concluded that the cis isomer was present in higher amount than the trans isomer.
1H NMR (400 MHz, CDCl3): δ 7.90–7.10 (m, Ph, 24H), 0.75 (s, cis-CH3, 6H), 0.60 (s, trans-CH3, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 145.70, 140.09, 140.03, 135.47, 133.87, 133.70, 133.02, 132.09, 131.71, 129.83, 128.70, 128.40, 127.87, 126.87, 126.87, 126.61, 125.00, 123.89, −5.71, −6.09. 29Si{1H} NMR (79.5 MHz, CDCl3): δ −22.90, −23.33. EI: found for C24H22Si2: 366.12 m/z. calc'd: 366.16 m/z.
Preparation of 3a and 3b
3a and 3b were synthesized via a literature procedure and the NMR data was in good agreement with what was reported.19Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the support provided by the School of Chemical Sciences, University of Auckland and the MacDiarmid Institute. VBK, SM, PAH, and EML thank Royal Society of New Zealand Marsden Fast-Start grant for the financial support and doctoral scholarships for VBK and SM. NJLKD acknowledges research funding from the Victoria Research Trust, the Science for Technological Innovation Science Challenges, the Marsden Fund, the Ministry of Business, Innovation and Employment and the Royal Society of New Zealand. We thank Tatiana Grousto for collecting the single crystal X-ray diffraction data and Dr David Ware and David Goodman for their assistance with the CV measurements. The theoretical studies were enabled by the services provide by New Zealand e-Science Infrastructure (NeSI).Notes and references
- R. West, L. D. David, P. I. Djurovich, K. L. Stearley, K. S. V. Srinivasan and H. Yu, J. Am. Chem. Soc., 1981, 103, 7352–7354 CrossRef CAS.
- E. Orti, R. Crespo, M. C. Piqueras, F. Tomas and J. L. Bredas, Synth. Met., 1993, 57, 4419–4424 CrossRef CAS.
- R. G. Kepler, Synth. Met., 1989, 28, 573–580 CrossRef.
- J. Michl, Acc. Chem. Res., 1990, 23, 127–128 CrossRef CAS.
- R. D. Miller and J. Michl, Chem. Rev., 1989, 89, 1359–1410 CrossRef CAS.
- M. J. Barnes, R. Conroy, D. J. Miller, J. S. Mills, J. G. Montana, P. K. Pooni, G. A. Showell, L. M. Walsh and J. B. H. Warneck, Bioorg. Med. Chem. Lett., 2007, 17, 354–357 CrossRef CAS PubMed.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- S. M. Sieburth, T. Nittoli, A. M. Mutahi and L. Guo, Angew. Chem., Int. Ed., 1998, 37, 812–814 CrossRef CAS PubMed.
- V. B. Kumar and E. M. Leitao, Appl. Organomet. Chem., 2020, 34, e5402 CrossRef CAS.
- K. Amro, S. Clément, P. Déjardin, W. E. Douglas, P. Gerbier, J.-M. Janot and T. Thami, J. Mater. Chem., 2010, 20, 7100–7103 RSC.
- S. J. Toal, D. Magde and W. C. Trogler, Chem. Commun., 2005, 5465–5467 RSC.
- W. Shu, C. Guan, W. Guo, C. Wang and Y. Shen, J. Mater. Chem., 2012, 22, 3075–3081 RSC.
- Y. Nakayama, A. Saito, T. Fujii and S. Akita, Photocarrier generation in polysilane films doped with and without fullerene, 1999 Search PubMed.
- R. G. Kepler, J. Zeigler, L. A. Harrah and S. R. Kurtz, Photocarrier generation and transport in -bonded polysilanes, 1987 Search PubMed.
- A. O. Kawashima, T. Oku, A. Suzuki, K. Kikuchi and S. Kikuchi, Mater. Sci. Appl., 2012, 3, 557–561 Search PubMed.
- P. Trefonas Iii, R. West, R. D. Miller and D. Hofer, J. Polym. Sci., Polym. Lett. Ed., 1983, 21, 823–829 CrossRef.
- H. Sakurai, Y. Nakadaira, M. Kira, H. Sugiyama, K. Yoshida and T. Takiguchi, J. Organomet. Chem., 1980, 184, C36–C40 CrossRef CAS.
- R. S. Klausen, J. R. Widawsky, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, J. Am. Chem. Soc., 2012, 134, 4541–4544 CrossRef CAS PubMed.
- K. M. Rabanzo-Castillo, M. Hanif, T. Söhnel and E. M. Leitao, Dalton Trans., 2019, 48, 13971–13980 RSC.
- W. Ando, T. Wakahara, T. Akasaka and S. Nagase, Organometallics, 1994, 13, 4683–4685 CrossRef CAS.
- N. Lühmann, H. Hirao, S. Shaik and T. Müller, Organometallics, 2011, 30, 4087–4096 CrossRef.
- S. A. Boer, R. P. Cox, M. J. Beards, H. Wang, W. A. Donald, T. D. M. Bell and D. R. Turner, Chem. Commun., 2019, 55, 663–666 RSC.
- M. L'Her, Y. Atoini, J. Fouchet, B. Heinrich, N. Del-Giudice, E. Scrafton, E. Bordes, L. Karmazin, L. Charbonière, L. De Cola and L. Douce, New J. Chem., 2019, 43, 12529–12532 RSC.
- S. Asir, A. S. Demir and H. Icil, Dyes Pigm., 2010, 84, 1–13 CrossRef CAS.
- M. Pandeeswar and T. Govindaraju, RSC Adv., 2013, 3, 11459–11462 RSC.
- H. Maeda, T. Maeda and K. Mizuno, Molecules, 2012, 17, 5108–5125 CrossRef CAS PubMed.
- T. Karatsu, T. Shibata, A. Nishigaki, A. Kitamura, Y. Hatanaka, Y. Nishimura, S.-i. Sato and I. Yamazaki, J. Phys. Chem. B, 2003, 107, 12184–12191 CrossRef CAS.
- T. Karatsu, T. Nakamura, M. Terasawa, S. Yagai, A. Kitamura, Y. Nishimura and I. Yamazaki, Res. Chem. Intermed., 2013, 39, 347–357 CrossRef CAS.
- T. Karatsu, T. Shibata, A. Nishigaki, K. Fukui and A. Kitamura, Chem. Lett., 2001, 30, 994–995 CrossRef.
- T. Karatsu, J. Photochem. Photobiol., C, 2008, 9, 111–137 CrossRef CAS.
- M. Shimada, Y. Yamanoi, T. Ohto, S.-T. Pham, R. Yamada, H. Tada, K. Omoto, S. Tashiro, M. Shionoya, M. Hattori, K. Jimura, S. Hayashi, H. Koike, M. Iwamura, K. Nozaki and H. Nishihara, J. Am. Chem. Soc., 2017, 139, 11214–11221 CrossRef CAS PubMed.
- T. Usuki, M. Shimada, Y. Yamanoi, T. Ohto, H. Tada, H. Kasai, E. Nishibori and H. Nishihara, ACS Appl. Mater. Interfaces, 2018, 10, 12164–12172 CrossRef CAS PubMed.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, J. R. C. M. A. Robb, G. Scalmani, V. Barone, G. A. P. B. Mennucci, H. Nakatsuji, M. Caricato, H. P. H. X. Li, A. F. Izmaylov, J. Bloino, G. Zheng, M. H. J. L. Sonnenberg, M. Ehara, K. Toyota, R. Fukuda, M. I. J. Hasegawa, T. Nakajima, Y. Honda, O. Kitao, T. V. H. Nakai, J. A. Montgomery Jr, J. E. Peralta, M. B. F. Ogliaro, J. J. Heyd, E. Brothers, K. N. Kudin, R. K. V. N. Staroverov, J. Normand, K. Raghavachari, J. C. B. A. Rendell, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. M. N. Rega, M. Klene, J. E. Knox, J. B. Cross, C. A. V. Bakken, J. Jaramillo, R. Gomperts, O. Y. E. Stratmann, A. J. Austin, R. Cammi, C. Pomelli, R. L. M. J. W. Ochterski, K. Morokuma, G. A. V. V. G. Zakrzewskil, P. Salvador, J. J. Dannenberg, A. D. D. S. Dapprich, Ö. Farkas, J. B. Foresman and J. C. a. D. J. F. J. V. Ortiz, Gaussian 09 Rev. D.01, Wallingford, CT, 2013 Search PubMed.
- K. Takeda, H. Teramae and N. Matsumoto, J. Am. Chem. Soc., 1986, 108, 8186–8190 CrossRef CAS.
- J.-L. Bredas, Mater. Horiz., 2014, 1, 17–19 RSC.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- N. T. Kim, H. Li, L. Venkataraman and J. L. Leighton, J. Am. Chem. Soc., 2016, 138, 11505–11508 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2060937 and 2060939. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra02961d |
| This journal is © The Royal Society of Chemistry 2021 |