
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of anisotropic fluorescent nanofibers assisted by electro-spinning and its optical sensing applications†
Fa-Heng
Zhang
a,
Rui-Xue
Jiang
c,
Wei
Cao
b,
Bin
Du
 a,
Ding-Yuan
Cao
d,
Zhi-Jun
Ding
*ab and
Zhi-Jun
Li
*a
a,
Ding-Yuan
Cao
d,
Zhi-Jun
Ding
*ab and
Zhi-Jun
Li
*a
aResearch Institute of Chemical Defense, Beijing 102205, China. E-mail: zhijunding01@gmail.com
bState Key Laboratory of NBC Protection for Civilian, Beijing 102205, China
cCollege of Chemical Engineering, China University of Petroleum Huadong, Qingdao Campus, Qingdao 266580, China
dNankai University, Tianjin 300071, China
First published on 23rd April 2019
Abstract
Fixing the gap between “nano-scaled” pieces and “product-scale” materials, devices or machines is an ineluctable challenge that people have to tackle. Herein, we show that combining self-assembly and electrospinning processes results in the fabrication of anisotropic fluorescent nanofibers (PDI@PVDF) in which the well-defined rod-like perylene bisimide derivative assemblies are embedded in a highly oriented way along the axis of the poly(vinylidene fluoride) (PVDF) fiber. Compared to fragile individual PDI assemblies, the electrospinning anisotropic fluorescent PDI@PVDF nanofibers not only maintain high sensitivity for aniline vapour but also exhibit an unexpected short response time for both quenching and recovering. The results demonstrate that electrospinning assistance is a versatile and effective strategy to maintain the anisotropy of fluorescent nanomaterials, building a bridge between self-assembled nano-rods and practical materials.
1. Introduction
For decades, scientists have paid more and more attention to developing technologies and processes to assemble nanometer-scale pieces into systems, components, or materials.1 As a basic structure among these micro/nanostructures, 1-dimensional (1D) nanostructures such as nanorods,2a nanotubes,3 nanowires4 and nanostrands2b exhibit unique and attractive “nano-scale” behaviors5 including quantized current–voltage behavior,6 dramatically lower melting points and significantly higher specific heats, highly anisotropic geometry, large surface-to-volume ratio, and carrier and photon confinement. However, they tend to lose these potentially beneficial traits when they are manufactured at larger “product-scale” dimensions, failing integration into devices and machines. This is mainly because the loss is in terms of the structural integrity and geometric uniformity.5a The bottom-up approach, through self-assembly and supramolecular chemistry, provides an exciting alternative route for the fabrication of various artificial materials with special micro/nanostructures in a hierarchical way.2 Up to now, solution-based self-assembly is indeed the main strategy for producing 1D structures.2b The main challenge4a is that the thermodynamic assembly processes7 usually take quite a long time (measured in hours even days) to achieve equilibrium.On the other hand, a number of techniques, including, self-assembling,2a,2b,2d nanolithography,8 electrospinning2c,9 and physical drawing,10 have currently been developed for the fabrication of 1D structure4 at “product-scale”. Especially, the electrospinning technique can boost the fabricating of polymer fibers with micro- or nanometer diameters within milliseconds from a broad range of polymer materials.2c The millisecond time-scale could prevent excessive growth of self assemblies by shortening (disrupt) the assembly process meanwhile the micro/nano-scaled diameter provides axial confining enclosure (interspace). For this reason, electrospinning shows potential applications in rapid self assembly of building blocks into high-ordered nanostructure.11 However, in most electrospun nanofibers, the dopants dispersed randomly in the polymer nanofibers without obvious ordered structures, wherein the functional dopants are not located in an ordered manner that like in their individual self assemblies.12 This is mainly because that, due to the loss in terms of the structural integrity and geometric uniformity, electrospinning process usually lead to some degree of function degradation that we have demonstrated above. In this context, how to maintain the anisotropy of dopants in electrospinning assistant 1D nanomaterials is another challenge (Scheme 1).
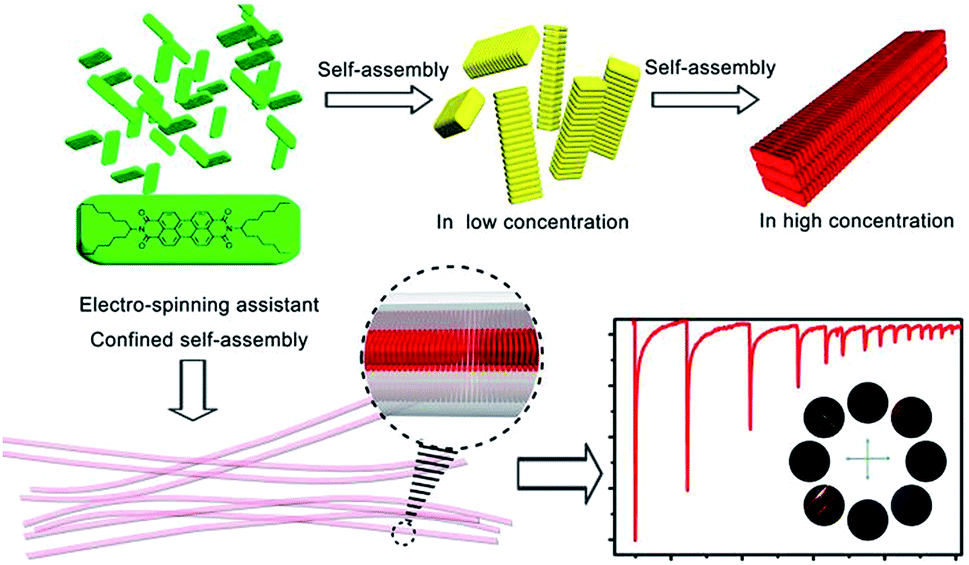 | ||
| Scheme 1 Schematic representation for the hierarchical assembly in solid-state and electro-spinning assistant confined self-assembly of N,N′-bis(1-hexylheptyl)perylene-3,4,9,10-tetracarboxylbisimide (PDI) for optical sensing applications. | ||
Based on their high thermal stability and photostability, perylene bisimide derivatives have been proven as excellent supramolecular building blocks and widely used as liquid crystals,11c,14 molecular wires,15 semiconductor,16 and transistors.17 Optical sensing behaviors18 of perylene bisimide nano-architectures in both solution and solid-state had been detailed reviewed by Zang and coworkers.2b Herein we report a strategy that combine self-assembly and electrospinning processes to construct a type of PDI@PVDF nanofibers. Structurally, the bricks (the organic assembly of PDI) are arranged in an ordered manner. Functionally, the response time of the nanofiber towards aniline was boosted from minutes to seconds.
2. Results and discussion
2.1 Self-assembly of PDI
We investigated the concentration dependent hierarchical self-assembly behavior of individual PDI in solution. The UV/vis absorption and fluorescence emission spectra of PDI were recorded in chloroform at different concentrations. The absorption spectrum of PDI at lower concentration ranges from 0 to 1.07 × 10−5 M showed sharp absorption bands at 458, 491, and 526 nm (Fig. S2a, ESI†), indicating that the PDI molecules remain monomeric phase at the relative lower concentration ranges. Higher concentration leaded dramatically rise of UV/vis absorption spectra and finally resulted a saturated spectrum.The fluorescence spectrum has a mirror-image relationship to the absorption spectrum19 and provides more information of the aggregate (Fig. S3, ESI†). At the lower concentration ranges the intensity of the emission increased upon gradual augmentation of the concentration, while the intensity of the emission decreased due to π–π electronic coupling as free molecules transforming into self-assemblies at the higher concentration ranges13 (Fig. S4, ESI†). The normalized emission spectrum showed a continuous red shift indicating the formation of the PDI aggregation underwent an assembly process (Fig. S2b, ESI†). Another evidence of the self-assembly process was the concentration dependent color change. The emission color changed gradually from bright green (for the totally free dispersed molecules) to bright red (for the self-assemblies) (Fig. S4 inset, ESI†).
This self-assembly process was further investigated in solid-state. As demonstrated by scanning electron microscopy (SEM) image in Fig. 1, when the solvent was evaporated in vacuum to give a red large agglomerates in which the sub-micrometer aggregates were randomly embedded (Fig. 1a and S5, ESI†). We attempted to maintain the thermodynamic process in order to get well-ordered PDI self-assemblies by slowing the evaporation of the solvent. As shown in Fig. 1b and S6 (ESI†), the cross section enlarged to 1 × 1 μm while the length was more than 40 μm. A further slow growing process over several days, in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 chloroform/acetonitrile solvent resulted the formation of micron-scale rod structure. Most of the rods are more than 100 μm in length, with a 5 × 5 μm cross section (Fig. 1c, d and S7, ESI†). These results indicated that the rod like structure undergo a bottom-up growing process from molecular to well-ordered rod structure. Powder X-ray powder diffraction (PXRD) measurements of PDI showed a typical π–π stacking peak (with d-spacing 3.5 Å), which indicated that the building blocks form a well-ordered crystalline arrangement19 (Fig. S8, ESI†). According to the reported literature,20 the strong π–π interaction between the PDI scaffolds played the key role in the formation of the rod like aggregates. The anisotropy character of the extended rod-like aggregate was examined by polarized light microscopy. The alternate appearance of birefringence when the rod was aligned at 45° to the polarizer implied that the optical axis is along the orientation of π–π stacking (Fig. 1e and f). No significant birefringence was observed when the agglomerates were examined under crossed polarizer conditions (Fig. S10, ESI†), indicating the randomly embedded submicrometer aggregate reduced the overall anisotropy of the agglomerates.16
:1 chloroform/acetonitrile solvent resulted the formation of micron-scale rod structure. Most of the rods are more than 100 μm in length, with a 5 × 5 μm cross section (Fig. 1c, d and S7, ESI†). These results indicated that the rod like structure undergo a bottom-up growing process from molecular to well-ordered rod structure. Powder X-ray powder diffraction (PXRD) measurements of PDI showed a typical π–π stacking peak (with d-spacing 3.5 Å), which indicated that the building blocks form a well-ordered crystalline arrangement19 (Fig. S8, ESI†). According to the reported literature,20 the strong π–π interaction between the PDI scaffolds played the key role in the formation of the rod like aggregates. The anisotropy character of the extended rod-like aggregate was examined by polarized light microscopy. The alternate appearance of birefringence when the rod was aligned at 45° to the polarizer implied that the optical axis is along the orientation of π–π stacking (Fig. 1e and f). No significant birefringence was observed when the agglomerates were examined under crossed polarizer conditions (Fig. S10, ESI†), indicating the randomly embedded submicrometer aggregate reduced the overall anisotropy of the agglomerates.16
 | ||
| Fig. 1 SEM images of PDI. (a) The solvent was evaporated in vacuum, (b) the solvent was naturally evaporated in several hours, (c) and (d) the solvent was slowly evaporated in several days, (e) PDI rods under cross-polarized microscope, (f) consecutive rotation of a single PDI rod showed alternate appearance of birefringence as the rod was aligned at 45° to the polarizer. | ||
2.2 Construction of anisotropic fluorescent nanofiber
However, our goal was to fabricate the prepared bricks (PDI assemblies) into second level structure in a high oriented way which would enable more expedient construction of integrated 1D nanofibers material. Since uniaxial optical property has a potential influence in the sensitivity of optical sensors, so the main problem to be faced was to keep the PDI assemblies in an ordered way while shorten the time of self-assembly process. An easy one-step method was developed to arrange the prepared aggregation in a neat second level row. The PDI aggregation was fabricated in a nanoscale limited space during the electrospinning process and then immediately encapsulated in the electrospun PVDF fiber. The whole process was limited in several milliseconds. As we had already mentioned that the millisecond time-scale could control the aggregate in a nano-scale size by shortening (disrupt) the assembly process meanwhile the micro/nano-scaled diameter provides axial confining enclosure (interspace) for arranging the assemblies in an order way.The microscope images of the electrospun PVDF fiber (PDI@PVDF) are presented in Fig. 2. The characteristic SEM image (Fig. 2a and b) reveals the randomly-arranged fibers were gathered by a flat collector. From these images we can see that the diameters of these fibers (0.5–1μm) are comparable to or less than the cross section of the rod-like PDI aggregate as shown in Fig. 1. The typical TEM images (Fig. 2c and d) show that the PDI aggregates are confined in the PVDF matrix, of which the longitudinal axis paralleled to the fiber axis. An aligned fibers was obtained by using a rotating cylindrical mandrel collector as shown in Fig. 2e and f. The different emission color of the fiber indicated that the confined formation of the PDI aggregate in the PVDF fiber shared the same hierarchical assembly process which had been discussed above. The hierarchical PDI aggregate with different emission color were frozen in the PVDF matrix during the fiber drawing process. The anisotropy character of the PDI@PVDF fibers depends on not only the orientation of the fibers, but also the orientation of PDI assemblies in it. The approximate paralleled fibers were used in the fluorescent microscopy examination. When rotating the fibers under crossed polarization, the same result was obtained in Fig. 2f implied that the optical axis is along the fiber axis.21 The anisotropy characters of both pure PDI assemblies (Fig. 1f) and PDI@PVDF fiber (Fig. 2f) further confirmed the longitudinal axis of the PDI assemblies paralleled to the PDI@PVDF fiber axis.21
 | ||
| Fig. 2 (a) SEM (scale bar = 20 μm), (b) enlarged SEM (scale bar = 5 μm), (c) TEM (scale bar = 2 μm), (d) enlarged TEM (scale bar = 500 nm), (e) confocal microscopy and (f) cross-polarized microscope images of PDI@PVDF fiber formed from PDI and PVDF. | ||
2.3 Optical sensing applications
As previously reported, PDI nanofibers with enhanced long-range exciton migration property rising from intermolecular π-electronic coupling can act as molecular wires for the fluorescence sensing of organic vapor.4–6 The prepared 1D PDI@PVDF fibril film (Fig. S11e and f, ESI†) in which the PDI assemblies was arranged along the long axis of the fiber, may also worked in a “molecular wire” mode. For ease of comparison, herein, we also chose the frequently-used aniline as analyte molecules to investigate the amplified quenching behavior.The PDI@PVDF film was fixed on a special designed scaffold then covered with a quartz cell (Fig. S12, ESI†). A 365 nm LED lamp was used to excite the film, and the fluorescence variation in the presence of organic amines (aniline) was continuously monitored by a spectrometer. As Fig. 3 showed, the fluorescence emission was significantly quenched by increasing aniline vapor concentration. Comparing with previous results, although the PDI was not attentional designed just like Liu5 and Fang6 reported, the PDI@PVDF fibril film still shown unexpected high sensitivity for aniline vapor. Concentration of aniline low to 0.4–50 ppb could induce observable fluorescence quenching (Fig. 3). Increased concentration up to 1250 ppb did result in further significant quenching (Fig. S13, ESI†). As an important factor for the practical use of a sensing film (Fig. S11e and f, ESI†), reversibility of the PDI@PVDF fibril film was investigated in detail. As shown in Fig. 3, the emission intensity of the film was monitored every 0.3 s. After being exposed to various concentrations of aniline vapors (Fig. 3 inset), the emission immediately quenched in less than 1 second. Additionally, the fluorescence quenching induced by amines was highly reversible. Upon exposed to 50 ppb aniline, the quenched fluorescence of the film could fully recover by re-exposing it to an ambient air-flow for only 20 seconds. However, the recovering time after exposure do has dependence on the aniline concentration. As shown in Fig. S13 (ESI†), the recovering time extended to 40 seconds when the concentration of aniline vapor reached 1250 ppb. This shorted response time for both quenching and recovering, in contrast to the other reversible PDI based sensor systems, in which the recovering time usually need 5 to 30 minutes due to the stable chemical binding between amines and the sensing material,15,22 indicated excellent reversibility of our hierarchical nanofiber.
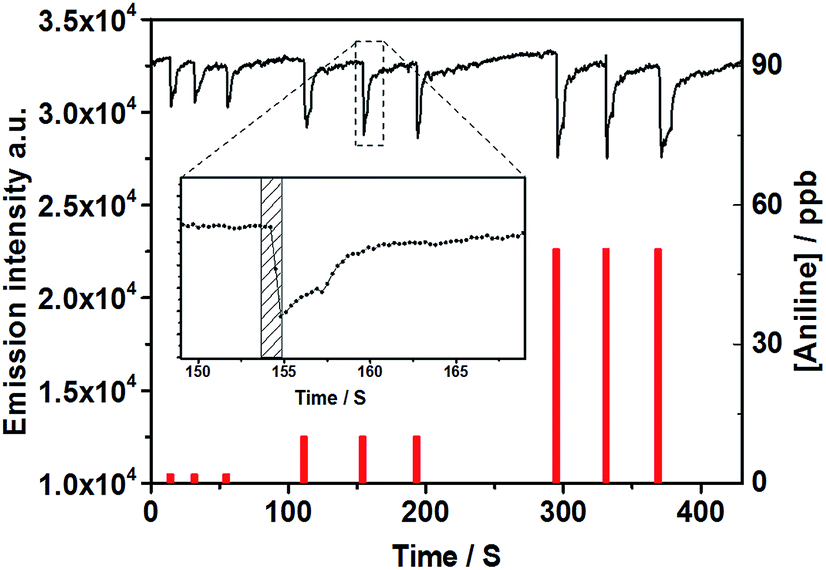 | ||
| Fig. 3 Time-resolved fluorescence intensity (black line) of a PDI@PVDF fibril film upon consecutive exposures to various concentrations of aniline vapors. (1 s exposures, concentrations indicated by the red columns). Inset: fluorescence change upon exposure to 10 ppb aniline, and the filled rectangle indicated the exposure times (the emission intensity at 630 nm was measured for every 300 ms, λex = 365 nm). | ||
Since the sensing behavior of PDI@PVDF fiber is mainly determined by PDI self-assembly, the fiber shows the same selectivity to organic amine. Common solvents like chloroform, toluene and methanol have little effect on the fluorescence emission of the film (Fig. S14, ESI†). Furthermore, the sensing behavior of PDI@PVDF fiber to different types of organic amines was investigated. As shown in Fig S14 (ESI†), the quenching efficiency induced by aromatic amines is higher than that induced by aliphatic amines, which might be attributed to the stronger electron-donating ability of the conjugated structure of aromatic amines. Additionally 4-methylaniline exhibited stronger quenching ability, while 4-nitroaniline did not cause significant quenching indicating that strong electron-withdrawing group on aniline might significantly reduce the electron cloud density of amino group and weaken the electron-donating ability of amino group. The significant quenching observed with phenol is likely due to its strong reducing power.22 Without specific modification of the molecular structure of PDI, it is difficult to distinguish phenolic compounds from organic amines only by fluorescence quenching. The static measurement and the fluorescence lifetime measurement has been performed in solution state to validate the quenching mechanism (Fig. S15 and S16, ESI†). According to the previous literature,15 present results (Fig. S17, ESI†) indicate that the fluorescence emission of PDI@PVDF fiber is quenched by both complex formation with aniline as well as by collision with aniline.
The rapid response behaviour of PDI@PVDF fibril film towards aniline can be attributed to the hierarchical assembly strategy. PDI firstly self-assembles into to well-ordered rod structure driven by the strong π–π interaction, resulting the sensing behaviour to aniline which depends on the photoinduced electron transfer from aniline to PDI probe.6 According to the literature,1b the long-range molecular arrangement leads to 1D enhanced exciton migration (via intermolecular π-electronic coupling) along the long axis of the assemblies, enabling amplified fluorescence quenching by the surface adsorbed fewer aniline molecules (quencher molecules). Subsequent electrospinning the fluorescent PDI@PVDF into nanofibers not only maintain the anisotropy of PDI in the thin film, but also the porosity between nanofibers facilitate the diffusion of gaseous analyte molecules throughout the film matrix, thus leading to instant capture (and accumulation) or release of the vapor species. To verify our hypothesis, control experiments were carried out. We prepared dense thin film via physical doping PDI into PVDF matrix (denoted as PDI⊂PVDF). The fluorescence emission of PDI⊂PVDF and pure PDI molecular lamellae was also significantly quenched by aniline vapor. Compare to PDI@PVDF fibril film, the even higher quenching efficiency might be attributed to the much longer long-range molecular arrangement. Time-resolved fluorescence experiment showed that the recovery time of PDI⊂PVDF and pure PDI molecular lamellae towards aniline vapor are 6 min and 19 min (Fig. S18, ESI†), respectively, both significant longer than PDI@PVDF fibril film. These results demonstrate that electrospinning assistant is a versatile and effective strategy to simultaneously maintain the anisotropy of 1D nanomaterials and accelerate the speed of their response to external stimulus. The ultra-sensitivity, the rapid sensing response, excellent reversibility along with the simple and economical preparation make the nanofibril PDI@PVDF film an ideal sensing material for designing trace amine onsite monitoring device.
3. Conclusions
We prepared a simple electrospun fiber for organic amines sensing, in which PVDF was chosen as mortar, meanwhile a simple perylene bisimide derivative PDI was used as the functional bricks. The well-defined PDI assemblies were embedded in the PVDF fiber in a highly oriented way along the axis of the fiber. Profiting from the structural integrity and geometric uniformity of the PDI assemblies in nanoscale and the 3D continuous, the fibroid electrospun PDI@PVDF film with micrometer scaled mesh-like porous secondary structure not only maintain high sensitivity for aniline vapour but also exhibit un-expected short response time for both quenching and recovering. Compared to fragile intrinsic PDI rod-like assemblies, the electrospun fiber PDI@PVDF film also shows greater flexibility originating from the soft PVDF scaffold. This report may provide an innovative approach to construction 1D fibers with desired size and morphology via appropriate choice of various lime mortar (alternative polymer) and brick (assembly blocks).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support for this work from the National Natural Science Foundation of China (21402237).References
- (a) G. Yuan, H. Jiang, L. Zhang, Y. Liu and Y. Cui, Coord. Chem. Rev., 2019, 378, 483–499 CrossRef CAS; (b) F. Wurthner, C. R. Moller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed; (c) X. Hu, X. Wu, S. Wang, D. Chen, W. Xia, C. Lin, Y. Pan and L. Wang, Polym. Chem., 2013, 4, 4292–4297 RSC; (d) Z. Li, Z. Hou, H. Fan and H. Li, Adv. Funct. Mater., 2017, 27, 1604379 CrossRef.
- (a) Y. Chen and Y. Liu, Adv. Mater., 2015, 27, 5403–5409 CrossRef CAS; (b) L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596–1608 CrossRef CAS; (c) Y. J. Kim, D. Y. Kim, J. H. Lee, C. Nah, J. H. Lee, M. H. Lee, H. Y. Kim, S. W. Kuo, S. Shin and K. U. Jeong, J. Mater. Chem., 2012, 22, 13477–13480 RSC; (d) L. Liu, X. Chen, K. Liu, M. He, G. Wang, X. Chang and Y. Fang, ACS Appl. Mater. Interfaces, 2015, 7, 21364–21372 CrossRef CAS; (e) X. Chang, Z. Zhou, C. Shang, G. Wang, Z. Wang, Y. Qi, Z. Li, H. Wang, L. Cao, X. Li, Y. Fang and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 1757–1765 CrossRef CAS.
- Y. Zhao and L. Jiang, Adv. Mater., 2009, 21, 3621–3638 CrossRef CAS.
- (a) J. F. Fennell Jr, S. F. Liu, J. M. Azzarelli, J. G. Weis, S. Rochat, K. A. Mirica, J. B. Ravnsbaek and T. M. Swager, Angew. Chem., Int. Ed. Engl., 2016, 55, 1266–1281 CrossRef; (b) R. Agarwal, Small, 2008, 4, 1872–1893 CrossRef CAS.
- (a) P. Wang, Y. Wang and L. Tong, Light: Sci. Appl., 2013, 2, e102 CrossRef; (b) A. K. Feldman, M. L. Steigerwald, X. Guo and C. Nuckolls, Acc. Chem. Res., 2008, 41, 1731–1741 CrossRef CAS; (c) Q. H. Cui, Y. S. Zhao and J. Yao, Chem. Sci., 2014, 5, 52–57 RSC; (d) S. Guo, Y. Song, Y. He, X. Hu and L. Wang, Angew. Chem., 2018, 130, 3217–3221 CrossRef.
- (a) S. Wang, M. Qiao, Z. Ye, D. Dou, M. Chen, Y. Peng, Y. Shi, X. Yang, L. Cui, J. Li, C. Li, B. Wei and W.-Y. Wong, iScience, 2018, 9, 532–541 CrossRef CAS; (b) T. S. Kang, A. P. Smith, B. E. Taylor and M. F. Durstock, Nano Lett., 2009, 9, 601–606 CrossRef CAS.
- Y. Wang, J. He, C. Liu, W. H. Chong and H. Chen, Angew. Chem., Int. Ed., 2015, 54, 2022–2051 CrossRef CAS.
- X. Ma, F. Li, Z. Xie, M. Xue, Z. Zheng and X. Zhang, Soft Matter, 2017, 13, 3685–3689 RSC.
- J. Yong, F. Chen, Q. Yang, J. Huo and X. Hou, Chem. Soc. Rev., 2017, 46, 4168–4217 RSC.
- H. W. Liang, S. Liu and S. H. Yu, Adv. Mater., 2010, 22, 3925–3937 CrossRef CAS.
- (a) C. L. Zhang and S. H. Yu, Chem. Soc. Rev., 2014, 43, 4423–4448 RSC; (b) S. Maity, K. A. Kozek, W. C. Wu, J. B. Tracy, J. R. Bochinski and L. I. Clarke, Part. Part. Syst. Charact., 2013, 30, 193–202 CrossRef CAS; (c) H. J. Kim, W. B. Jung, H. S. Jeong and H. T. Jung, J. Mater. Chem. C, 2017, 5, 12241–12248 RSC.
- E. Schoolaert, R. Hoogenboom and K. De Clerck, Adv. Funct. Mater., 2017, 27, 1702646 CrossRef.
- K. Sano, Y. Ishida and T. Aida, Angew. Chem., Int. Ed. Engl., 2018, 57, 2532–2543 CrossRef CAS.
- A. M. Kendhale, A. P. H. J. Schenning and M. G. Debije, J. Mater. Chem. A, 2013, 1, 229–232 RSC.
- H. Peng, L. Ding, T. Liu, X. Chen, L. Li, S. Yin and Y. Fang, Chem. - Asian J., 2012, 7, 1576–1582 CrossRef CAS.
- K. Balakrishnan, A. Datar, R. Oitker, H. Chen, J. M. Zuo and L. Zang, J. Am. Chem. Soc., 2005, 127, 10496–10497 CrossRef CAS.
- S. Prasanthkumar, S. Ghosh, V. C. Nair, A. Saeki, S. Seki and A. Ajayaghosh, Angew. Chem., Int. Ed. Engl., 2015, 54, 946–950 CrossRef CAS.
- (a) Y. Liu, K. R. Wang, D.-S. Guo and B. P. Jiang, Adv. Funct. Mater., 2009, 19, 2230–2235 CrossRef CAS; (b) D. J. Yang, I. Kamienchick, D. Y. Youn, A. Rothschild and I. D. Kim, Adv. Funct. Mater., 2010, 20, 4258–4264 CrossRef CAS; (c) K. Liu, C. Shang, Z. Wang, Y. Qi, R. Miao, K. Liu, T. Liu and Y. Fang, Nat. Commun., 2018, 9, 1695 CrossRef.
- N. Wu, Y. Zhang, C. Wang, P. M. Slattum, X. Yang and L. Zang, J. Phys. Chem. Lett., 2017, 8, 292–298 CrossRef CAS.
- K. Balakrishnan, A. Datar, T. Naddo, J. L. Huang, R. Oitker, M. Yen, J. C. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS.
- Y. J. Kim, D. Y. Kim, J. H. Lee, C. Nah, J. H. Lee, M. H. Lee, H. Y. Kim, S. W. Kuo, S. Shin and K. U. Jeong, J. Mater. Chem., 2012, 22, 13477–13480 RSC.
- Y. Che, X. Yang, S. Loser and L. Zang, Nano Lett., 2008, 8(8), 2219–2223 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9ra00502a |
| This journal is © The Royal Society of Chemistry 2019 |