
Photoelectron spectroscopy of cryogenically cooled NiO2−via slow photoelectron velocity-map imaging†
Mark C.
Babin‡
 a,
Martin
DeWitt
a,
Jascha A.
Lau
a,
Marissa L.
Weichman§
a,
Jongjin B.
Kim¶
a,
Lan
Cheng
*b and
Daniel M.
Neumark
*ac
a,
Martin
DeWitt
a,
Jascha A.
Lau
a,
Marissa L.
Weichman§
a,
Jongjin B.
Kim¶
a,
Lan
Cheng
*b and
Daniel M.
Neumark
*ac
aDepartment of Chemistry, University of California, Berkeley, CA 94720, USA. E-mail: dneumark@berkeley.edu
bDepartment of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, USA. E-mail: lcheng24@jhu.edu
cChemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
First published on 7th July 2022
Abstract
High-resolution anion photoelectron spectra of cryogenically cooled NiO2− anions, obtained using slow photoelectron velocity-map imaging (cryo-SEVI), are presented in tandem with coupled cluster electronic structure calculations including relativistic effects. The experimental spectra encompass the ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 1Σg+ ← 2Πg, ã3Πg ← 2Πg, and Ã1Πg ← 2Πg photodetachment transitions of linear ONiO0/−, revealing previously unobserved vibrational structure in all three electronic bands. The high-resolution afforded by cryo-SEVI allows for the extraction of vibrational frequencies for each state, consistent with those previously measured in the ground state and in good agreement with scalar-relativistic coupled-cluster calculations. Previously unobserved vibrational structure is observed in the ã3Πg and Ã1Πg states and is tentatively assigned. Further, a refined electron affinity of 3.0464(7) eV for NiO2 is obtained as well as precise term energies for the ã and à states of NiO2 of 0.3982(7) and 0.7422(10) eV, respectively. Numerous Franck–Condon forbidden transitions involving the doubly degenerate ν2 bending mode are observed and ascribed to Herzberg-Teller coupling to an excited electronic state.
1Σg+ ← 2Πg, ã3Πg ← 2Πg, and Ã1Πg ← 2Πg photodetachment transitions of linear ONiO0/−, revealing previously unobserved vibrational structure in all three electronic bands. The high-resolution afforded by cryo-SEVI allows for the extraction of vibrational frequencies for each state, consistent with those previously measured in the ground state and in good agreement with scalar-relativistic coupled-cluster calculations. Previously unobserved vibrational structure is observed in the ã3Πg and Ã1Πg states and is tentatively assigned. Further, a refined electron affinity of 3.0464(7) eV for NiO2 is obtained as well as precise term energies for the ã and à states of NiO2 of 0.3982(7) and 0.7422(10) eV, respectively. Numerous Franck–Condon forbidden transitions involving the doubly degenerate ν2 bending mode are observed and ascribed to Herzberg-Teller coupling to an excited electronic state.
I. Introduction
Nickel oxides are an important class of catalytic materials with a wide range of industrial applications including the formation of syngas,1,2 oxidative dehydrogenation of alkanes,3,4 and carbon monoxide oxidation.5 Therefore, understanding their properties is of both technological and fundamental importance. While much of the emphasis of nickel oxide catalysis involves condensed phase experiments, the gas phase study of isolated mass-selected metal oxide clusters provides a complementary molecular-level understanding of the structure and reactivity of these species.6,7 Here, we report high-resolution anion photoelectron spectra of cryogenically cooled NiO2−, revealing a wealth of vibronic structure throughout the three lowest-lying electronic states of NiO2.There are relatively few experimental studies concerning the structure8–12 and reactivity of NiO20/−.13–16 Neutral NiO2 was first studied in gas matrices, where three isomers were identified in an Ar matrix:9 cyclic Ni(O2), bent NiOO, and linear ONiO (referred to as NiO2 hereafter). Subsequent work in Ne matrices found preferential formation of NiO2 with trace Ni(O2), allowing for the extraction of two and three vibrational frequencies for these species, respectively.12 In the gas phase, anion photoelectron spectroscopy (PES) has been used to probe the geometric and electronic structure of these species. The first PES study of NiO2− was performed by Wu and coworkers,10 who observed photodetachment from both the bent Ni(O2) and linear NiO2 anions, finding the electron affinity (EA) of Ni(O2) to be 0.82(3) eV, considerably lower than that of NiO2 at 3.05(1) eV. The NiO2 band showed partially resolved vibrational structure with a frequency of 750 cm−1. This work also found transitions to two low-lying electronic states of NiO2 with term energies of 0.40(2) and 0.77(3) eV. Subsequent anion PES work performed by Ramond et al.11 reexamined the ground state spectrum of NiO2 with improved resolution, yielding a refined EA of 3.043(5) eV as well as the v1 symmetric stretch frequency of 745(30) cm−1 from a progression in this mode.
The electronic and vibrational structure of NiO2 pose a considerable challenge to theory owing to its multireference nature.17,18 Treatments using density functional theory,9,12,19–21 coupled cluster methods,21 and multireference methods22 have found a Σg+ ground electronic term for linear NiO2 that lies 0.7–1.5 eV below the Ni(O2) structure. In both structures, there are numerous low-lying singlet and triplet species calculated to reside within ∼2 eV of the ground state. The most definitive of these works is a joint multiconfiguration self-consistent field (MCSCF) and multireference configuration interaction (MRCI) study by Hübner,22 which finds a 1Σg+ ground state with 3Πg and 1Πg states lying 0.537 and 0.943 eV higher in energy, in good agreement with the observed term energies from anion PES. Less work has been done on the NiO2− anion, with an early DFT study by Gustev et al.20 finding a 2A2 bent ground state and more recent DFT work by Deng and coworkers19 finding a linear 2Πg ground state.
Here, we utilize slow electron velocity-map imaging spectroscopy of cryogenically-cooled anions (cryo-SEVI), a high-resolution variant of anion photoelectron spectroscopy, to study the photodetachment of NiO2−. In this method, cold ions are detached with a tunable laser and the resultant electron kinetic energy distribution is analyzed by a velocity-map imaging (VMI) spectrometer that is optimized for the detection of slow electrons, yielding photoelectron spectra with sub-meV resolution.23,24 Revisiting the photoelectron spectra of NiO2− with cryo-SEVI provides substantial improvements in resolution over previous work. Beyond well-resolved vibrational structure in the ground and excited states, we observe a number of Franck–Condon (FC)-forbidden transitions in the ground state that display behavior distinct from that of the FC-allowed transitions near the photodetachment threshold. These features are ascribed to vibronic coupling to an excited electronic state. Our findings are supported by scalar-relativistic coupled-cluster calculations, with computed term energies and vibrational frequencies in good agreement with our observed spectra.
II. Experimental methods
The cryo-SEVI method has been described in detail previously.23,25,26 Here, NiO2− anions are formed via laser ablation using a rotating and translating nickel disk onto which a frequency-doubled Nd:YAG is focused. The resulting plasma is entrained within a pulse of He carrier gas from an Even-Lavie solenoid valve,27 with residual O2 in this carrier gas serving to form NiO2− as it passes through a narrow channel to foster collisional cooling and cluster formation. The ions then pass through a skimmer before entering a radiofrequency (RF) hexapole ion guide and RF quadrupole mass filter that direct them into a linear RF octupole ion trap held at 5 K and filled with a buffer gas mixture of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :80 H2:He. Collisions with this cold buffer gas mixture result in effective vibrational, rotational, and electronic cooling of the ions, leading to internal temperatures of around 10 K.26,28–30
:80 H2:He. Collisions with this cold buffer gas mixture result in effective vibrational, rotational, and electronic cooling of the ions, leading to internal temperatures of around 10 K.26,28–30
The ions are held approximately 40 ms in the trap before being extracted into an orthogonal Wiley-McLaren time-of-flight mass spectrometer31 and focused into the interaction region of a standard three-plate Eppink-Parker VMI electrostatic lens assembly.32 In the VMI spectrometer, the ions are photodetached using the frequency-doubled output of a dye laser pumped by the second harmonic of a Nd:YAG laser operating at 20 Hz.
The resulting photoelectrons are projected onto a 2D detector comprising two chevron-stacked microchannel plates coupled to a phosphor screen, which is photographed by a CCD camera after each laser shot.33 Each image is analyzed for individual electron events for which the centroids are calculated and binned into a 1024 × 1024 grid.34 The three-dimensional electron velocity distribution is reconstructed from the accumulated images using an inverse-Abel method.35 The radial position of features in the reconstructed image is related to electron kinetic energy (eKE) by acquiring VMI images for detachment from atomic F− at several photon energies.36
The VMI spectrometer has an approximately constant resolving power, ΔeKE/eKE,32 yielding the highest eKE resolution for slow photoelectrons. As such, a SEVI spectrum is acquired by first taking an overview spectrum at a sufficiently high photon energy to access all (or most) of the photodetachment transitions before tuning the detachment laser to energies slightly above features of interest. This procedure results in the collection of high-resolution spectra over narrow energy windows that are then concatenated and scaled to match intensities in the overview spectrum, which is less sensitive to variation of the photodetachment cross section with photon energy. Spectra are plotted as a function of electron binding energy (eBE), given by eBE = hv − eKE.
III. Computational methods
Electronic structure calculations for the2Πg state of NiO2− and the 1Σg+ state of NiO2 have been carried out at the coupled-cluster singles doubles augmented with a noniterative triples [CCSD(T)]37 level of theory to determine the equilibrium structures and the harmonic vibrational frequencies of these states as well as to enable Franck–Condon simulation for the electron detachment transitions from the electronic ground state of NiO2− to that of NiO2. Scalar-relativistic effects have been taken into account using spin-free exact two-component theory in its one-electron variant (SFX2C-1e)38,39 together with the aug-cc-pVTZ basis set40–42 recontracted for SFX2C-1e calculations. Calculations using a hierarchy of CC methods including CCSD,43 CC singles doubles triples (CCSDT),44,45 and CC singles doubles triples quadruples (CCSDTQ)46,47 have also been performed for the 1Σg+ state of NiO2 to examine the reliability of the CCSD(T) results, as this state exhibits strong electron-correlation effects.17,18 The results of this analysis are outlined in Table S1 of the ESI.† Harmonic frequency calculations have been performed by means of numerical differentiation of analytically evaluated gradients using the analytic-gradient techniques for the CC methods and the SFX2C-1e scheme as implemented in the CFOUR program package.48–55 FC simulations presented here employ the double-harmonic approximation using the fcsquared module56 of the CFOUR program.
We have also performed SFX2C-1e equation-of-motion coupled-cluster singles doubles (EOM-CCSD)57 and EOM-CC singles doubles triples (EOM-CCSDT)58,59 calculations to determine the excitation energies of NiO2 in the anionic equilibrium structure. These are combined with the computed vertical detachment energy for the ground 1Σg+ state of NiO2 to describe the vertical detachment energies from the ground state of NiO2− to these excited states of NiO2. Here, aug-cc-pVQZ basis sets were used for EOM-CCSD calculations. The triples corrections have been obtained as the differences between EOM-CCSDT and EOM-CCSD results using cc-pVDZ basis sets. All calculations have been performed using the CFOUR program package,53,55 except that the EOM-CCSDT calculations of triplet excited states have been carried out using the MRCC program package.60–62
IV. Results and discussion
The cryo-SEVI spectrum of NiO2− is presented in Fig. 1. Fig. 1a shows the lower-eBE region of structure, spanning 24400–28000 cm−1 and covering the ground state band of the NiO2 ← NiO2− photodetachment transition previously observed.10,11 The higher-energy region of the spectra shown in Fig. 1b spans 27550–32500 cm−1 and encompasses detachment transitions to excited electronic states of NiO2 as will be discussed in Sections IV.A and IV.D. In both figures, a low-resolution overview spectrum (blue) is displayed above high-resolution composite spectra (black) taken at several photon energies.
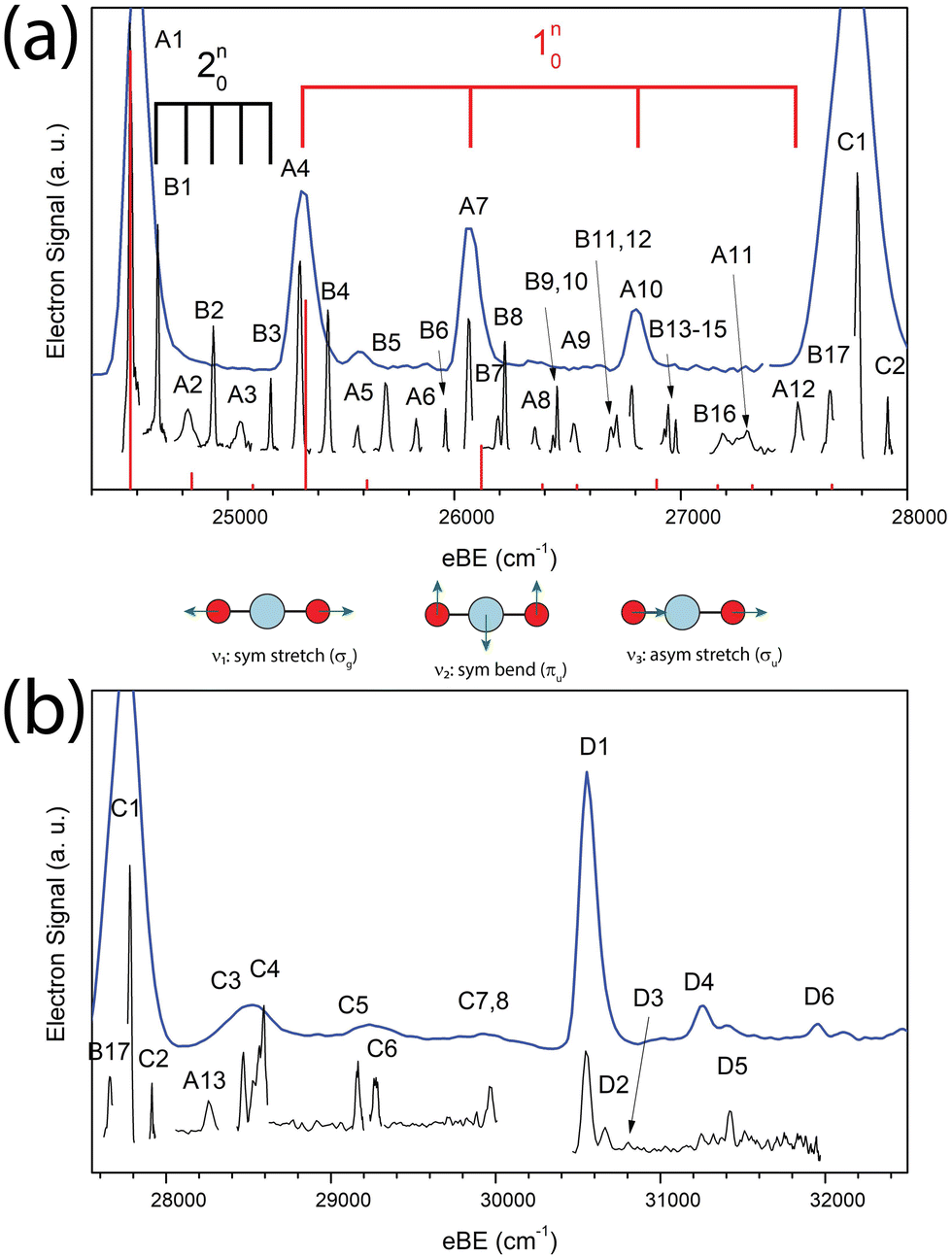 | ||
| Fig. 1 Cryo-SEVI spectra of NiO2− showing detachment to the (a) 1Σg+ ground state and (b) ã3Πg and Ã1Πg excited states. Blue traces are low-resolution overview scans taken with photon energies of 30394 and 35025 cm−1 for panels (a) and (b), respectively. Black traces are high-resolution scans taken at variable photon energies, while red stick spectra in (a) represent FC simulations for the 1Σg+ ← 2Πg photodetachment transition from NiO2−. Vibrational modes for the ground state are depicted below the ground-state spectrum. | ||
These spectra represent a significant improvement over previous anion photoelectron spectroscopy studies.10,11 The improved resolution afforded by the cryo-SEVI method reveals not only the electronic transitions to the three lowest-lying states of NiO2 (features A1, C1, D1, respectively) previously observed, but also a manifold of transitions to vibrationally excited states of the neutral species. Peak assignments, electron binding energies (eBEs), as well as shifts from peak A1 are presented in Tables 1–3, while extracted experimental parameters are presented in Table 4.
1Σg+ ← 2Πg detachment transitions of NiO2−. Uncertainty in the peak positions correspond to one standard deviation from a Gaussian fit to the highest-resolution experimental trace for each feature
| Peak | eBE | Shift | Assignment |
|---|---|---|---|
| A1 | 24571(6) |
— | 000 |
| B1 | 24691(7) |
120 | 210 |
| A2 | 24815(13) |
244 | 220 |
| B2 | 24936(8) |
365 | 230 |
| A3 | 25061(12) |
490 | 240 |
| B3 | 25190(7) |
619 | 250 |
| A4 | 25316(6) |
745 | 110 |
| B4 | 25442(9) |
871 | 110210 |
| A5 | 25572(9) |
1001 | 110220 |
| B5 | 25700(8) |
1129 | 110230 |
| A6 | 25832(9) |
1261 | 110240 |
| B6 | 25961(7) |
1390 | 110250 |
| A7 | 26065(8) |
1494 | 120 |
| B7 | 26192(9) |
1621 | 110270 |
| B8 | 26223(7) |
1652 | 120210 |
| A8 | 26356(9) |
1786 | 120220 |
| B9 | 26436(6) |
1865 | 110290 |
| B10 | 26454(7) |
1883 | 120230 |
| A9 | 26529(11) |
1957 | 320 |
| B11 | 26692(9) |
2121 | 210320 |
| B12 | 26716(9) |
2145 | 120250 |
| A10 | 26783(9) |
2212 | 130 |
| B13 | 26928(10) |
2357 | 110210 |
| B14 | 26945(7) |
2374 | 120270 |
| B15 | 26978(7) |
2406 | 130230 |
| B16 | 27187(28) |
2616 | 120290 |
| A11 | 27283(35) |
2712 | 110320 |
| A12 | 27519(11) |
2948 | 140 |
| B17 | 27660(11) |
3089 | 140210 |
| A13 | 28260(11) |
3689 | 150 |
2Πg detachment transitions of NiO2−. Uncertainty in the peak positions correspond to one standard deviation from a Gaussian fit to the highest-resolution experimental trace for each feature
| Peak | eBE | Shift | Assignment |
|---|---|---|---|
| C1 | 27783(3) |
— | ã3Πg000 |
| C2 | 27913(5) |
131 | 210 |
| C3 | 28468(11) |
685 | 110 |
| C4 | 28589(13) |
807 | 110210 |
| C5 | 29162(12) |
1380 | 120 |
| C6 | 29271(17) |
1488 | 120210 |
| C7 | 29883(9) |
2100 | 130 |
| C8 | 29968(15) |
2186 | 130210 |
2Πg detachment transitions of NiO2−. Uncertainty in the peak positions correspond to one standard deviation from a Gaussian fit to the highest-resolution experimental trace for each feature
| Peak | eBE | Shift | Assignment |
|---|---|---|---|
| D1 | 30557(7) |
— | Ã1Πg000 |
| D2 | 30669(12) |
112 | 210 |
| D3 | 30814(10) |
257 | 220 |
| D4 | 31263(19) |
706 | 110 |
| D5 | 31422(16) |
865 | 110210 |
| D6 | 31944(9) |
1275 | 120 |
| Parameter | Calculated | SEVI | Literature | |
|---|---|---|---|---|
| a Ref. 11. b Ref. 12. c Ref. 10. | ||||
| 1Σg+ |
||||
| EA (eV) | 3.0464(7) | 3.043(5)a | ||
| ω 1 (cm−1) | 774 | 745(6) | 749b | |
| ω 2 (cm−1) | 135 | 120(7) | ||
| ω 3 (cm−1) | 985 | 978(16) | 977.5b | |
| ã3Πg | ||||
| T e (eV) | 0.56 | 0.3982(7) | 0.40(2)c | |
| ω 1 (cm−1) | 685(11) | |||
| ω 2 (cm−1) | 131(5) | |||
| Ã1Πg | ||||
| T e (eV) | 0.88 | 0.7422(10) | 0.77(3)c | |
| ω 1 (cm−1) | 706(19) | |||
| ω 2 (cm−1) | 112(12) | |||
In the case of the NiO2 ground state band, assignments of features are facilitated by our SFX2C-1e-CCSD(T)/aug-cc-pVTZ calculations, which allow for FC simulations to be performed for detachment transitions terminating in the 1Σg+ state of NiO2. This simulated spectrum facilitates the definitive vibrational assignments in this state and suggests vibrational assignments for the ã and à states of NiO2. Notably, there are several strong transitions (B1–17) observed in this spectrum that are not reproduced by our simulations. Moreover, these features (B1–B5, for example) are clearly not present in the overview spectrum and retain considerable intensity near detachment threshold. This trend is depicted in Fig. 2, in which spectra collected at three photon energies highlight the differing behavior of these features as the photodetachment threshold is approached. As discussed in Section IV.C, our assignments for the B peaks all involve odd changes of vibrational quanta in the non-totally symmetric ν2 bending mode and are hence Franck–Condon forbidden.
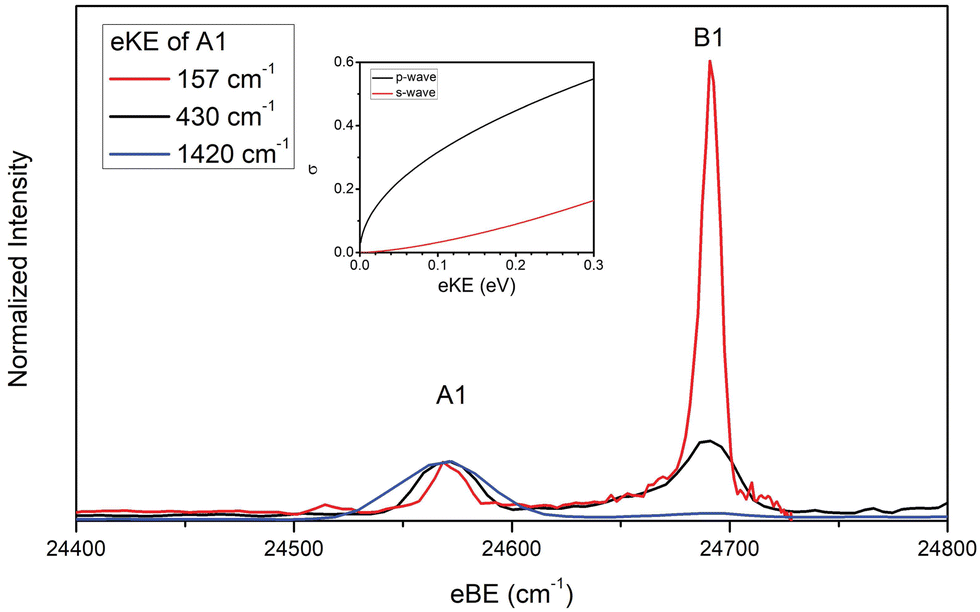 | ||
| Fig. 2 Cryo-SEVI spectra of NiO2− at three photon energies illustrating the differing signal attenuation for features A1 and B1 as eKE decreases. The intensity of each scan has been normalized to feature A1. Photon energies employed are 25991 (blue), 25001 (black), and 24728 cm−1 (red). Insert: photodetachment cross-section as a function of kinetic energy for s- and p-wave detachment as outlined by the Wigner threshold law.67 | ||
Vibrational frequencies were not calculated for the two observed excited states of NiO2, though the even spacing of levels and intensity patterns in the cryo-SEVI spectra lend themselves to tentative assignments to different vibrational levels of these two states of NiO2 (Section IV.D).
IV.A Computational results
Our coupled-cluster calculations confirm that the electronic ground state of NiO2 is the X1Σg+ state, while the ground state of NiO2− is determined as the2Πg state. The valence orbitals of NiO2 are composed of the Ni 4s and 3d orbitals and the O 2s and 2p orbitals, as shown in Fig. 3. The corresponding orbital compositions are summarized in Table 5. The Ni 3dxz and 3dyz orbitals together with the O 2px and 2py orbitals form the 1πg bonding orbitals and the 2πg anti-bonding orbitals. The Ni 3dx2−y2 and 3dxy orbitals do not mix with oxygen valence orbitals and form the non-bonding 1δg orbitals. The 2σg+ orbitals consist of contributions from the Ni orbitals and the O 2s and 2pz orbitals. The 1σg+ orbitals are mainly composed of the O 2s and 2pz orbitals with a small contribution from the Ni 4s orbitals. The ground state of NiO2 takes a closed-shell configuration (1σg+)2(1σu+)2(2σg+)2(1δg)4(3σg+)2(2σu+)2(1πu)4(1πg)4(2πg)0, with the anti-bonding 2πg orbitals left unoccupied. Since the 1σg+, 2σg+, 1δg, 3σg+, 1πg orbitals are fully occupied for all electronic states discussed here, we will use the occupation numbers for the 3σg+, 2σu+, 1πu, and 2πg orbitals to denote electronic states. In this notation the ground state of NiO2 is represented as (3σg+)2(2σu+)2(1πu)4(2πg)0. The ground 2Πg state of NiO2− places the excess electron in the 2πg orbital, which is expected to weaken the Ni–O bond.
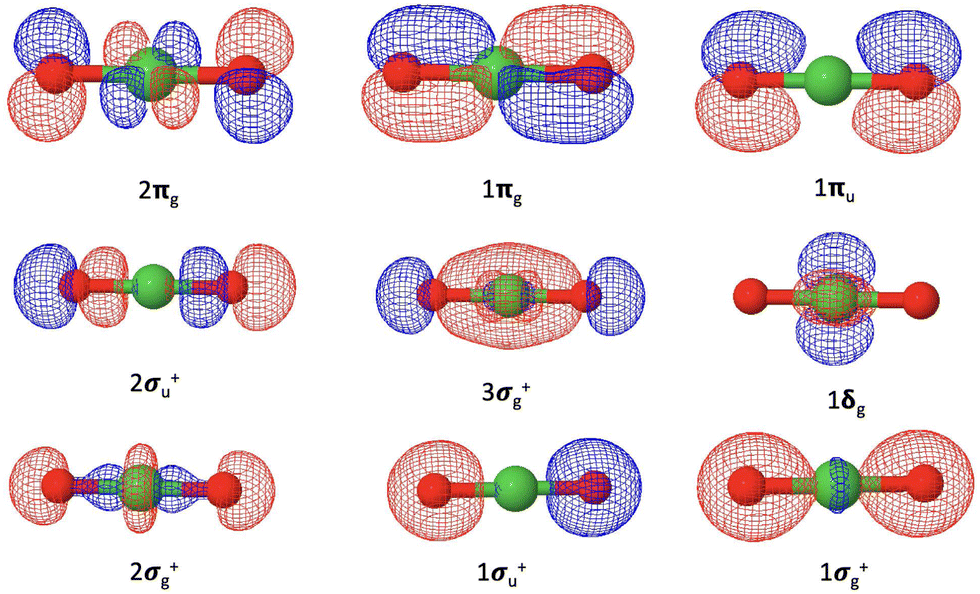 | ||
| Fig. 3 Valence molecular orbitals of NiO2 originating from the Ni 4s and 3d orbitals and the O 2s and 2p orbitals. | ||
| Molecular orbitals | Compositions |
|---|---|
| 1σg+ | O 2s |
| 1σu+ | O 2s |
| 2σg+ | Ni 3dz2 [O 2s and 2pz] |
| 1δg | Ni 3dx2−y2 and 3dxy |
| 3σg+ | O 2s, 2pz [Ni 4s, 3d0] |
| 2σu+ | O 2s, 2pz |
| 1πu | O 2px and 2py |
| 1πg | O 2px and 2py [Ni 3dxz and 3dyz] |
| 2πg | Ni 3dxz and 3dyz [O 2px and 2py] |
Although the ground state wave function of NiO2 is dominated by a closed shell (3σg+)2(2σu+)2(1πu)4(2πg)0 configuration, this electronic state exhibits strong electron correlation because of the low-lying anti-bonding 2πg orbitals. An analysis in the ESI† shows that CCSD(T) calculations provide reasonably accurate structure and frequencies, in spite of the slow convergence of the CC series. The CCSD(T) results perhaps slightly overestimate the vibrational frequency of the bending mode ω2 and underestimate those of the antisymmetric stretching and symmetric stretching modes (ω3 and ω1). The ground state of NiO2− is dominated by the (3σg+)2 (2σu+)2 (1πu)4 (2πg)1 configuration. As discussed in the ESI,† the CCSD(T) results are also reasonably accurate for this electronic state.
We have summarized the adiabatic electron affinity, equilibrium structures, and harmonic frequencies computed at the SFX2C-1e-CCSD(T)/aug-cc-pVTZ level of theory in Table 6 and have utilized these parameters in the FC simulations presented in Fig. 1. As expected, the occupation of an anti-bonding 2πg orbital in NiO2− significantly increases the Ni–O bond length, e.g., the difference between the Ni–O bond lengths in NiO2 and NiO2− computed at the CCSD(T) level amounts to 0.034 Å. The population of this orbital also reduces the asymmetric stretching frequency of NiO2− by around 70 cm−1. The large change in the Ni–O bond length leads to a significant FC progression for the photodetachment transitions from the 2Πg state of NiO2− to the 1Σg+ of NiO2 due to the FC activity of the symmetric stretch ω1 (σg) mode, which is responsible for much of the vibrational structure observed in the photoelectron spectrum of NiO2−.
2Πg state of NiO2−. Scalar-relativistic effects have been taken into account using the SFX2C-1e scheme. The aug-cc-pVTZ basis sets recontracted for the SFX2C-1e scheme have been used. Reported values are in cm−1 except for bond length, which is reported in Å
| R(Ni–O) | ω 1 (σg) | ω 2 (πu) | ω 3 (σu) | EAa | |
|---|---|---|---|---|---|
| NiO2 | 1.609 | 774 | 135 | 985 | 24523 |
| NiO2− | 1.643 | 772 | 96 | 915 | — |
Excitation of a 3σg+, 1πu or 2σu electron into an anti-bonding 2πg orbital gives rise to several low-lying excited states of NiO2. The lowest excited state of NiO2 is a 3Πg state with the leading configuration (3σg+)1(2σu+)2(1πu)4(2πg)1, which resides 0.56 eV above the ground 1Σg+ state. The corresponding singlet 1Πg state lies 0.88 eV above the ground state. Both states can be accessed from the ground 2Πg state of NiO2− by detaching a 3σg+ electron. Meanwhile, the detachment of a 1πu electron or a 2σu electron leads to a manifold of (3σg+)2(2σu+)2(1πu)3(2πg)1 states and a manifold of (3σg+)2(2σu+)1(1πu)4(2πg)1 states lying around 1.1 eV and 2 eV above the ground state, respectively. These findings are in good agreement with the MRCI calculations of Hübner,22 and are outlined in Table S2 (ESI†). Details about the EOM-CC computations for excitation energies are documented in the ESI.†
IV.B Ground state Franck–Condon allowed transitions
Table 1 lists the vibrational assignments of features arising from the1Σg+ ← 2Πg transition. These assignments are informed by the results of our FC simulations as well as the previously reported photoelectron spectrum of Ramond et al.11 As features A1–13 are largely reproduced in the simulated spectra, they are assigned to Franck–Condon allowed transitions within the 1Σg+ ← 2Πg photodetachment band of NiO2. FC-allowed transitions include all Δv transitions in totally symmetric vibrational modes (σg for linear species) and even Δv transitions along non-totally symmetric modes.63
Most prominent among these features is A1, here assigned as the 000 vibrational origin, allowing for the extraction of a refined electron affinity for NiO2 and providing an order of magnitude improvement in the precision of this measurement of 3.0464(7) eV, compared to 3.043(5) eV in ref. 11. The measured EA here is in good agreement with our calculated value of 3.0405 eV.
Beyond the vibrational origin is a wealth of vibrational structure, owing to an extended progression in the v1 symmetric stretch of NiO2, where there 110, 120, 130, 140, and 150 transitions are observed (A4, A7, A10, A12, A13, respectively). Weak but allowed transitions involving the non-totally symmetric v2 bending and v3 anti-symmetric stretching modes contribute to the remainder of the A-features in this band, with a short progression in the v2 bending coordinate (220 and 240, corresponding to A2 and A3, respectively), the appearance of the FC-allowed 320 transition (A9), and several combination bands including A5, A6, and A8 (see Table 1).
From the harmonic progressions along v1 and v2 and the appearance of the 320 transition, we obtain the frequencies of all three vibrational modes of the 1Σg+ state of NiO2. These values, v1 = 745(6), v2 = 122(18), and v3 = 978(16) cm−1, are in good agreement with those previously reported from photoelectron spectroscopy (v1 = 745(30) cm−1)11 and FTIR studies of NiO2 in a Ne matrix (v1 = 749 and v3 = 977.5 cm−1),12 as well as our calculated values (Table 6) of v1 = 774, v2 = 135, and v3 = 985 cm−1. Further, the observed frequency for the v2 bending mode matches well with an observed but unassigned feature in the Ne matrix data at 129.9 cm−1,9 suggesting that this peak was from IR excitation of the v2 mode.
IV.C Ground state Franck–Condon forbidden transitions
We now consider the assignments to the features in the1Σg+ ← 2Πg band that are not reproduced by our FC simulations, peaks B1–B17 (Table 1). The spacing of these features is consistently ∼120 cm−1 above that of an allowed transition (as is the case for B1–B9, B13, B17) or ∼240 cm−1 above another “B” feature (as is the case for B9 → B11, B10 → B12, B12 → B14, B13 → B15, B14 → B16). The similarity of these spacings with that of our measured value for the v2 bending frequency (122(18) cm−1) suggests these features arise from transitions terminating in odd quanta of the non-totally symmetric v2 mode. We thus assign peaks B1–B17 to transitions of this type to neutral levels with πu vibrational symmetry.
These transitions are Franck–Condon forbidden but can arise through Herzberg-Teller (HT) coupling to an excited electronic state with the appropriate symmetry.29,64–66 Briefly, two vibronic states, a and b, can mix through HT-coupling provided their electronic and vibrational symmetries Γelec and Γvib, respectively, satisfy63
| Γaelec ⊗ Γavib ⊗ Γbelec ⊗ Γbvib ⊃ ΓTS | (1) |
In the present case, the electronic and vibrational symmetries of the final states in features B1–B17, Γaelec and Γavib, are Σg+ and πu, respectively. These states can only be observed if they mix with a state b that is FC-allowed for detachment from the anion, i.e. Γbvib = σg, thereby requiring that the excited electronic state HT-coupled to this state be of Πu symmetry. Our calculations (Table S2, ESI†) find that such an excited state resides 2.24 eV above the neutral ground state.
The presence of vibronic coupling here is affirmed by the differing behavior of the FC-forbidden and FC-allowed features as the photon energy is changed, as shown in Fig. 2. Here, cryo-SEVI spectra are plotted at three photon energies, showing that as the photon energy is lowered and approaches the detachment threshold, the intensity of features A1 and A2 drops precipitously relative to that of B1 and B3. This effect is ascribed to a reduced photodetachment cross section for feature A1 and A3 at low eKE. Such a difference can be related to the relative scaling of the detachment cross sections at low eKE's, given by the Wigner threshold law:67
| σ ∝ (eKE)l+1/2, | (2) |
For detachment to the 1Σg+ state of NiO2, selection rules for molecular photodetachment prohibit detachment of l = 0 electrons, and p-wave detachment dominates.68 Conversely, transitions terminating in odd quanta along the v2 normal coordinate do so in a neutral level with a contribution from an excited state of Πu symmetry, for which detachment can proceed via l = 0 (s-wave) transitions.
The disparity in near-threshold cross-section between the “A” and “B” features is a tell-tale sign of HT coupling in cryo-SEVI spectra,29,64–66 as this indicates that “B” features correspond to detachment with lower values of l than the “A” features, reflecting the electronic character of the Πu electronic state that lends intensity to these transitions. Consequently, spectra for the “B” peaks can be obtained closer to photodetachment threshold, where cryo-SEVI resolution is improved, leading to narrower features as was observed in the cryo-SEVI spectra of the indenyl and nitrate anions.66,69 As such, we are able to refine our determined value for the v2 bending mode of NiO2 from the position of B1 as 120(7) cm−1.
IV.D Excited state transitions
Beyond the strongest vibrational transitions in the1Σg+ manifold reside the strong features C1 and D1. The positions of these features (3.4447(4) and 3.7886(9) eV, respectively) coincide with the previously observed onset of transitions assigned to the ã1Δg and ![[b with combining tilde]](https://www.rsc.org/images/entities/char_0062_0303.gif) 1Σg+ states (3.45(2) and 3.82(3) eV, respectively) of NiO2 based on a molecular orbital picture.10 Subsequent theoretical work, however, has determined the low-lying states of NiO2 as the ã3Πg and Ã1Πg states.22 These assignments agree with our EOM-CC calculations, which find the same state ordering. We thus assign C1 and D1 as the vibrational origins of the ã3Πg ← 2Πg and Ã1Πg ← 2Πg transitions, with term energies of 0.3982(7) and 0.7422(10) eV, respectively, determined by the shift in peak location from A1. These reported term energies are more precise than those previously reported (0.40(2) and 0.77(3) eV, respectively),10 and are in good agreement with our computed term energies of 0.56 and 0.88 eV, validating this reassignment.
1Σg+ states (3.45(2) and 3.82(3) eV, respectively) of NiO2 based on a molecular orbital picture.10 Subsequent theoretical work, however, has determined the low-lying states of NiO2 as the ã3Πg and Ã1Πg states.22 These assignments agree with our EOM-CC calculations, which find the same state ordering. We thus assign C1 and D1 as the vibrational origins of the ã3Πg ← 2Πg and Ã1Πg ← 2Πg transitions, with term energies of 0.3982(7) and 0.7422(10) eV, respectively, determined by the shift in peak location from A1. These reported term energies are more precise than those previously reported (0.40(2) and 0.77(3) eV, respectively),10 and are in good agreement with our computed term energies of 0.56 and 0.88 eV, validating this reassignment.
Numerous weaker features reside beyond peaks C1 and D1 (C2–C8, D2–D6) that report on the vibrational structure of the ã3Πg and Ã1Πg excited states (Tables 2 and 3). In the ã3Πg state, there appears to be a progression of a vibrational feature with a frequency of 685(11) cm−1 as well as the appearance of the vibrational fundamental and combination bands involving a mode with frequency of 130(5) cm−1. Similarly, in the Ã1Πg state, there appear to be two progressions in features with average spacings of 112(12) and 705(19) cm−1. While we do not have theoretical calculations to make definitive assignments to these features, their relative spacing are close to those of the v1 and v2 vibrational modes that dominate the structure in the ground state of spectrum NiO2. We thus tentatively assign the observed structure to activity along the v1 and v2 vibrational modes of NiO2 in the ã3Πg and Ã1Πg excited states with vibrational frequencies of 685(11) and 130(5) cm−1 (ã3Πg) as well as 705(19) and 112(12) cm−1 (Ã1Πg), respectively (outlined in Table 4).
Conclusion
Here, we report high-resolution cryo-SEVI spectra of NiO2− showing photodetachment to the1Σg+, ã3Πg, and Ã1Πg states of NiO2. In the ground state, we observe FC-allowed transitions involving the v1 symmetric stretch, v2 bending, and v3 anti-symmetric stretch from which we extract vibrational frequencies of v1 = 745(6), v2 = 122(18), and v3 = 978(16) cm−1, in good agreement with previously reported values. These assignments are facilitated by FC simulations using the SFX2C-1e-CCSD(T)/aug-cc-pVTZ geometries and vibrational frequencies for NiO20/−. Notably, we observe extended FC-forbidden progressions to final states with odd quanta in the v2 bending coordinate that we ascribe to vibronic coupling to the Πu excited state of NiO2, calculated to reside 2.24 eV above the ground state. From the onset of structure in the ground and excited states, we are able to refine the electron affinity and term energies of NiO2 and its first two excited states (EA = 3.0464(7) eV, Te(ã3Πg) = 0.3982(7) eV, Te(Ã1Πg) = 0.7422(10) eV), in good agreement with our calculated values.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research conducted at UC Berkeley is funded by the Air Force Office of Scientific Research under Grant No. FA9550-19-1-0051 and the work done at Johns Hopkins University is supported by Department of Energy Early Career Research Program under Award Number DE-SC0020317. M. C. B. thanks the Army Research Office for a National Defense Science and Engineering Graduate fellowship. J. A. L. thanks the Alexander von Humboldt Foundation for a Feodor Lynen Research Fellowship.References
- N. A. K. Aramouni, J. G. Touma, B. Abu Tarboush, J. Zeaiter and M. N. Ahmad, Renewable Sustainable Energy Rev., 2018, 82, 2570–2585 CrossRef CAS
.
- C. Alvarez-Galvan, M. Melian, L. Ruiz-Matas, J. L. Eslava, R. M. Navarro, M. Ahmadi, B. Roldan Cuenya and J. L. G. Fierro, Front. Chem., 2019, 7, 104 CrossRef CAS PubMed
- E. Heracleous, A. F. Lee, K. Wilson and A. A. Lemonidou, J. Catal., 2005, 231, 159–171 CrossRef CAS
- Y. L. Zhou, F. F. Wei, J. Lin, L. Li, X. Y. Li, H. F. Qi, X. L. Pan, X. Y. Liu, C. D. Huang, S. Lin and X. D. Wang, ACS Catal., 2020, 10, 7619–7629 CrossRef CAS
- S. Dey, G. C. Dhal, D. Mohan and R. Prasad, Adv. Compos. Hybrid Mater., 2019, 2, 626–656 CrossRef CAS
- A. W. Castleman, Catal. Lett., 2011, 141, 1243–1253 CrossRef CAS
- J. L. Mason, C. N. Folluo and C. C. Jarrold, J. Chem. Phys., 2021, 154, 200901 CrossRef CAS PubMed
- H. Huber and G. A. Ozin, Can J. Chem., 1972, 50, 3746–3747 CrossRef CAS
- A. Citra, G. V. Chertihin, L. Andrews and M. Neurock, J. Phys. Chem. A, 1997, 101, 3109–3118 CrossRef CAS
- H. B. Wu and L. S. Wang, J. Chem. Phys., 1997, 107, 16–21 CrossRef CAS
- T. M. Ramond, G. E. Davico, F. Hellberg, F. Svedberg, P. Salen, P. Soderqvist and W. C. Lineberger, J. Mol. Spectrosc., 2002, 216, 1–14 CrossRef CAS
- F. Allouti, L. Manceron and M. E. Alikhani, Phys. Chem. Chem. Phys., 2006, 8, 448–455 RSC
- W. D. Vann, R. L. Wagner and A. W. Castleman, J. Phys. Chem. A, 1998, 102, 1708–1718 CrossRef CAS
- M. C. Oliveira, J. Marcalo, M. C. Vieira and M. A. A. Ferreira, Int. J. Mass Spectrom., 1999, 185, 825–835 CrossRef
- J. U. Reveles, G. E. Johnson, S. N. Khanna and A. W. Castleman, J. Phys. Chem. C, 2010, 114, 5438–5446 CrossRef CAS
- C. Salvitti, M. Rosi, F. Pepi, A. Troiani and G. de Petris, Chem. Phys. Lett., 2021, 776, 138555 CrossRef CAS
- M. R. A. Blomberg, E. M. Siegbahn and A. Strich, Chem. Phys., 1985, 97, 287–301 CrossRef CAS
- C. W. Bauschlicher, J. Phys. Chem. A, 2004, 108, 2871–2873 CrossRef CAS
- K. Deng, J. L. Yang and Q. S. Zhu, J. Chem. Phys., 2003, 118, 6868–6873 CrossRef CAS
- G. L. Gutsev, B. K. Rao and P. Jena, J. Phys. Chem. A, 2000, 104, 11961–11971 CrossRef CAS
- E. L. Uzunova, H. Mikosch and G. S. Nikolov, J. Chem. Phys., 2008, 128, 094307 CrossRef PubMed
- O. Hubner and H. J. Himmel, J. Phys. Chem. A, 2012, 116, 9181–9188 CrossRef PubMed
- D. M. Neumark, J. Phys. Chem. A, 2008, 112, 13287–13301 CrossRef CAS PubMed
- M. L. Weichman and D. M. Neumark, Annu. Rev. Phys. Chem., 2018, 69, 101–124 CrossRef CAS PubMed
- A. Osterwalder, M. J. Nee, J. Zhou and D. M. Neumark, J. Chem. Phys., 2004, 121, 6317–6322 CrossRef CAS PubMed
- C. Hock, J. B. Kim, M. L. Weichman, T. I. Yacovitch and D. M. Neumark, J. Chem. Phys., 2012, 137, 244201 CrossRef PubMed
- U. Even, J. Jortner, D. Noy, N. Lavie and C. Cossart-Magos, J. Chem. Phys., 2000, 112, 8068–8071 CrossRef CAS
- J. B. Kim, C. Hock, T. I. Yacovitch and D. M. Neumark, J. Phys. Chem. A, 2013, 117, 8126–8131 CrossRef CAS PubMed
- J. A. DeVine, M. L. Weichman, B. Laws, J. Chang, M. C. Babin, G. Balerdi, C. J. Xie, C. L. Malbon, W. C. Lineberger, D. R. Yarkony, R. W. Field, S. T. Gibson, J. Y. Ma, H. Guo and D. M. Neumark, Science, 2017, 358, 336–339 CrossRef CAS PubMed
- M. C. Babin, J. A. DeVine, M. L. Weichman and D. M. Neumark, J. Chem. Phys., 2018, 149, 174306 CrossRef PubMed
- W. C. Wiley and I. H. Mclaren, Rev. Sci. Instrum., 1955, 26, 1150–1157 CrossRef CAS
- A. T. J. B. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS
- D. W. Chandler and P. L. Houston, J. Chem. Phys., 1987, 87, 1445–1447 CrossRef CAS
- M. B. Doyle, C. Abeyasera and A. G. Suits, NuACQ, http://faculty.missouri.edu/suitsa/NuAqc.html.
- E. W. Hansen and P. L. Law, J. Opt. Soc. Am. A, 1985, 2, 510–520 CrossRef
- C. Blondel, C. Delsart and F. Goldfarb, J. Phys. B: At. Mol. Phys., 2001, 34, L281–L288 CrossRef CAS
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS
- K. G. Dyall, J. Chem. Phys., 2001, 115, 9136–9143 CrossRef CAS
- W. J. Liu and D. L. Peng, J. Chem. Phys., 2009, 131, 031104 CrossRef PubMed
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS
- N. B. Balabanov and K. A. Peterson, J. Chem. Phys., 2005, 123, 064107 CrossRef PubMed
- G. D. Purvis and R. J. Bartlett, J. Chem. Phys., 1982, 76, 1910–1918 CrossRef CAS
- J. Noga and R. J. Bartlett, J. Chem. Phys., 1987, 86, 7041–7050 CrossRef CAS
- G. E. Scuseria and H. F. Schaefer, Chem. Phys. Lett., 1988, 152, 382–386 CrossRef CAS
- S. A. Kucharski and R. J. Bartlett, Theor. Chim. Acta, 1991, 80, 387–405 CrossRef CAS
- N. Oliphant and L. Adamowicz, J. Chem. Phys., 1991, 95, 6645–6651 CrossRef CAS
- J. Gauss, J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1991, 95, 2623–2638 CrossRef CAS
- J. F. Stanton, J. Gauss, J. D. Watts and R. J. Bartlett, J. Chem. Phys., 1991, 94, 4334–4345 CrossRef CAS
- J. D. Watts, J. Gauss and R. J. Bartlett, Chem. Phys. Lett., 1992, 200, 1–7 CrossRef CAS
- L. Cheng and J. Gauss, J. Chem. Phys., 2011, 135, 084114 CrossRef PubMed
- D. A. Matthews and J. F. Stanton, J. Chem. Phys., 2015, 142, 064108 CrossRef PubMed
- D. A. Matthews, L. Cheng, M. E. Harding, F. Lipparini, S. Stopkowicz, T. C. Jagau, P. G. Szalay, J. Gauss and J. F. Stanton, J. Chem. Phys., 2020, 152, 214108 CrossRef CAS PubMed
- D. A. Matthews, J. Chem. Theory Comput., 2020, 16, 6195–6206 CrossRef CAS PubMed
- J. G. J. F. Stanton, L. Cheng, M. E. Harding, D. A. Matthews, P. G. Szalay, CFOUR, coupled-cluster techniques for computational chemistry, a quantum-chemical program package, with contributions from, A. A. Auer, R. J. Bartlett, U. Benedikt, C. Berger, D. E. Bernholdt, S. Blaschke, Y. J. Bomble, S. Burger, O. Christiansen, D. Datta, F. Engel, R. Faber, J. Greiner, M. Heckert, O. Heun, M. Hilgenberg, C. Huber, T.-C. Jagau, D. Jonsson, J. Jusélius, T. Kirsch, K. Klein, G. M. Kopper, W. J. Lauderdale, F. Lipparini, T. Metzroth, L. A. Mück, T. Nottoli, D. P. O'Neill, D. R. Price, E. Prochnow, C. Puzzarini, K. Ruud, F. Schiffmann, W. Schwalbach, C. Simmons, S. Stopkowicz, A. Tajti, J. Vázquez, F. Wang, J. D. Watts and the integral packages MOLECULE (J. Almlöf and P. R. Taylor), PROPS (P. R. Taylor), ABACUS (T. Helgaker, H. J. Aa. Jensen, P. Jørgensen, and J. Olsen) and ECP routines by, A. V. Mitin and C. van Wüllen, For the current version, see (http://www.cfour.de).
- S. M. Rabidoux, V. Eijkhout and J. F. Stanton, J. Chem. Theory Comput., 2016, 12, 728–739 CrossRef CAS PubMed
- J. F. Stanton and R. J. Bartlett, J. Chem. Phys., 1993, 98, 7029–7039 CrossRef CAS
- K. Kowalski and P. Piecuch, J. Chem. Phys., 2001, 115, 643–651 CrossRef CAS
- S. A. Kucharski, M. Wloch, M. Musial and R. J. Bartlett, J. Chem. Phys., 2001, 115, 8263–8266 CrossRef CAS
- M. Kallay and P. R. Surjan, J. Chem. Phys., 2001, 115, 2945–2954 CrossRef CAS
- M. Kallay and J. Gauss, J. Chem. Phys., 2004, 121, 9257–9269 CrossRef CAS PubMed
- M. Kallay, P. R. Nagy, D. Mester, Z. Rolik, G. Samu, J. Csontos, J. Csoka, P. B. Szabo, L. Gyevi-Nagy, B. Hegely, I. Ladjanszki, L. Szegedy, B. Ladoczki, K. Petrov, M. Farkas, P. D. Mezei and A. Ganyecz, J. Chem. Phys., 2020, 152, 074107 CrossRef CAS PubMed
-
G. Herzberg, Electronic spectra and electronic structure of polyatomic molecules, Van Nostrand Reinhold Company, Princeton, NJ, 1966 Search PubMed
- J. A. DeVine, A. Abou Taka, M. C. Babin, M. L. Weichman, H. P. Hratchian and D. M. Neumark, J. Chem. Phys., 2018, 148, 222810 CrossRef PubMed
- M. L. Weichman, L. Cheng, J. B. Kim, J. F. Stanton and D. M. Neumark, J. Chem. Phys., 2017, 146, 224309 CrossRef PubMed
- M. C. Babin, J. A. DeVine, M. DeWitt, J. F. Stanton and D. M. Neumark, J. Phys. Chem. Lett., 2020, 11, 395–400 CrossRef CAS PubMed
- E. P. Wigner, Phys. Rev., 1948, 73, 1002–1009 CrossRef CAS
- K. J. Reed, A. H. Zimmerman, H. C. Andersen and J. I. Brauman, J. Chem. Phys., 1976, 64, 1368–1375 CrossRef CAS
- J. B. Kim, M. L. Weichman, T. I. Yacovitch, C. Shih and D. M. Neumark, J. Chem. Phys., 2013, 139, 104301 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp02396b |
| ‡ Current address: Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford St, Cambridge, MA 02138, USA. |
| § Current address: Department of Chemistry, Princeton University, Princeton, New Jersey 08544, USA. |
| ¶ Current address: KLA Corporation, 1 Technology Dr, Milpitas, CA 95035. |
| This journal is © the Owner Societies 2022 |