
Chemical reactions of graphitic carbon nitride films with glass surfaces and their impact on photocatalytic activity†
Kouki
Akaike
 *a,
Ayako
Hosokai
a,
Hiroki
Nagashima
b,
Qingshuo
Wei
a and
Takuya
Hosokai
c
*a,
Ayako
Hosokai
a,
Hiroki
Nagashima
b,
Qingshuo
Wei
a and
Takuya
Hosokai
c
aNanomaterials Research Institute, National Institute of Advanced Industrial Science and Technology, Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan. E-mail: kouki.akaike@aist.go.jp
bInterdisciplinary Research Center for Catalytic Chemistry, National Institute of Advanced Industrial Science and Technology, Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
cNational Metrology Institute of Japan, National Institute of Advanced Industrial Science and Technology, Central 5, 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
First published on 4th July 2022
Abstract
Thin films of graphitic carbon nitride (g-CN), a visible-light-driven photocatalyst, have recently attracted interest for application in photoelectrochemical cells for water splitting and high-throughput photocatalysis. In typical syntheses, g-CN films are formed by heating the nitrogen-rich precursor and substrate to 500–600 °C. The heated substrate should affect the polycondensation of the precursor and thereby alter the properties of the g-CN film. In this paper, we demonstrate that soda-lime glass, such as commercial glass slides, modifies the chemical structure of g-CN. The terminal amino groups of g-CN are partially substituted with cyanamide and hydroxyl groups. The electron-withdrawing groups provide the energy offsets of the frontier orbitals between the modified and unmodified molecules, facilitating exciton dissociation. After alkali metals are removed, the modified g-CN film exhibits a faster photodegradation of methyl orange compared with a melon film. The simple protocol to activate a g-CN film without co-catalysts paves a new way to enhance photocatalytic activity via selections of substrates, including waste glass.
Introduction
Photocatalysts are the key to creating a sustainable society through utilizing renewable solar energy. Graphitic carbon nitride (g-CN) is a heptazine-based polymer demonstrating a wide range of photocatalytic applications under visible-light irradiation like water photolysis,1–7 CO2 reduction,8,9 and degradation of organic pollutants.10,11 For repeated practical use, it is necessary to immobilize g-CN to avoid the need for filtering off and recapturing the photocatalyst. Vapor deposition polymerization (VDP)12 and the direct growth method13 were used for coating desired substrates, like photoanodes and reactors, with g-CN for hydrogen evolution6,13,14 and high-throughput photocatalysis,15 respectively. To prepare a g-CN film, a substrate is placed together with a precursor in a reactor, or its surface is covered with a precursor by a solution process. Heating the samples to a high temperature of 500–600 °C induces polycondensation of the precursor to form g-CN in bulk and on a substrate surface. Since the substrate becomes thermally activated during film formation, the surface reactivity of the chemical species is enhanced.16–18 Hence, the substrate can affect the chemical, physical, and photocatalytic properties of the g-CN film grown on it, thereby determining the applications of the g-CN film. The photocatalytic applications of g-CN films require extensive photo-absorption in the solar spectrum, efficient exciton dissociation into free charges, and efficient charge transport toward the film surface where redox reactions occur. The introduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C π bonds by annealing the film19 and making a composite with reduced graphene oxide14 enhance photon harvesting, electron diffusion length, and surface area. However, the potential of a used substrate in boosting photocatalytic activities of a g-CN film is elusive.
C π bonds by annealing the film19 and making a composite with reduced graphene oxide14 enhance photon harvesting, electron diffusion length, and surface area. However, the potential of a used substrate in boosting photocatalytic activities of a g-CN film is elusive.
This study demonstrates that commercial soda-lime glass slides modify the chemical structure of g-CN in a well-oriented film prepared by VDP. We found that alkali metal hydroxides were formed on the glass surface and that they substituted terminal amino groups in g-CN with cyanamide and hydroxyl groups. Ion exchange experiments showed that Na+ and K+ that diffused from the substrates were ionically bound to the nitrogen atoms bridging the monomers and to the cyanamide groups. The g-CN film modified with the functional groups shows enhanced photoluminescence (PL) quenching, indicating efficient free-charge generation owing to the incorporation of the electron-withdrawing groups into the polymer. Soaking in aqueous acid removed the alkali metals from the film, thereby improving film crystallinity along the surface normal, which improved vertical charge transport toward the film surface. The facilitated charge generation and transport in the g-CN film on glass resulted in over 17-fold higher photocatalytic degradation of methyl orange than that over the untreated film, and the activity was found to be 12-fold higher than the performance of an unmodified g-CN (melon) film synthesized under the same conditions with no cocatalysts. We also demonstrated that a piece of broken glass, usually regarded as waste glass, could be used as a substrate to activate g-CN for photocatalytic applications.
Experimental
Materials
Melamine (99.0%), sodium hydroxide (97.0%), and aqueous hydrochloric acid (0.02 M) were purchased from FUJIFILM Wako Pure Chemical Corp. Citric acid (CA) was purchased from Tokyo Chemical Industry Corp. Glass slides were purchased from Matsunami Glass Ind., Ltd (S1112, glass #1), Paul Marienfeld GmbH & Co. KG (1005412, glass #2), Ci Medical Co., Ltd (7101, glass #3), and Leona Co., Ltd (0302-0005, glass #4). Unless otherwise noted, we used glass #1 to prepare g-CN films. Glasses #2–4 were used to examine the generality of the chemical modification of g-CN films on commercial glass slides. The quartz substrate was purchased from SEIREN KST Corp. Alkali-free glass (AFG, EAGLEXG) was purchased from Corning, Inc. A glass substrate coated with a transparent electrode (TE) was purchased from Geomatec Co., Ltd. Prior to use, the substrate was ultrasonicated in pure water, acetone, and isopropanol for 15 min each, followed by UV–ozone treatment for 20 min. A piece of broken glass (surface area ∼5.3 cm2) was also used to form a g-CN film. The cleaning procedure was the same as that for the glass slides. For comparison of photocatalytic activity, a 750 nm-thick TiO2 film was formed on the cleaned AFG by sputtering.Preparation of melem, melon, and g-CN films
The melem, melon, and g-CN films were synthesized in a quartz tube in a commercial tube furnace (KTF035N1, Koyo Thermo Systems Co., Ltd) under a nitrogen atmosphere. Details of the synthesis of melem are described elsewhere.20 To obtain bulk melon, melamine (1 g) was placed at the bottom of a test tube (14-961-31, Thermo Fisher Scientific Inc.) capped with aluminum foil with a pinhole. The test tube was heated to 500 °C at a rate of 10 °C min−1, and kept at 500 °C for 2 h. The reactor was cooled at a rate of 2 °C min−1 to room temperature. g-CN films were prepared in a similar manner. Melamine was placed at the bottom of a test tube, together with a cleaned substrate 7.5 cm from the aluminum cap. Calcination conditions except for the g-CN film on a transparent electrode were the same as for bulk melon. The g-CN film on a transparent electrode-covered glass was synthesized at 450 °C. Heating at a temperature over 450 °C drastically reduces the electrical conductivity of the electrode. The average thickness of g-CN films was controlled by the amount of melamine. Calcination of 20 and 100 mg of melamine leads to 150 and 393 nm-thick films on glass #1, respectively. The g-CN film was also synthesized on glass #1 at 310 °C for 2 h using pure melem as a precursor instead of melamine. Melem (20 mg) was placed in the bottom of the test tube, and the heating and cooling rates were 10 °C min−1 and 2 °C min−1, respectively. Prior to film formation for photocatalytic experiments, glass slides were heated at 500 °C for 20 h in air and cleaned with UV-ozone treatment for 40 min. We denote the g-CN films deposited on quartz, AFG, glass, TE, and heated glass as g-CN/Q, g-CN/AFG, g-CN/G, g-CN/TE, and g-CN/hG, respectively.Activation of g-CN films by acid treatment
g-CN films on glass slides were soaked in 0.02 M aqueous HCl for 15 min. The treated films were rinsed with copious amounts of pure water until the pH was neutral. The film was then annealed at 100 °C for 10 min in air to remove adsorbed water. For CA treatment, we also used 2 wt% aqueous CA instead of HCl. The methods for rinsing and drying the CA-treated g-CN films were the same as those for HCl treatment. For denoting the acid-treated g-CN films, the acid name is added after the film name with a hyphen.Ion exchange experiments
HCl-treated g-CN/G (g-CN/G-HCl) was further soaked in 14 wt% NaCl aqueous solution for 3 days. Then, the film was soaked in 2.0 M aqueous NaOH for 15 min. After rinsing thoroughly with pure water, the film was subjected to ATR-FTIR measurements at each step to investigate the change in vibrational absorptions of functional groups where alkali metals are involved.Characterization
Attenuated total reflection (ATR)-Fourier transform infrared (FTIR) measurements were performed using a Ge prism in an FTIR spectrometer (FT/IR-6600, JASCO). When performing the measurements, the IR beam was incident to the prism at an angle of 45°. XRD analyses of g-CN films were carried out with a diffractometer (SmartLab, Rigaku) using Cu Kα as a radiation source. Film thicknesses were evaluated using a surface profiler (Dektak XT-E, Bruker).UV-vis absorption spectra were collected using a spectrophotometer (V-750, JASCO) fitted with an integrating sphere. g-CN films were peeled off from the glass substrates by soaking them in boiled water for 30 min. The small pieces of the film were collected and reflectance measurements were conducted. The reflection spectra were converted to absorption spectra using the Kubelka–Munk transformation.
Analyses with X-ray photoelectron spectroscopy (XPS) were carried out using a photoelectron spectrometer (PHI 5000 VersaProbe, ULVAC-PHI) with monochromatic Al Kα as an excitation source and a neutralizer to compensate for hole accumulation. To measure the XPS spectra of g-CN/G treated with HCl (Fig. 2) and with boiled water (Fig. S5, ESI†), the films were transferred to gold-coated glass substrates, otherwise charging distorted the spectra even with electron irradiation from a neutralizer. The photoelectron yield spectroscopy spectrum of g-CN/G-HCl was recorded under a pressure of less than 5 × 10−3 Pa with a spectrometer (BIP-KV200, Bunkou Keiki).
DNP-enhanced solid-state 13C, 15N, and 23Na magic angle spinning (MAS) NMR experiments were performed at 9.4 T on a DNP-NMR spectrometer (Avance-NEO, Bruker) equipped with a 263 GHz gyrotron and a double-resonance 1H/X 3.2 mm low-temperature MAS probe at 110 K. The g-CN sample was impregnated with 16 mM TEKPOL/1,1,2,2-tetrachloroethane (TCE) solution in a microtube, transferred to a 3.2 mm sapphire rotor, and closed with a Teflon insert and a zirconia cap. The film sample (approximately 5 mg) peeled off a glass slide (glass #1) in boiled water was impregnated with 16 mM TEKPOL/TCE solution directly in a 3.2 mm sapphire rotor. Soaking affected the alkali metal concentrations, and thus the ATR-FTIR and XPS spectra. Details are provided in Fig. S4 and S5 in the ESI.† All samples were spun at a MAS frequency of 10 kHz. 13C CP MAS DNP enhancement ε(on/off) was 52 for g-CN and 3.0 for the film sample. For all experiments, the 1H relaxation delay was fixed as 1.3 times the effective build-up time of DNP-enhanced 1H polarization, TB,on(1H), with microwave irradiation. DNP-enhanced 1H polarization was transferred to 13C and 15N by CP MAS and to 23Na by a dipolar-mediated RINEPT sequence.21 SPINAL-64 decoupling with an rf field of ν1,dec = 100 kHz was applied during the acquisition. For the 15N experiment on the film sample, CPMG acquisition was used due to the low sensitivity. The conditions for 15N CPMG were a number of echoes of 12 and an echo duration of 4.4 ms. CPMG echoes were summed before Fourier transform. The 13C spectra were referenced with respect to adamantane. The 15N spectra were referenced with respect to NH4Cl. The 23Na spectrum was referenced with respect to liquid NaCl.
1H fast MAS NMR experiments were performed at 9.4 T using a Bruker 1H/X 1.3 mm MAS probe at room temperature. 1H MAS 1D spectra were recorded by Hahn echo sequence spun at a MAS frequency of 50 kHz. The delay between π/2 and π pulses was 1τR = 20 μs. 1H–1H double quantum (DQ)–single quantum (SQ) 2D MAS spectra were recorded by using R1252 symmetry-based recoupling spun at a MAS frequency of 45 kHz. The recoupling time of R1252 was 88.8 μs for both the film and bulk samples. 1H spectra were referenced with respect to adamantane.
For sum frequency generation (SFG) spectroscopy, a mode-locked Nd:YAG laser (PL2231-50, EKSPLA) was used to generate a visible beam at 532 nm and to generate the frequency-tunable IR beam. The sample surface was irradiated with both beams at incident angles of 60° and 55°, respectively. The generated SFG signal was detected using a photomultiplier. The SFG spectra were acquired using the ssp polarization combination (s-polarized SF signal, s-polarized visible beam, and p-polarized IR beam).
PL measurements were performed in a homemade vacuum deposition system combined with time-resolved PL spectroscopy (FluoroCube, HORIBA) at room temperature (297 K).22 The PL spectra and TR-PLs were acquired at a measurement port in the vacuum chamber using a Duran glass jar (EVAC), which was placed at the center of the spectrometer sample chamber. A pulsed laser diode (NanoLED, HORIBA; wavelength: 342 nm, pulse width: 1.2 ns, pulse energy: 1–2 pJ, repetition rate: 100 kHz) was used as the excitation light source. The sample films were irradiated with the excitation light passed through a short-pass filter (cut-off wavelength: 360 nm) at 30° relative to the surface normal of the samples. The emitted PL was detected at 60° relative to the surface normal through a long-pass filter (cut-on wavelength: 370 nm). Photoluminescence quantum yield (PLQY) was measured with a commercial spectrometer (C9920-02G, Hamamatsu).
Evaluation of photocatalytic activity
The photocatalytic performance of the g-CN films was evaluated by the photodegradation of methyl orange aqueous solution (concentration = 3.06 × 10−6 M) at room temperature. The g-CN film formed on the glass substrates (surface area = 3.15 cm2) was placed at the inside edge of a quartz cell used for UV-vis absorption spectroscopy. With this setup, only one side of a substrate coated with g-CN contributes to photodegradation of methyl orange. The g-CN film formed on a piece of a broken glass cup was characterized by placing the sample in the middle of the cell. That is, both sides of the glass coated with g-CN contributed to the photodegradation of methyl orange. Before starting the photocatalytic reaction, a film sample was placed in the methyl orange solution for 30 min to stabilize the adsorption kinetics of the organic molecules on the film surface. Absorbance at 464 nm of methyl orange was monitored over 5 h in total under air mass 1.5 simulated solar light irradiation at an intensity of 100 mW cm−2 (HAL-320, Asahi Spectra). The light was incident on the g-CN surface used for the photocatalytic reactions. Degradation efficiency η of methyl orange was calculated by23where A0 and At denote absorbances at 0 h and reaction time t, respectively.
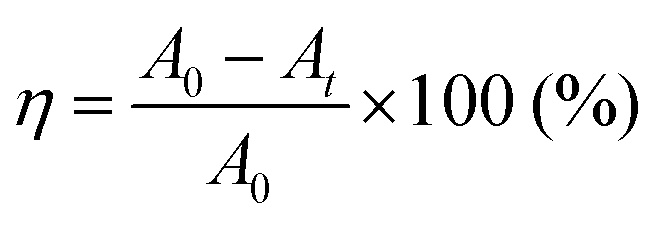
DFT calculations
Molecular orbital calculations were performed using the GAUSSIAN09 package. DFT calculations of melem dimers were performed using the B3LYP exchange–correlation function and 6-311G(d,p) basis set after structural optimization with the same calculation conditions.Results and discussion
Identification of the film product
g-CN films synthesized via VDP of a nitrogen-rich precursor like melamine are highly oriented irrespective of substrate type.12,20 Out-of-plane XRD profiles for g-CN films on a glass slide (g-CN/G) and a quartz substrate (g-CN/Q), recorded under grazing incident conditions to remove the background from the substrates, show diffraction only at around 2θ = 27.5° (Fig. S1 in the ESI†). This peak corresponds to π–π stacking of g-CN compounds.1,20,24,25 Thus, both g-CN/G and g-CN/Q adopt a flat-lying orientation on substrates in which the graphitic layers are parallel to the substrate surfaces. However, g-CN/G is less crystalline than g-CN/Q, as shown by the broader diffraction. This may be due to random chemical modification of g-CN during film formation (discussed later) and suppression of crystallization by alkali metals diffused from the glass slide. 23Na solid-state NMR proves the presence of Na+ in the film (Fig. 1a). Using the Scherrer equation, the crystallite size in the vertical direction for g-CN/G is calculated as 26.3 Å, whereas that for g-CN/Q is 79.1 Å.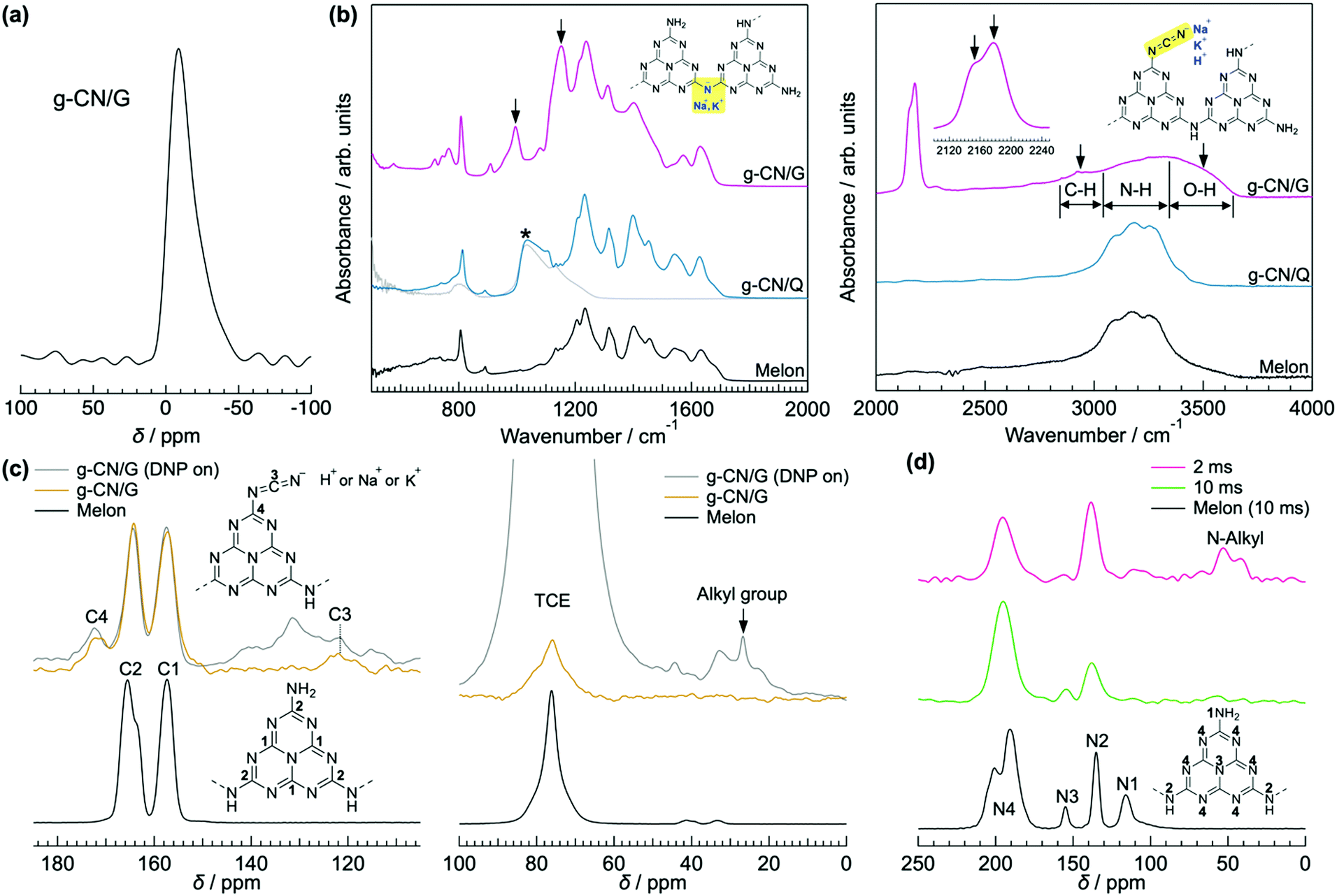 | ||
| Fig. 1 Characterization of g-CN/G. (a) 23Na solid-state NMR spectrum of g-CN/G peeled from a glass substrate by treatment with boiled water. Soaking the film in boiled water decreases concentrations of Na+ and K+ by 68%. See Table S2 and Fig. S4, S5 (ESI†), and related discussion for details. (b) ATR-FTIR spectra of g-CN/G, g-CN/Q, and bulk melon. The IR spectrum of quartz is also shown for reference. The downward arrows indicate specific absorptions observed for g-CN/G. Molecular structures in the left- and right-hand panels show melem dimers with metal-NC2 and cyanamide groups, respectively. Cyanamide absorptions are also magnified in the right-hand panel. (c) 13C solid-state NMR spectra of g-CN/G (top) and bulk melon (bottom). The DNP-enhanced spectrum of g-CN/G is shown in grey. The inset shows the numbering of respective carbon atoms for assignment. A signal from solvent 1,1,2,2-tetrachloroethane (TCE) appears at 78 ppm. (d) DNP-enhanced 15N solid-state NMR spectra of g-CN/G (top and middle) and bulk melon (bottom). The spectra of g-CN/G are acquired at contact times of 2 ms (top) and 10 ms (middle). The inset shows the numbering of the nitrogen atoms for assignment. | ||
ATR-FTIR, solid-state NMR, and XPS show that g-CN/G contains a heptazine backbone. The details to draw this conclusion together with the identification of bulk g-CN are described in greater detail in the ESI.† We identified bulk g-CN and g-CN/Q as melon. The extra vibrational feature observed for g-CN/G by ATR-FTIR (indicated by downward arrows in Fig. 1b) indicates the difference in chemical structure between g-CN/G and melon. In g-CN/G, new vibrational absorptions appear at 994, 1153, and 2175 cm−1 together with two broad bands at 2800–2900 cm−1 and 3400–3600 cm−1. The chemical modification is independent of glass slide suppliers (Fig. S2a, ESI†). Based on the chemical compositions given by suppliers (Table S1, except for glass #3, ESI†), the total concentrations of alkali metal oxides in the glasses exceed 13%. For g-CN/G prepared on glass #1, Na+ and K+ diffuse into the g-CN film from the substrate, as shown by XPS (Fig. 2a and b, respectively). Calcium is also present on bare glass with a surface concentration of 1.14%, but the concentrations of the element in g-CN films are negligible (Table S2, ESI†). The diffusion of alkali metals to the film surface from soda-lime glass, through upper layers is well known in copper indium gallium di-selenide (CIGS) solar cells.26,27 Calcination of nitrogen-rich precursors with alkali halides transforms g-CN to poly(heptazine imide) (PHI) salts;25,28 thus, the alkali metals present in g-CN/G are likely related to the change in the molecular structure indicated by the ATR-FTIR spectrum. Following a previous assignment,25 new absorptions at 994 and 1153 cm−1 are attributed to symmetric and asymmetric stretching of an NC2 group bound to an alkali metal, respectively.29 This result indicates the removal of a proton from NH groups bridging heptazine monomers (molecular structure shown in the right-hand panel of Fig. 1b). The sharp, asymmetrical absorption at around 2175 cm−1 is assigned to the stretching mode of the cyanamide moiety.30 Cyanamide carbons and the carbons bound to the cyanamide are detected at 120.8 and 172.3 ppm,30,31 respectively, in the 13C solid-state CP MAS NMR spectrum (left panel in Fig. 1c). The spectroscopic results demonstrate that cyanamide is introduced into g-CN/G. Compared with the spectrum for bulk melon measured for the same contact time, about one-sixth of the amino groups are substituted with a cyanamide moiety. The vibrational absorption attributed to the cyanamide moiety consists of two main components at 2151 and 2178 cm−1 (inset in the right-hand panel of Fig. 1b). These features of the g-CN/G ATR-FTIR spectrum are also observed for the film formed on soda-lime glass coated with a transparent electrode (Fig. S2b, ESI†). This fact suggests that reactants diffuse from the substrate through the overlayer and subsequently affect the g-CN film. In contrast, alkali-free glass (AFG) strongly suppresses the new vibrational absorptions observed for g-CN/G. The ATR-FTIR spectrum of g-CN/AFG is consistent with bulk melon (Fig. S2b, ESI†). This result also suggests that the alkali metals are related to the modification of the g-CN film.
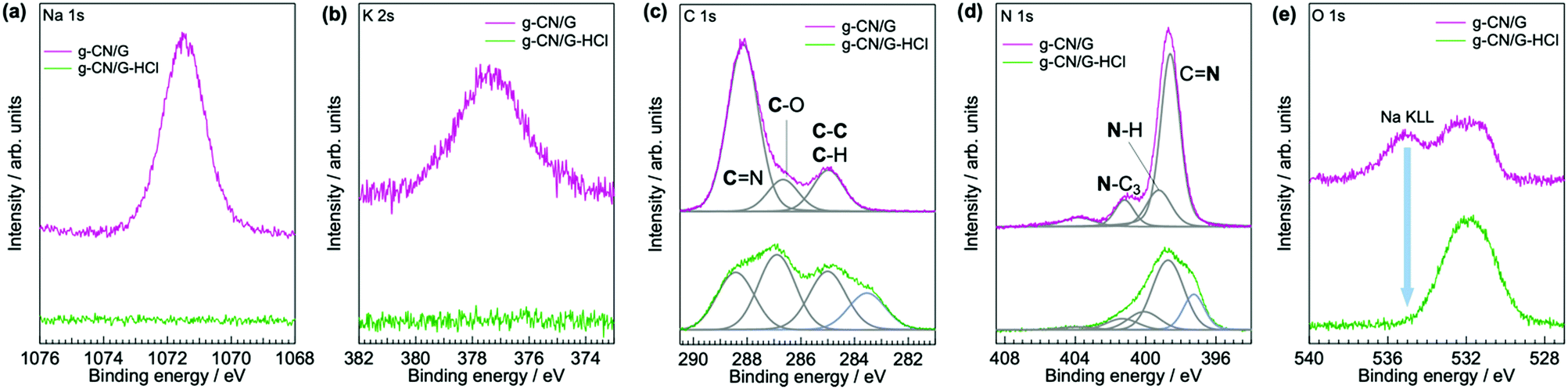 | ||
| Fig. 2 XPS spectra of g-CN/G (top) and g-CN/G-HCl (bottom) in the Na 1s (a), K 2s (b), C 1s (c), N 1s (d), and O 1s (e) regions. Gray curves in (c) and (d) indicate components separated by the fitting procedure. Blue components of g-CN/G-HCl in (c) and (d) are not identified. The higher energy component in the g-CN/G spectrum in (e) is attributed to Na KLL. | ||
DNP-enhanced 13C solid-state CP MAS NMR clarifies the origin of the C–H absorption in the ATR-FTIR spectrum of g-CN/G (Fig. 1b). With DNP, a sharp signal is detected at 27 ppm in the 13C solid-state NMR (Fig. 1c). Chemical shifts in this range are attributable to the carbons of normal alkanes.32 This is consistent with the C 1s XPS spectrum for g-CN/G, in which a photoemission attributed to C–C and/or C–H appears at 285.0 eV (Fig. 2c). In addition, the 15N solid-state DNP-NMR spectrum acquired at 2 ms (Fig. 1d) identifies alkyl amines in the range of 20–50 ppm.33,34 Therefore, the C–H stretching absorption originates from N-alkyl groups, such as –NH–CH3, presumably caused by hydrocarbon contaminants. Moreover, the C 1s and O 1s XPS spectra for g-CN/G confirm the presence of oxygen atoms in C–O bonds (Fig. 2c and e). Likely, O–H terminal groups are present, as indicated by a broad absorption at 3400–3600 cm−1 (Fig. 1a). Note that water molecules adsorbed on the g-CN film also give vibrational signals in this range. The 1H MAS NMR spectrum of g-CN/G demonstrates the presence of water in g-CN/G as evidenced by the chemical shift at 4.5 ppm along with the dramatic spectral change after drying the film at 120 °C under vacuum for two days (Fig. S3a, ESI†).31 Compared with the spectrum of the dried bulk melon (Fig. S3b, ESI†), the 1H signal intensity of NH2 lacking hydrogen bonds (H-bonds) (4.5 ppm)35 is enhanced. This result indicates poor molecular order in g-CN/G, supporting the lower crystallinity of g-CN/G than the melon film (g-CN/Q) (Fig. S1, ESI†).
Based on this analysis, the chemical modifications of g-CN/G are summarized in the schematics in Fig. 3. The observation that the g-CN film is concisely decorated with photocatalytically relevant moieties,30 cyanamide and hydroxyl groups, is important. In the present study, the selectivity of the chemical reaction cannot be controlled, and thus the modification should be random.
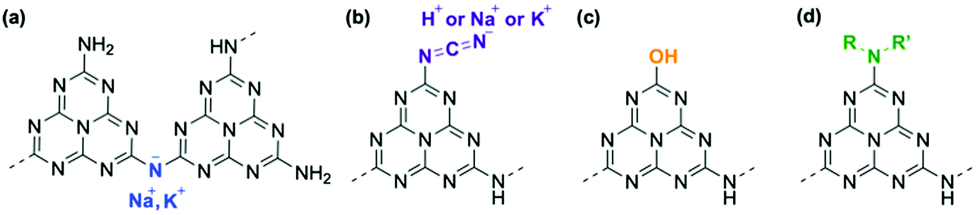 | ||
| Fig. 3 Summary of the chemical modifications of g-CN/G. (a) Removal of a proton from NH bridging heptazine units. Substitutions of –NH2 with cyanamide groups (b), hydroxyl groups (c), and alkyl amine groups (R,R′ = unknown alkyl groups) (d). | ||
Similar to the ion exchange properties of PHI salts,31,36 most of the alkali metals in g-CN/G are removed by soaking it in 0.02 M aqueous HCl for 15 min. The absorption of the cyanamide stretching at 2151 cm−1 (L, right-hand panel of Fig. 4a) decreases and the NC2 vibration modes disappear (left-hand panel of Fig. 4a). The ratio of L to the component at 2178 cm−1 (H, right-hand panel of Fig. 4a) decreases to 0.15 from 0.81 (Fig. 4b). The Na 1s and K 2s XPS spectra unambiguously show the removal of Na+ and K+ from g-CN/G by HCl treatment (Fig. 2a and b, respectively). Soaking HCl-treated g-CN/G (g-CN/G-HCl) in aqueous NaCl partially recovers the absorbance of cyanamide at 2150 cm−1. This means that some Na+ binds again to the cyanamide groups. However, the absorptions at 994 and 1153 cm−1 are not recovered by NaCl treatment. Subsequently, soaking g-CN/G in 2.0 M aqueous NaOH fully recovers these bands. This means that a strong base is necessary to deprotonate the bridging NH and to form alkali metal-NC2 groups. Conversely, the N–H in the cyanamide group is a strong acid due to the electron-withdrawing properties; the pKa of cyanamide is 1.1, whereas that of secondary amines is around 10.37 Therefore, the ion exchange between H+ and Na+ occurs more easily at cyanamide sites than at NC2 sites. We further noticed the appearance of a vibrational peak at 2250 cm−1 in g-CN/G and g-CN/G-HCl. The absorption is presumably attributed to the asymmetric stretching mode of isocyanate. The functional group is subject to hydrolysis and finally converted to amine after soaking the film in aqueous NaCl and NaOH.
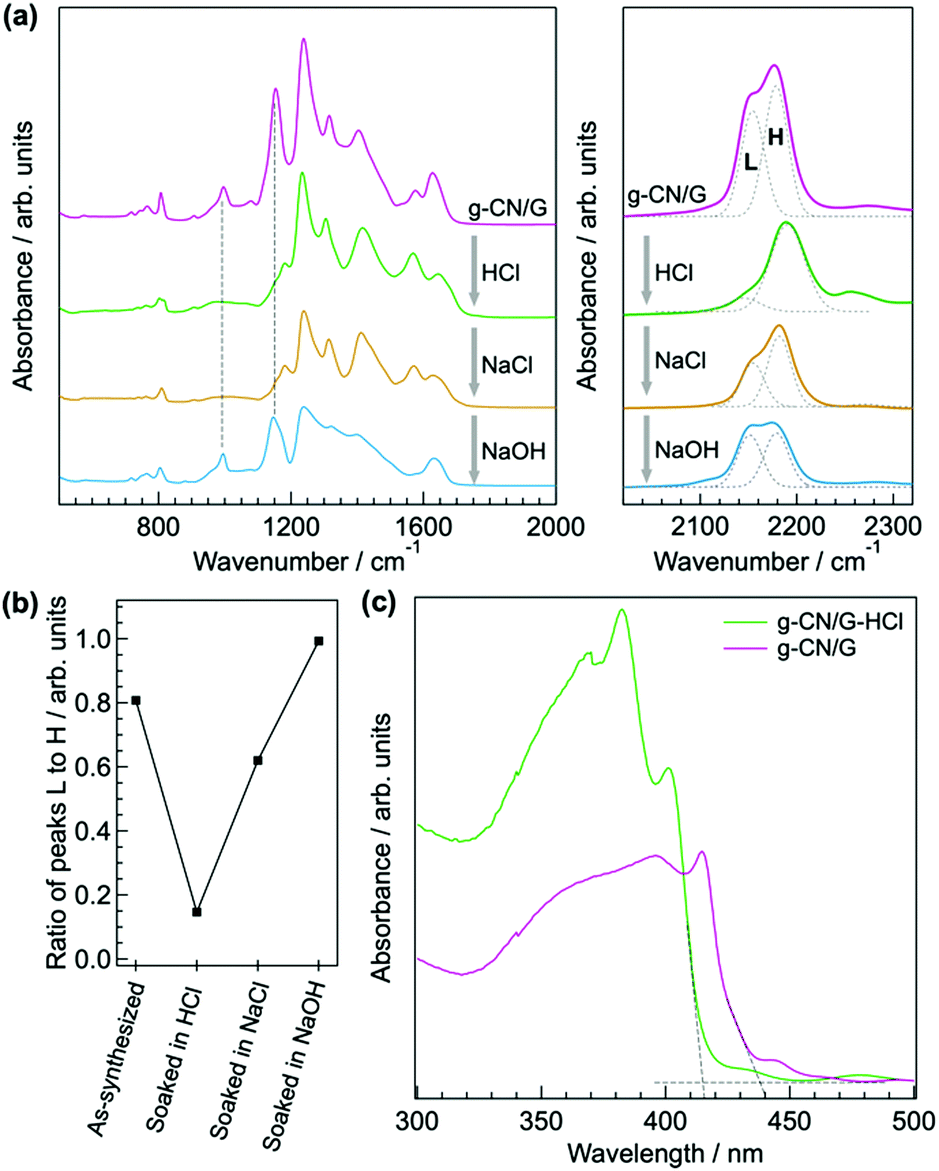 | ||
| Fig. 4 Ion exchange properties and UV-vis spectra of g-CN/G. (a) Evolution of ATR-FTIR spectra of g-CN/G upon soaking in HCl, NaCl, and NaOH aqueous solutions. Dashed lines in the left-hand panel indicate the vibrational absorptions of symmetric and asymmetric stretching of M–NC2 groups (M = Na or K). Gray dashed curves in the right-hand panel are peaks separated by Gaussian fitting. The components labelled L and H are attributed to the stretching mode of the cyanamide moieties bound to alkali metal cations and protons, respectively. (b) Evolution of the intensity ratio of the components L and H in the right-hand panel of (a). (c) UV-vis spectra of g-CN/G and g-CN/G-HCl peeled off from glass. The crossing point of the dashed tangent and parallel lines determines the onset of the optical absorption. Variation in the absorption at longer wavelengths beyond the absorption onsets is caused by interference of the incident and reflection light. | ||
We also conclude that g-CN/G prepared in this study is not PHI, despite the ion exchange properties. The optical gap of g-CN/G is 2.88 eV, and, after HCl treatment, the value increases to 2.95 eV (Fig. 4c). Because the optical gap of PHI is reported to be 2.1 eV,36 PHI is not formed on the glass slides.
Surface reaction mechanism
Our analyses demonstrate that using a glass slide introduces hydroxyl and cyanamide groups into a g-CN film upon VDP. To explore how the functional groups are formed, the surface properties of the glass are examined. The presence of adsorbed water molecules on the glass is confirmed by sum frequency generation (SFG) spectroscopy, which can probe vibrational modes of adsorbates (Fig. S6, ESI†). In addition to the surface hydroxyl group peak at 3775 cm−1,38 two broad absorptions are visible at 3270 and 3445 cm−1, which are attributed to the O–H stretching of ice-like and of liquid-like water molecules, respectively.39 In addition, Na+ and K+ are present likely as –SiO− Na+ and –SiO− K+ on the surface of the heated glass with concentrations of 10.82 and 1.80%, respectively (Table S2, ESI†). The adsorbed water and alkali metal ions undergo an ion exchange reaction via deprotonation of adsorbed water molecules by basic silanolate groups (Fig. 5a).40,41 Hence, an alkali metal hydroxide is generated as a product.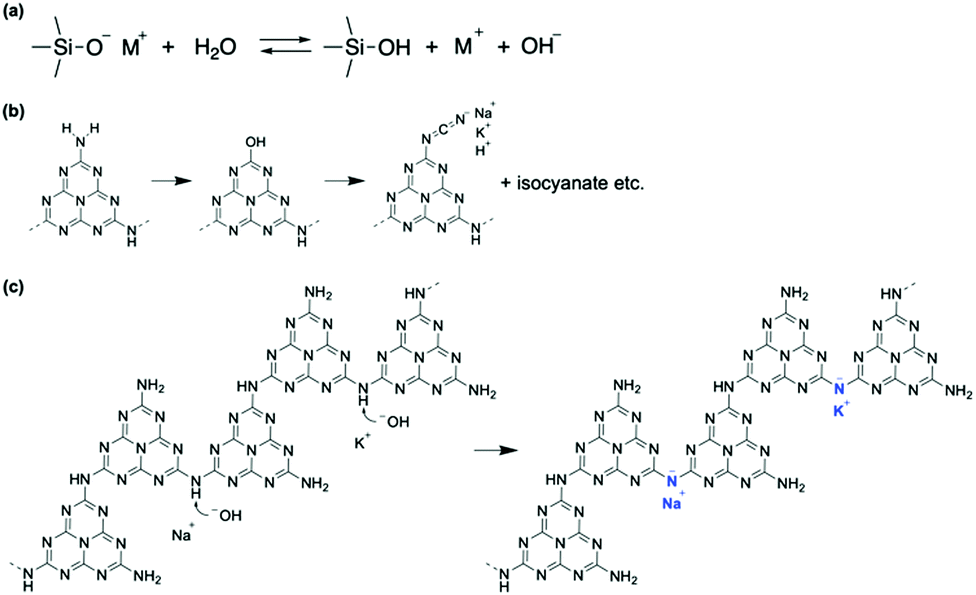 | ||
| Fig. 5 Reaction mechanisms occurring at the surface of a glass slide. (a) Surface exchange reaction. Alkali metal cations (M+, M = Na or K) are replaced with a proton from an adsorbed water molecule, generating OH−. (b) Substitution of –NH2 with –OH, followed by the formation of cyanamide-modified molecules and isocyanate. The bonding state of isocyanate is elusive. (c) OH− can remove a proton from NH groups bridging heptazine units, leading to M–NC2 groups (M = Na or K). | ||
The resultant MOH (M = K or Na) is responsible for cyanamide modification of a g-CN film because heating bulk melem and melon with NaOH (10 wt%) forms the cyanamide moiety (right-hand panel of Fig. 6a). Moreover, cyanamide groups are formed in g-CN/G made from melem at 310 °C, a temperature that is insufficient for condensation to melon (Fig. 6b). This also exemplifies the reaction occurring between heptazine cores and MOH during film formation.
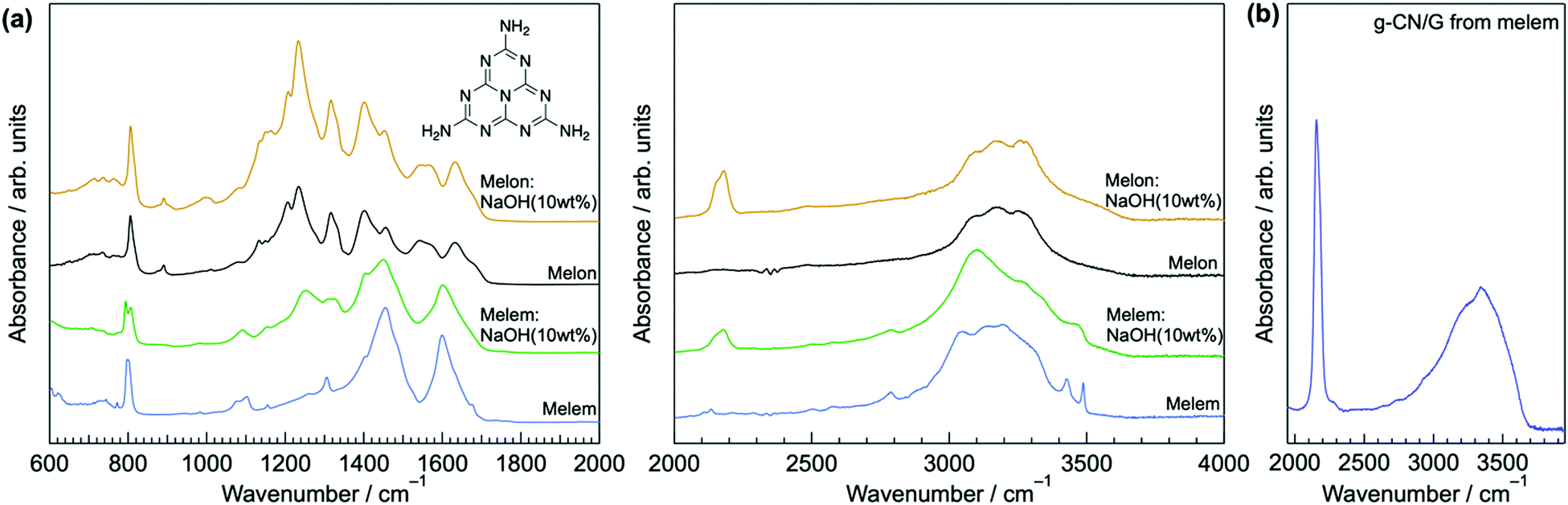 | ||
| Fig. 6 Demonstration of cyanamide formation. (a) ATR-FTIR spectra of powdery melem and melon calcined with NaOH (10 wt%). Inset in the left-hand panel shows the molecular structure of melem. (b) ATR-FTIR spectrum of g-CN/G synthesized from melem at 310 °C. | ||
A possible route to introducing a cyanamide moiety is shown in Fig. 5b. A fraction of terminal NH2 groups is substituted with OH, as shown by the presence of C–O (Fig. 2a) and O–H bonds (right-hand panel of Fig. 1b). This reaction is possible at high temperatures because alkali metal cyamelurate is obtained by refluxing melon with alkali metal hydroxides.42 Thermal decomposition of this moiety would lead to the cyanamide-substituted molecules and isocyanate (right panel of Fig. 4a),43 as IR analysis of sodium cyamelurate heated with base suggests the formation of these moieties (data not shown). The bonding state of isocyanate to the melem backbone is unclear at present. Yu et al. proposed that calcination of urea with KOH led to a ring-open structure with a cyanamide termination.44 However, according to theoretical calculations with DFT, the change in Gibbs free energy to produce this intermediate from a melem dimer and NaOH in the solid state is +546 kJ mol−1, indicating that the ring-opened form of a heptazine ring is thermodynamically unfavorable. Moreover, OH− could remove a hydrogen atom from a bridging N–H (Fig. 5c). This reaction is supported by the recovery of symmetric and asymmetric stretching of the NC2 bond of metal–NC2 groups after NaOH treatment (vibration absorptions at 998 and 1150 cm−1 in Fig. 4a).
Photocatalytic activity
We finally examine the effects of the chemical modification on the photocatalytic performance of the g-CN film by measuring the photodegradation of methyl orange in aqueous solution. g-CN films are formed on a pre-heated glass slide (denoted as g-CN/hG). The pre-heating treatment dramatically improves the adhesion strength of the film to the substrate. Otherwise, the g-CN film partially peels off from the substrate during the photocatalytic experiments. g-CN/hG contains hydroxyl and cyanamide groups, and Na+ and K+ that are ionically bound to nitrogen atoms in the NC2 bond (Fig. S7a, ESI†). Treating g-CN/hG with HCl eliminates the alkali metals from the film (Fig. S7b and c, ESI†). In Fig. 7a, the degradation efficiency (η, see the Experimental section for calculations) of methyl orange, an organic dye, for g-CN/hG is plotted (red open circles). After 5 h, η for g-CN/hG reaches only 0.98%. The observed activity is inferior to g-CN/AFG (melon film, η = 2.0% after 5 h). Soaking g-CN/hG in 0.02 M aqueous HCl (denoted as g-CN/hG-HCl) dramatically facilitates the photodegradation (Fig. 7b). η of g-CN/G-HCl after 5 h reaches 16.6%, which is 17-fold higher than the performance of g-CN/hG. The degradation rates of g-CN/AFG, g-CN/hG and g-CN/hG-HCl, which were estimated based on a first-order model, were found to be 0.0038, 0.0020, and 0.042 h−1, respectively (Fig. S8a, ESI†). The adsorption manner of methyl orange is not largely different for g-CN/AFG and g-CN/hG-HCl, because the change in the blank data is negligible (Fig. S8b, ESI†). All g-CN/hG films prepared on four types of glass slides show an increase in η after the HCl treatment (Fig. S9a, ESI†). It is remarkable that g-CN/hG-HCl shows superior photocatalytic activity to g-CN/AFG prepared under the same synthesis conditions. In contrast, the HCl treatment results in low η for g-CN/AFG (plot for g-CN/AFG-HCl in Fig. 7a). This result rules out the possibility that protonation activates g-CN.45 Rather, the removal of the alkali metals contributes to enhanced photodegradation. The addition of isopropanol (IPA) to the methyl orange solution further increases η (Fig. 7b), which indicates that IPA acts as a hole scavenger46,47 rather than a quencher of OH radicals.48 The performance reaches about two-thirds of the degradation efficiency of the TiO2 film (anatase) (Fig. S10c, ESI†). The degradation rate of g-CN/hG-HCl + IPA is 0.052 h−1 (Fig. S8a, ESI†). Furthermore, treating g-CN/hG with aqueous citric acid (CA), an acid ubiquitous in nature, increases the photodegradation by a factor of 8 (Fig. S9b, ESI†). Interestingly, a g-CN film prepared on a piece of a broken glass exhibits comparable performance after CA treatment (Fig. S9b, ESI†). The results demonstrate that waste glass and a natural acid could be used to activate a g-CN photocatalyst in the film form.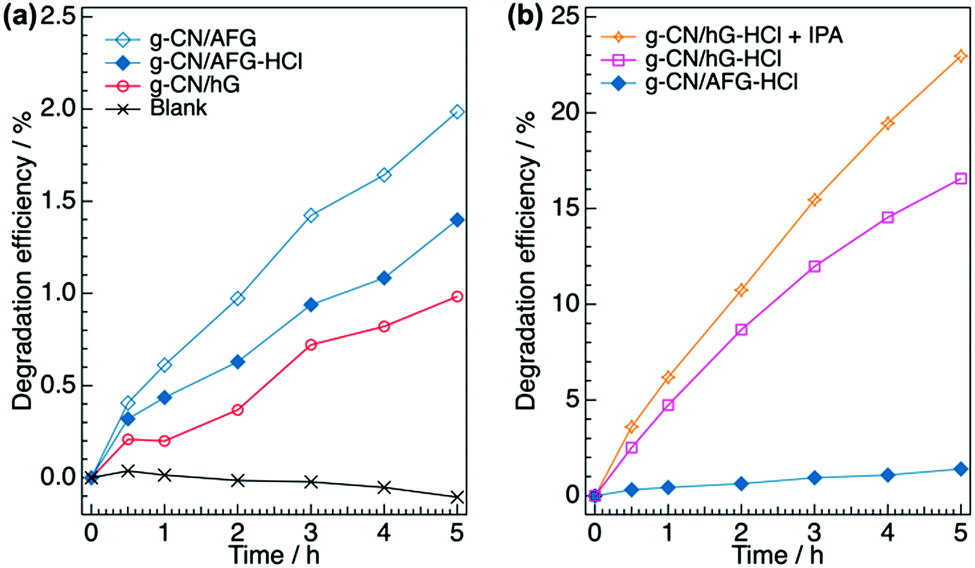 | ||
| Fig. 7 Photocatalytic degradation of methyl orange in aqueous solution using g-CN films. (a) Evolution of degradation efficiency of methyl orange (η) over 5 h using g-CN/AFG, g-CN/AFG-HCl, and g-CN/hG. (b) Evolution of η for g-CN/AFG-HCl, g-CN/hG-HCl, and g-CN/hG-HCl with the addition of 0.99% IPA (denoted as g-CN/hG-HCl + IPA). The values are determined from the absorbance at 464 nm in the UV-vis spectra shown in Fig. S12 (ESI†). | ||
PL spectroscopy provides clues to understand the enhanced photocatalytic activity of g-CN/hG-HCl. The steady-state PL spectrum of g-CN/AFG (melon film) shows a broad peak at 450 nm with a shoulder structure at around 525 nm, which is consistent with typically synthesized melon (Fig. 8a).49–51 The band originates from the recombination of singlet excitons.52 The photoluminescence quantum yield (PLQY) of this film is 3.9% (Table 1), again in good agreement with the literature.49 The PL spectrum of g-CN/hG is more distinct than that of g-CN/AFG (Fig. 8b), but the PLQY of g-CN/hG is only 0.8%. This result implies that excitons generated in a g-CN film on a glass slide are quenched more efficiently than those generated in g-CN/AFG. Exciton recombination is suppressed further by HCl treatment (Fig. 8b) and PLQY drops to less than 0.5% (Table 1). The faster decay of excitons is demonstrated for g-CN/hG and g-CN/hG-HCl via time-resolved PL spectroscopy (TR-PL). Fig. 8c shows the decay profiles of PL at around 450 nm for g-CN/AFG, g-CN/hG, and g-CN/hG-HCl on a nanosecond scale. The measured decay profiles, a(t), are fitted with a tri-exponential function,53a(t) = bf![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/tf) + bmexp(−t/tm) + bsexp(−t/ts), where bi and ti are the fraction and decay time of the excited states, respectively (i = f, m, s). The parameters obtained by the fitting are summarized in Table 1. tf (1.5 ns) and tm (6.1 ns) for g-CN/AFG agree well with non-radiative quenching and recombination of free excitons in bulk g-CN.54 The lifetimes decrease for g-CN/hG (tf = 0.82 ns, tm = 3.3 ns). In addition, bf increases by a factor of 2.5, whereas bm and bs decrease and ts also decreases (33 ns for g-CN/AFG to 15 ns for g-CN/hG). These results suggest that the recombination of generated excitons is decreased in g-CN/hG; that is, exciton dissociation is facilitated, and hence more charges are generated. The HCl treatment increases bm and bs, and it decreases bf. As we show later, this effect is attributed to the longer exciton diffusion length in g-CN/hG-HCl due to the decrease in d-spacing of graphitic sheets and improved mosaicity.
exp(−t/tf) + bmexp(−t/tm) + bsexp(−t/ts), where bi and ti are the fraction and decay time of the excited states, respectively (i = f, m, s). The parameters obtained by the fitting are summarized in Table 1. tf (1.5 ns) and tm (6.1 ns) for g-CN/AFG agree well with non-radiative quenching and recombination of free excitons in bulk g-CN.54 The lifetimes decrease for g-CN/hG (tf = 0.82 ns, tm = 3.3 ns). In addition, bf increases by a factor of 2.5, whereas bm and bs decrease and ts also decreases (33 ns for g-CN/AFG to 15 ns for g-CN/hG). These results suggest that the recombination of generated excitons is decreased in g-CN/hG; that is, exciton dissociation is facilitated, and hence more charges are generated. The HCl treatment increases bm and bs, and it decreases bf. As we show later, this effect is attributed to the longer exciton diffusion length in g-CN/hG-HCl due to the decrease in d-spacing of graphitic sheets and improved mosaicity.
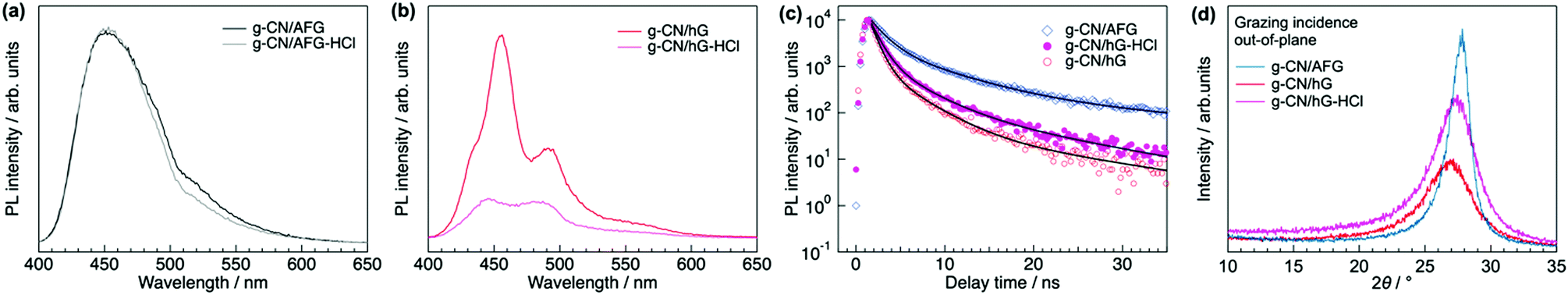 | ||
| Fig. 8 Steady-state PL spectra and TR-PL profiles of g-CN/hG and g-CN/hG-HCl compared with g-CN/AFG. (a) Comparison of steady-state PL spectra of g-CN/AFG with and without HCl treatment. (b) Steady-state PL spectra of g-CN/hG and g-CN/hG-HCl. (c) TR-PL profiles of g-CN/hG, g-CN/hG-HCl and g-CN/AFG. The black curves are the fitting of the measured data with tri-exponential functions. The extracted parameters are summarized in Table 1. (d) Grazing incidence XRD profiles of g-CN/hG, g-CN/hG-HCl, and g-CN/AFG. | ||
| Sample | η 5/% | PLQY/% | b f/104 | t f/ns | b m/103 | t m/ns | b s/102 | t s/ns | d g/Å | l g/Å |
|---|---|---|---|---|---|---|---|---|---|---|
| g-CN/AFG | 2.0 | 3.9 | 2.2 | 1.5 | 3.3 | 6.1 | 2.6 | 33 | 3.18 | 50.0 |
| g-CN/hG | 0.98 | 0.8 | 5.5 | 0.82 | 1.7 | 3.3 | 0.59 | 15 | 3.36 | 19.9 |
| g-CN/hG-HCl | 16.6 | <0.5 | 4.0 | 0.99 | 2.2 | 3.5 | 1.7 | 13 | 3.29 | 21.5 |
The enhanced, faster PL quenching in g-CN/hG can be attributed to the chemical modification by the glass. Molecular orbital calculations with DFT for melem dimers with cyanamide and hydroxyl groups suggest that the modification with the electron-withdrawing groups increases the ionization energy and electron affinity (Fig. 9b and c), compared with an unmodified melem dimer (Fig. 9a). The threshold ionization energy (Ith) of g-CN/G-HCl is measured by photoelectron yield spectroscopy as 6.99 eV (Fig. S13, ESI†), which is 0.19 eV larger than the Ith of a melon film determined by ultraviolet photoelectron spectroscopy.20 However, only a fraction of the molecules possesses these electron-withdrawing functional groups because ATR-FTIR, NMR, and XPS show that g-CN/G contains unreacted NH2 and bridging NH. From these results, we expect that the energy offset is present at the interface between unmodified and cyanamide or hydroxyl-modified molecules (Fig. 10). The energy offsets of the HOMO and/or LUMO facilitate exciton dissociation into free charges in the g-CN film. By comparing the potentials of g-CN films calculated from the solid-state ionization energy55 with redox potentials of related reactions vs. NHE taken from the literature,56 the photogenerated electrons on the LUMO of the modified molecules can produce O2 radicals with sufficient driving force. The reactants degrade methyl orange via the subsequent formation of OH radicals.48 Here, the LUMO position of the unmodified molecule agrees well with the data reported in the literature.57
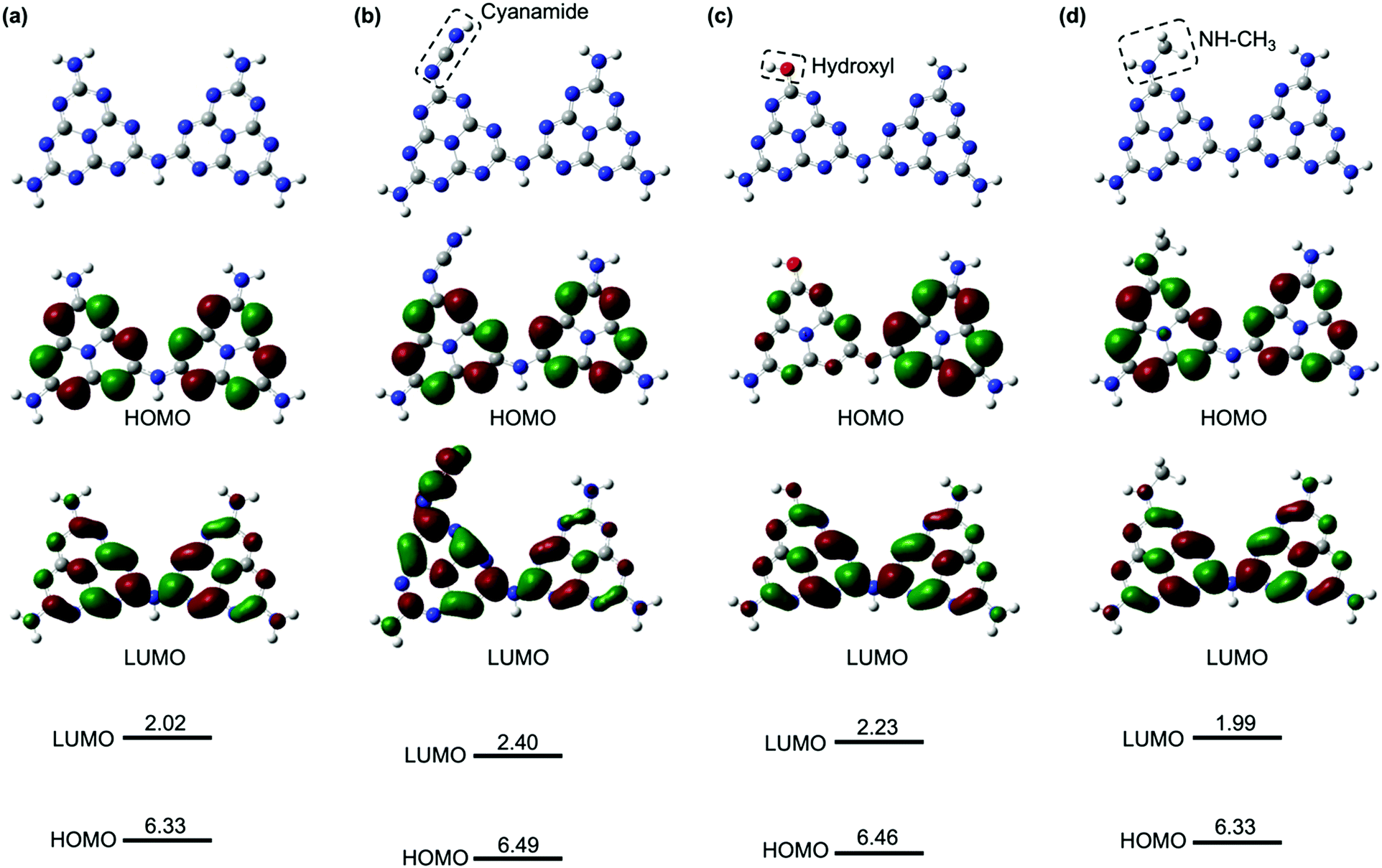 | ||
| Fig. 9 Characters and energies of the HOMO and LUMO with respect to the vacuum level for melem dimers. (a) Unmodified melem dimer. (b) Melem dimer with a cyanamide group in the left heptazine ring. (c) Melem dimer with a hydroxyl group. (d) Melem dimer with an N-methyl group as an example of alkyl amine substitution. | ||
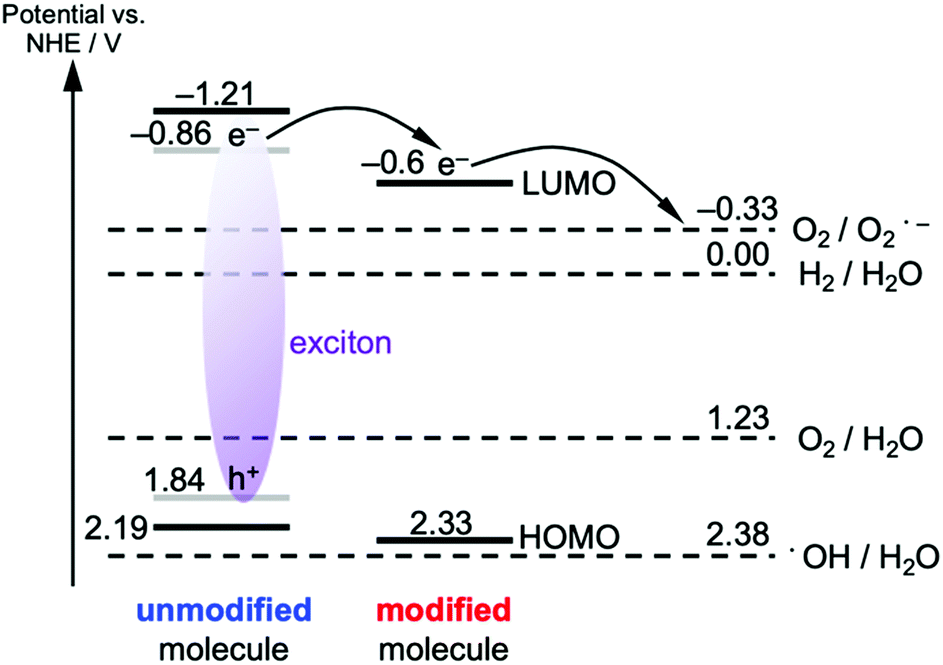 | ||
| Fig. 10 Energy diagram of the g-CN film deposited on the glass. The optical excitation of the unmodified molecule is considered. The HOMO and LUMO (black lines) of the unmodified molecule assume the same values of the melon film, which are obtained from the solid-state ionization energy and electron affinity. To convert the ionization energy obtained by ultraviolet photoelectron spectroscopy to electrical potential vs. NHE, eqn (4) in ref. 55 was used. The frontier orbitals of the excited molecule have lower energies due to the exciton binding energy (0.7 eV for the case of melon film). The grey lines for the excited unmodified molecule implement this effect under the assumption of the equal energy correction for the hole and electron. The HOMO energy for the modified molecule is obtained from the PYS data of g-CN/hG-HCl (Fig. S13, ESI†), whereas the LUMO potential is estimated from the optical gap of g-CN/hG-HCl and the HOMO energy. Potential vs. NHE for each redox reaction is taken from ref. 56. | ||
It should also be pointed out that the HOMO of the modified molecules is very close to the redox potential of the generation of OH radicals, but the driving force to directly generate OH radicals by the photogenerated holes on the HOMO of the modified molecule is insufficient. Note that the NH–CH3 group, a model of alkyl-amine substitution, does not affect the energies of the HOMO and LUMO greatly (Fig. 9d).
Efficient free-charge generation is indicated by PL spectroscopy and DFT calculations. Nevertheless, g-CN/hG exhibits poor photodegradation of methyl orange (Fig. 7a). Other elementary processes after charge generation, namely charge transport and chemical reactions, would hamper the photocatalytic activity of g-CN/hG, and the film needs acid treatment.
The molecular order along the surface normal, which is critical for charge transport toward reaction sites at the surface, varies with substrate type and HCl treatment. Grazing incidence XRD (Fig. 8d) shows that the d-spacing between graphitic sheets (dg) for g-CN/hG is 3.36 Å, which is 0.18 Å wider than that of g-CN/AFG (Table 1). The HCl treatment of g-CN/hG decreases the value to 3.29 Å. The XRD data explains the increase in η after the acid treatment of g-CN/hG: a shorter dg improves charge transport along the surface normal due to efficient overlaps of wavefunctions between graphitic sheets. In particular, the unoccupied molecular orbital (LUMO) is responsible for electron transport toward the surface to degrade methyl orange with OH radicals.48 Furthermore, the mosaicity of graphitic sheets is increased, as shown by the increase in diffraction intensity (Fig. 8d), which also contributes to better charge diffusion toward the film surface. The improvement in crystallinity by HCl treatment facilitates exciton diffusion between the graphitic layers, explaining the increase in the middle and slow components, bm and bs, of the PL for g-CN/hG-HCl (Fig. 8c and Table 1). g-CN/AFG exhibits the shortest dg and largest crystallite size (lg in Table 1), but the photodegradation is poor, indicating that increased exciton dissociation in g-CN/hG-HCl leads to superior photodegradation compared with g-CN/AFG.
The acid-treated g-CN/hG film shows superior photodegradation of methyl orange compared with the melon film. That is, film formation on a glass slide, followed by acid treatment, produces a g-CN photocatalyst film superior to melon without using a cocatalyst, like MoO3,58 due to the enhanced exciton dissociation and transport toward the film surface. This result contrasts with the case of the TiO2 film prepared on crown glass, where the maximum photocatalytic activity is comparable to that of the film formed on glasses with little or no Na+.59 However, adequate care must be taken to compare photocatalytic activity between dissimilar samples directly. The average thickness of g-CN/hG-HCl (393 nm) is 2.31 times larger than that of g-CN/AFG (170 nm), presumably due to increased adhesion of the g-CN film through the reaction between the carbon nitrides and the glass. Because a thicker film absorbs more photons, g-CN/hG-HCl can generate more excitons and, in principle, free carriers, than g-CN/AFG. However, only free carriers within the carrier diffusion length can contribute to photocatalytic reactions at the film surface. The carrier diffusion length of typical organic semiconductors is estimated to be 50–125 nm.60 By assuming this value for g-CN, free electrons generated only in the surface area of g-CN/hG-HCl and g-CN/AFG participate in the photodegradation reactions. Accordingly, the difference in film thickness does not explain the enhanced photodegradation of methyl orange for g-CN/hG-HCl. The photocatalytic activity is indeed unchanged even when the average thickness of g-CN-HCl decreases down to 151 nm (Fig. S9c, ESI†), the value which is close to g-CN/AFG. Moreover, even if the absorbance of g-CN/hG-HCl is 2.31 times larger than that of g-CN/AFG, g-CN/hG-HCl still exhibits higher η than g-CN/AFG. Thus, we conclude that chemical modification by the glass, followed by the acid treatment, boosts the photocatalytic performance of g-CN films.
The acid treatment to activate g-CN/hG could exfoliate graphitic sheets from the layered g-CN, which increases the surface area, thereby boosting photocatalytic activity.51,61–63 However, because the film thickness of g-CN/hG is unchanged by soaking it in an acid solution, the increase in the surface area by acid treatment is not the reason for the improvement in η. Instead, almost complete removal of the alkali metals from cyanamide moieties is essential. The LUMO of a cyanamide-modified g-CN spreads over heptazine rings as well as the electron-withdrawing groups (Fig. 9b and c). The presence of alkali metals with positive charges near cyanamide can trap photogenerated electrons. The acid treatment both increases film crystallinity (Fig. 8d) and reduces the electron trapping effect of the alkali metal cations.
For practical use of the g-CN film deposited on glass, its repeatability and stability are important. We found that the photodegradation rate of g-CN/hG-HCl did not decrease after three-cycle runs of the photodegradation experiments over 16 h (Fig. 11). The result suggests that the repeated use of the g-CN film is possible. A long-term durability test of the film is ongoing, and corresponding results will be reported elsewhere.
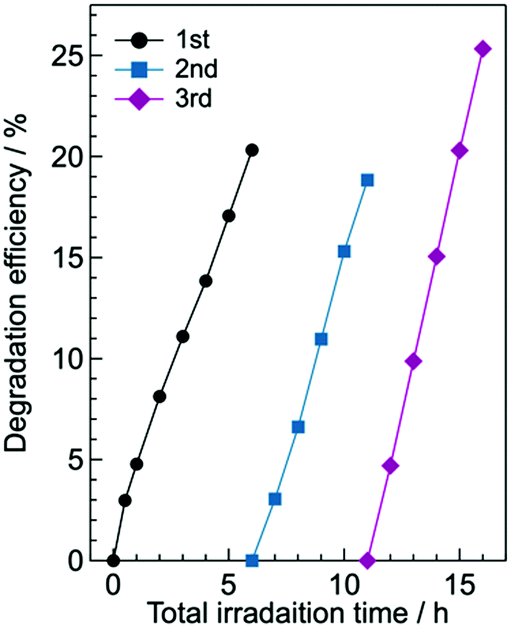 | ||
| Fig. 11 Cycling run of the photodegradation experiment of aqueous methyl orange with g-CN/hG-HCl. 0.99% IPA is added to the solution. The g-CN film used is synthesized in another batch and thus different from the sample used to obtain the data shown in Fig. 7b. | ||
Conclusions
In summary, this study demonstrates that the interface reaction with soda-lime glass drastically modifies the chemical structure of g-CN with electron-withdrawing groups. The enhanced photocatalytic behavior of the g-CN films on glasses was attributed to the increased free-carrier generation caused by the HOMO/LUMO offsets and improved crystallinity along the surface normal owing to the removal of the alkali metals from the films. Even waste glass from tableware and a natural, organic acid activated the g-CN films. Our findings will advance the development of scalable photocatalytic reactions with high activity using low-cost soda-lime glass for capillaries15 or glass beads48 with a large surface area. More generally, the modulation of the substrate surface provides a new way to develop efficient g-CN film photocatalysts, together with efforts to decrease the optical gap for efficiently harvesting photons over a wider range of the solar spectrum.64–66Conflicts of interest
There are no conflicts to declare.Acknowledgements
K. A. acknowledges the Leading Initiative for Excellent Young Researchers from the Ministry of Education, Culture, Sports, Science and Technology. H. N. acknowledges the JSPS Grant-in-Aid for Early-Career Scientists (JP20K15319) for the support of this research.Notes and references
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Di, X. Wang, A. Thomas and M. Antonietti, ChemCatChem, 2010, 2, 834–838 CrossRef CAS.
- R. Malik and V. K. Tomer, Renewable Sustainable Energy Rev., 2021, 135, 110235 CrossRef CAS.
- Z. Zhao, Y. Sun and F. Dong, Nanoscale, 2015, 7, 15–37 RSC.
- L. Jiang, X. Yuan, Y. Pan, J. Liang, G. Zeng, Z. Wu and H. Wang, Appl. Catal., B, 2017, 217, 388–406 CrossRef CAS.
- N. Karjule, J. Barrio, L. Xing, M. Volokh and M. Shalom, Nano Lett., 2020, 20, 4618–4624 CrossRef CAS PubMed.
- R. Shen, K. He, A. Zhang, N. Li, Y. H. Ng, P. Zhang, J. Hu and X. Li, Appl. Catal., B, 2021, 291, 120104 CrossRef CAS.
- Q. Lu, K. Eid, W. Li, A. M. Abdullah, G. Xu and R. S. Varma, Green Chem., 2021, 23, 5394–5428 RSC.
- S. Bhowmik, S. J. Phukan, N. K. Sah, M. Roy, S. Garai and P. K. Iyer, ACS Appl. Nano Mater., 2021, 4, 12845–12890 CrossRef CAS.
- Y. Shen, A. J. Dos santos-Garcia and M. J. Martín de Vidales, Processes, 2021, 9, 66 CrossRef CAS.
- J. Tan, Z. Li, J. Li, J. Wu, X. Yao and T. Zhang, Chemosphere, 2021, 262, 127675 CrossRef CAS PubMed.
- H. Arazoe, D. Miyajima, K. Akaike, F. Araoka, E. Sato, T. Hikima, M. Kawamoto and T. Aida, Nat. Mater., 2016, 15, 1084–1089 CrossRef CAS PubMed.
- J. Qin, J. Barrio, G. Peng, J. Tzadikov, L. Abisdris, M. Volokh and M. Shalom, Nat. Commun., 2020, 11, 4701 CrossRef PubMed.
- G. Peng, M. Volokh, J. Tzadikov, J. Sun and M. Shalom, Adv. Energy Mater., 2018, 8, 1800566 CrossRef.
- S. Mazzanti, G. Manfredi, A. J. Barker, M. Antonietti, A. Savateev and P. Giusto, ACS Catal., 2021, 11, 11109–11116 CrossRef CAS.
- P. V. Kamat, Chem. Rev., 1993, 93, 267–300 CrossRef CAS.
- M. A. Henderson, Surf. Sci. Rep., 2002, 46, 1–308 CrossRef CAS.
- S. M. George, A. W. Ott and J. W. Klaus, J. Phys. Chem., 1996, 100, 13121–13131 CrossRef CAS.
- J. Bian, L. Xi, J. Li, Z. Xiong, C. Huang, K. M. Lange, J. Tang, M. Shalom and R.-Q. Zhang, Chem. – Asian J., 2017, 12, 1005–1012 CrossRef CAS PubMed.
- K. Akaike, K. Aoyama, S. Dekubo, A. Onishi and K. Kanai, Chem. Mater., 2018, 30, 2341–2352 CrossRef CAS.
- H. Nagashima, J. Trébosc, Y. Kon, K. Sato, O. Lafon and J.-P. Amoureux, J. Am. Chem. Soc., 2020, 142, 10659–10672 CrossRef CAS PubMed.
- T. Hosokai, T. Nakanishi, A. Honda, K. Akaike, R. Tsuruta, H. Itoh, H. Nakanotani and Y. Nakayama, J. Phys. Chem. C, 2020, 124, 24919–24929 CrossRef CAS.
- S. Xie, P. Huang, J. J. Kruzic, X. Zeng and H. Qian, Sci. Rep., 2016, 6, 21947 CrossRef CAS PubMed.
- F. Fina, S. K. Callear, G. M. Carins and J. T. S. Irvine, Chem. Mater., 2015, 27, 2612–2618 CrossRef CAS.
- Z. Chen, A. Savateev, S. Pronkin, V. Papaefthimiou, C. Wolff, M. G. Willinger, E. Willinger, D. Neher, M. Antonietti and D. Dontsova, Adv. Mater., 2017, 1700555 CrossRef PubMed.
- D. Braunger, D. Hariskos, G. Bilger, U. Rau and H. W. Schock, Thin Solid Films, 2000, 6 Search PubMed.
- W. Li, X. Yan, A. G. Aberle and S. Venkataraj, Sci. Rep., 2019, 9, 2637 CrossRef PubMed.
- W. Xu, X. Zhao, X. An, S. Wang, J. Zhang, Z. Li, W. Wu and M. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 48526–48532 CrossRef CAS PubMed.
- D. C. Bradley and M. H. Gitlitz, J. Chem. Soc. A, 1969, 980–984 RSC.
- V. W. Lau, I. Moudrakovski, T. Botari, S. Weinberger, M. B. Mesch, V. Duppel, J. Senker, V. Blum and B. V. Lotsch, Nat. Commun., 2016, 7, 12165 CrossRef CAS PubMed.
- H. Schlomberg, J. Kröger, G. Savasci, M. W. Terban, S. Bette, I. Moudrakovski, V. Duppel, F. Podjaski, R. Siegel, J. Senker, R. E. Dinnebier, C. Ochsenfeld and B. V. Lotsch, Chem. Mater., 2019, 31, 7478–7486 CrossRef CAS PubMed.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- C. Perinu, G. Saramakoon, B. Arstad and K.-J. Jens, Energy Proc., 2014, 63, 1144–1150 CrossRef CAS.
- R. Marek and A. Lycka, COC, 2002, 6, 35–66 CrossRef CAS.
- X. Li, I. V. Sergeyev, F. Aussenac, A. F. Masters, T. Maschmeyer and J. M. Hook, Angew. Chem., Int. Ed., 2018, 57, 6848–6852 CrossRef CAS PubMed.
- A. Savateev, S. Pronkin, M. G. Willinger, M. Antonietti and D. Dontsova, Chem. – Asian J., 2017, 12, 1517–1522 CrossRef CAS PubMed.
- S. Soloway and H. J. Lipschitz, Org. Chem., 1958, 23, 613–615 CrossRef CAS.
- N. Sheth, D. Ngo, J. Banerjee, Y. Zhou, C. G. Pantano and S. H. Kim, J. Phys. Chem. C, 2018, 122, 17792–17801 CrossRef CAS.
- L. Zhang, C. Tian, G. A. Waychunas and Y. R. Shen, J. Am. Chem. Soc., 2008, 130, 7686–7694 CrossRef CAS PubMed.
- Y. Li, T. F. Mehari, Z. Wei, Y. Liu and R. G. Cooks, Angew. Chem., Int. Ed., 2021, 60, 2929–2933 CrossRef CAS PubMed.
- Y. Li, K.-H. Huang, N. M. Morato and R. G. Cooks, Chem. Sci., 2021, 12, 9816–9822 RSC.
- E. Horvath-Bordon, E. Kroke, I. Svoboda, H. Fueß, R. Riedel, S. Neeraj and A. K. Cheetham, Dalton Trans., 2004, 3900–3908 RSC.
- A. Schwarzer, T. Saplinova and E. Kroke, Coord. Chem. Rev., 2013, 257, 2032–2062 CrossRef CAS.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- Y. Zhang, A. Thomas, M. Antonietti and X. Wang, J. Am. Chem. Soc., 2009, 131, 50–51 CrossRef CAS PubMed.
- G. R. Dey, A. D. Belapurkar and K. Kishore, J. Photochem. Photobiol., A, 2004, 163, 503–508 CrossRef CAS.
- B. Ohtani, M. Kakimoto, H. Miyadzu, S. Nishimoto and T. Kagiya, J. Phys. Chem., 1988, 92, 5773–5777 CrossRef CAS.
- J. Hui, C. J. Pestana, M. Caux, H. Q. N. Gunaratne, C. Edwards, P. K. J. Robertson, L. A. Lawton and J. T. S. Irvine, J. Photochem. Photobiol., A, 2021, 405, 112935 CrossRef CAS.
- Z. Zhou, X. Niu, L. Ma and J. Wang, Adv. Theory Simul., 2019, 2, 1900074 CrossRef.
- L. He, M. Fei, J. Chen, Y. Tian, Y. Jiang, Y. Huang, K. Xu, J. Hu, Z. Zhao, Q. Zhang, H. Ni and L. Chen, Mater. Today, 2019, 22, 76–84 CrossRef CAS.
- X. Zhang, X. Xie, H. Wang, J. Zhang, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18–21 CrossRef CAS PubMed.
- B. Choudhury, K. K. Paul, D. Sanyal, A. Hazarika and P. K. Giri, J. Phys. Chem. C, 2018, 122, 9209–9219 CrossRef CAS.
- A. Y. Kobitski, R. Scholz, I. Vragović, H. P. Wagner and D. R. T. Zahn, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 153204 CrossRef.
- W. Xie, L. Tian, K. Wu, B. Guo and J. R. Gong, J. Catal., 2021, 395, 91–104 CrossRef CAS.
- B. Dandrade, S. Datta, S. Forrest, P. Djurovich, E. Polikarpov and M. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef CAS.
- S. Trasatti, Pure Appl. Chem., 1986, 58, 955 CrossRef CAS.
- T. Giannakopoulou, I. Papailias, N. Todorova, N. Boukos, Y. Liu, J. Yu and C. Trapalis, Chem. Eng. J., 2017, 310, 571–580 CrossRef CAS.
- Y. He, L. Zhang, X. Wang, Y. Wu, H. Lin, L. Zhao, W. Weng, H. Wan and M. Fan, RSC Adv., 2014, 4, 13610–13619 RSC.
- H.-J. Nam, T. Amemiya, M. Murabayashi and K. Itoh, J. Phys. Chem. B, 2004, 108, 8254–8259 CrossRef CAS.
- C. Longeaud, A. Fath Allah, J. Schmidt, M. El Yaakoubi, S. Berson and N. Lemaitre, Org. Electron., 2016, 31, 253–257 CrossRef CAS.
- T. Y. Ma, Y. Tang, S. Dai and S. Z. Qiao, Small, 2014, 10, 2382–2389 CrossRef CAS PubMed.
- J. Tian, Q. Liu, C. Ge, Z. Xing, A. M. Asiri, A. O. Al-Youbi and X. Sun, Nanoscale, 2013, 5, 8921 RSC.
- S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452–2456 CrossRef CAS PubMed.
- S. Zuluaga, L.-H. Liu, N. Shafiq, S. M. Rupich, J.-F. Veyan, Y. J. Chabal and T. Thonhauser, Phys. Chem. Chem. Phys., 2015, 17, 957–962 RSC.
- P.-K. Chuang, K.-H. Wu, T.-F. Yeh and H. Teng, ACS Sustainable Chem. Eng., 2016, 4, 5989–5997 CrossRef CAS.
- D. Vidyasagar, S. G. Ghugal, S. S. Umare and M. Banavoth, Sci. Rep., 2019, 9, 7186 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01677j |
| This journal is © the Owner Societies 2022 |