
Engineered manganese oxide nanocrystals for enhanced uranyl sorption and separation†
Seung Soo
Lee
a,
Wenlu
Li
a,
Changwoo
Kim
a,
Minjung
Cho
b,
Jeffrey G.
Catalano
c,
Brandon J.
Lafferty
d,
Paolo
Decuzzi
b and
John D.
Fortner
*a
aDepartment of Energy, Environmental, and Chemical Engineering, Washington University, St. Louis, MO63130, USA. E-mail: jfortner@wustl.edu
bDepartment of Translational Imaging and Department of Nanomedicine, Houston Methodist Research Institute, Houston, TX77030, USA
cDepartment of Earth and Planetary Sciences, Washington University, St. Louis, MO63130, USA
dU.S. Army Corps of Engineers, Engineer Research and Development Center, Vicksburg, MS39180, USA
First published on 26th August 2015
Abstract
For the first time, this work develops and demonstrates precisely engineered manganese oxide nanoscale particles for the sorption of uranium, as uranyl, in water. Size controlled monodisperse nanocrystalline manganese oxides (12 to 28 nm) were systematically synthesized via thermal decomposition of manganese oleate and phase-transferred into water by ligand exchange and bilayer stabilization methods. Resulting monodisperse suspensions demonstrate significantly enhanced uranyl adsorption as a function of size, surface coating chemistries, and solution pH. In particular, 12 nm particles coated with the unsaturated–unsaturated carbon chains linked bilayers, (e.g. oleic acid-oleyl phosphate linked bilayer coatings) have binding capacities well over 600 mg U per g of Mn, which is the highest reported uranium sorption capacity for any manganese based sorbent to date. Further, we spectrally identify significant uranyl reduction as part of the adsorption mechanism(s) for high capacity materials. Last, oleyl-based (phosphate and carboxylic) functionalized bilayered nanocrystals were extremely stable in the presence of high ionic strength/type (>800 mM); calcium (>19 mM), including the presence of uranyl cations (from 0.1 to 60 ppm). Taken together, these data demonstrate the potential for engineered monodisperse manganese oxide nanocrystals as ultra-high capacity platform sorbent materials for uranium separation at environmentally relevant ionic strengths and pH.
Nano impactEnvironmental radionuclide detection, separation, and treatment remain widespread, global challenges. Advances in nanoscale engineering now allows for precisely tuned sorbent materials with extreme surface to volume ratios and specific bulk and surface chemistries for advanced environmental applications. In this work, for the first time, we have systematically developed and demonstrated manganese oxide nanocrystals as a platform material for ultra-high capacity uranium sorption and separation under environmentally relevant conditions. As an example, for 12 nm, surface optimized materials, we observe binding capacities well over 600 mg U per g of Mn, which is the highest reported uranium sorption capacity for any manganese based sorbent to date. |
Introduction
The potential of applying engineered nano-scale materials for environmental treatment and sensing applications has gained significant attention in recent years.1–8 As sorbents, the use of engineered nanoscale particles allows not only extreme surface area to volume ratios, but also for the enhancement of enthalpic interactions, resulting in ultra-high sorption capacities compared with bulk phase analogues.2,4,5,9–15 As an example, there are a number of reports exploring enhanced uranium sorption processes to iron based metal oxide nanocrystals.4,9,11,12,16–18 Recently, manganese oxides based materials have gained attention due to potential redox reactions between uranium(IV and VI) and the variable oxidation sates of manganese(II, III and IV), which can enhance possible sorption capacity efficiencies via lowered U(IV) solubility.19,20 However, as engineered nanomaterials designed specifically for uranyl sorption and separation, manganese oxide based structures have not been thoroughly explored and optimized.Systematic evaluation of size and surface coating dependence of uranium sorption on manganese oxide nanocrystals necessitates precise control over both diameter (i.e. narrow distributions) and surface chemistry of the particles. Such controlled synthesis can be accomplished by various methods such as aqueous, hydrothermal, solvo-thermal, vapor phase growth, and non-hydrolytic routes.21–30 Among these processes, thermal decomposition routes are the only reliable strategies that allow for systematic development of uniform libraries of manganese oxide nanocrystals with narrow diameter distribution (σ < 10%).21,24,26,27,29,30 As synthesized, these nanocrystals, which are typically suspended in a hydrophobic phase, must be transferred to water using suitable surface transfer agents, that for application optimization, must simultaneously allow for colloidally stable suspensions and favorable uranium sorption.
In this work, libraries of precisely engineered monodisperse manganese oxide nanocrystals have been synthesized, stabilized in water, characterized, and demonstrated for uranium, as uranyl, sorption based applications. Monodisperse manganese oxide nanocrystals were synthesized through the thermal decomposition of manganese oleate with fine size control (12–28 nm) achieved through described combinations of reaction temperature, time, the ratio of metal precursor to surfactant, and monomer concentration(s). As-synthesized nanocrystalline manganese oxides were then systematically phase transferred into water, via a series of surface coating strategies, including organic acid bilayers (oleic acid base layer with oleyl phosphate, octadecylphosphonic acid, oleic acid, and stearic acid second layers interfacing the aqueous phase) and single layered, polyethylene glycol (PEG) of varied molecular weights (200–10k MW). The resulting colloidally stable nanocrystals were characterized and evaluated for uranium sorption capacities and aqueous stabilities under varied pH, ionic strengths, and types (Na+, Ca2+). Together, the results indicate that these materials are capable of ultra-high uranyl sorption capacities (>50% wt U/wt Mn, with partial U reduction from VI to IV observed) with simultaneously high aqueous stabilities when optimized via bilayer coating(s) with second (outer-facing) layers consisting of either carboxylic acid or phosphonic acid head groups.
Experimental
Materials
Manganese(II) chloride tetrahydrate (MnCl2·4H2O, 99.99%), oleic acid (90%), 1-octadecene (90%), poly (ethylene glycol) (PEG, Mw = 200, 1000, and 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), octadecylphosphonic acid (ODP, 97%), stearic acid (SA, 98.5%), manganese(II) oxide (MnO, 99%), iron(II, III) oxide (Fe3O4, 97%) were purchased from Sigma-Aldrich; oleyl phosphate (OP) and sodium oleate (97%) was purchased from TCI America; uranyl nitrate hexahydrate (UO2(NO3)2·6H2O) was purchased from Antec, Inc. All nanocrystals were synthesized under nitrogen condition (N2, 99.999%).
000), octadecylphosphonic acid (ODP, 97%), stearic acid (SA, 98.5%), manganese(II) oxide (MnO, 99%), iron(II, III) oxide (Fe3O4, 97%) were purchased from Sigma-Aldrich; oleyl phosphate (OP) and sodium oleate (97%) was purchased from TCI America; uranyl nitrate hexahydrate (UO2(NO3)2·6H2O) was purchased from Antec, Inc. All nanocrystals were synthesized under nitrogen condition (N2, 99.999%).
Synthesis of manganese oleate
Manganese oleate was synthesized by the method reported by An et al.21 Manganese chloride tetrahydrate (15.8 g, 80 mmol) reacted with oleic acid (45.2 g, 144 mmol) in 20 g of ethanol, 10 g of water, and 30 g of hexane at 60 °C for 4 h. The resulting pink colloidal suspension was washed by using water and ethanol. The purified manganese oleate was extracted by hexane.Synthesis of nanocrystalline manganese oxide
Manganese oleate was used as a manganese precursor for manganese oxide nanocrystal synthesis. 12.2 nm manganese oxides were prepared using 3.0 mmol manganese oleate, 2.0 mmol oleic acid in 5.0 g 1-octadecene at 320 °C for 1 h. 18.7 nm manganese oxide was synthesized by 6.0 mmol manganese oleate with 2 mmol oleic acid in 5.0 g 1-octadecene at 320 °C for 1 h. For 27.9 nm manganese oxide, 9.5 mmol manganese oleate was decomposed in the presence of 2.0 mmol oleic acid in 5.0 g 1-octadecene at 320 °C for 1 h. The resulting nanocrystals were purified using methanol, acetone, and hexane. More specifically, 1 g of the resulting colloidal nanocrystals was mixed with 10 ml of hexane, 20 ml of ethanol, and 20 ml of acetone and centrifuged at 7000 rpm for 20 min. This process was repeated three times. Final, cleaned colloidal nanocrystals were redispersed in hexane.Phase transfer of manganese oxide nanocrystals
As-synthesized nanocrystals were transferred to water by ligand encapsulation and exchange methods using probe sonication.31,32 For bilayer structures, manganese oxide nanocrystals, 0.05 mmol of surface stabilizer (oleyl phosphate, oleic acid, octadecylphosphonic acid, and stearic acid) was stirred with 0.5 ml of nanocrystal in hexane solution (concentration of manganese = 7.5 mg ml−1) and 10 ml of ultra-pure water (Millipore, 18.2 Ω). Aqueous and organic phases were mixed by application of a probe-sonicator (UP 50H, Dr. Hielscher, GMHB) for 5 min at 80% amplitude and full cycle. The resulting solution was further stirred for 1 day to evaporate hexane, resulting in a transparent, brown solution. To remove excess free phase transfer agent(s) left in the suspension, the phase transferred manganese oxide nanocrystals were further purified using ultracentrifugation (Sorvall WX Ultra 80, Thermo scientific) at 40000 rpm (ca. 150000 G-force) for 2 h, and membrane filtration (Ultrafiltration cellulose membranes, 100 kDa MWCO) using a stirred cell (Amicon), followed by syringe filtration (pore size of 0.22 μm, Whatman-NYL). The transfer yield of the final solution was typically ca. 70% by mass. For single layered polyethylene glycol coated manganese oxide nanocrystals, polyethylene glycol (PEG) (30.0 mg of PEG 200, 10.0 mg of PEG 1k, and 10 mg of PEG 10k) was mixed with 0.5 ml manganese oxide nanocrystal solution ([Mn] = 7.5 mg ml−1) in hexane and 10 ml of Milli-Q water. The aqueous and organic mixture was probe sonicated and purified by ultracentrifugation, filtered as done above. In all cases, the transfer yield of the purified nanocrystalline manganese oxide coated with PEG was over 60%.
Dynamic light scattering
The hydrodynamic diameters and surface charge of different sized manganese oxides coated with oleic acid, and poly (ethylene glycol) (PEG 200, 1k 10k) were measured at pH 7.2 and 25 °C, using a Malvern Nano ZS system by Malvern Instruments equipped with a HeNe 633 nm laser (Malvern Zetasizer Nanoseries, Malvern, UK). The average hydrodynamic diameters were obtained by the mean size of the first peak of the number distribution and the standard deviation was determined from triplicate measurements.Transmission electron microscope (TEM)
The diameters of the as-synthesized nanocrystals were directly measured by transmission electron microscope (TEM). TEM specimens were prepared using carbon support film on 300 mesh copper grids (Electron Microscopy Sciences). The TEM micrographs were taken by a Tecnai G2 Spirit Twin microscope (FEI, Hillsboro, OR) operated at 120 kV. HRTEM analysis was performed using a JEOL 2100F microscope (JEOL. Tokyo, Japan) operated at 200 kV. The size and size distribution data were obtained by counting over 1000 nanocrystals using Image-Pro Plus 6.0.33X-ray diffraction (XRD)
X-ray powder diffraction (XRD) patterns were obtained using a Bruker D8 Advance with a LynxEye XE detector. 2θ range was from 10 to 75 degree with a Cu Kα X-ray source (1.54 Å), generated at 40 kV and 40 mA.X-ray photoelectron spectroscope (XPS)
XPS data was collected using a PHI Quantera XPS with a monochromatic Aluminum 38.6 W X-ray source and 200.0 μm X-ray spot size with a pass energy of 26.00 eV at 45.0°.ATR-FTIR
FTIR spectra were recorded on a Nicolet Nexus 470 spectrometer equipped with a single bounce horizontal attenuated total reflection (HATR) accessory. A germanium crystal was used as the internal reflection element.Inductively coupled plasma optical emission spectroscopy (ICP-OES)
Elemental concentrations were measured by an inductively coupled plasma optical emission spectroscopy (ICP-OES, Perkin Elmer Optima 7300DV) instrument equipped with autosampler.Inductively coupled plasma mass spectroscopy (ICP-MS)
The uranium concentrations remained after uranium sorption measurement using manganese oxide nanocrystals were measured by an inductively coupled plasma mass spectroscopy (ICP-MS, Perkin Elmer ELAN DRC II) instrument equipped with autosampler.Uranium sorption measurements
Engineered nanocrystals (oleic acid coated and PEG 200 coated; 0.2 mg of manganese(II)) and commercial manganese oxides (typically as 6.7 mg of manganese(II)) were tested for uranium sorption at uranium concentrations ranging from 0.1 to 60 mg L−1 of uranium(VI) at various pH conditions (pH 5.6, pH 7.0, and pH 8.5). System pH was adjusted with HNO3 (0.05% by weight) and/or NaOH (0.02% by weight) before and after uranyl nitrate addition. The solution pH was further monitored and adjusted at least 3 times during each experiment (24 h). At equilibrium (24 h), nanocrystals were separated using ultracentrifugation at 40000 rpm for 2 h (ca. 150000 G-force) and the remaining concentrations of uranium(VI) in the supernatant solution were analyzed by ICP-MS. All measurements were conducted in triplicate. The measured uranium sorption density values (mass of sorbed uranium per mass of manganese) as a function of equilibrium concentration of uranium (mg L−1) were fitted by the Langmuir isotherm equation:where qe is the amount of adsorbed uranium at equilibrium concentration (mg g−1), k is the sorption constant, qmax is the maximum sorption density (mg g−1; mass of the sorbed uranium per mass of manganese), and Ce is the equilibrium concentration of uranium.
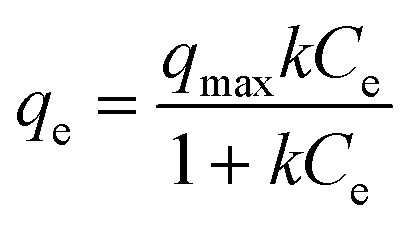
Critical coagulation concentration (CCC)
Nanocrystal aggregation kinetics were examined by monitoring hydrodynamic diameters of 12 nm manganese oxide nanocrystals (typically, 2.7 × 1012 nanocrystals per milliliter) coated with organic acids (bilayered structures) and PEG 200 (single layered structure) in varied concentration of NaCl (from 50 to 1000 mM) and CaCl2 (from 1 to 40 mM) at pH 5.6 for 40 min. The attachment efficiency of the nanocrystal aggregates, α, in the presence of monovalent or divalent salt suspension was calculated by the equation:where k is the initial aggregation rate constant at elevating salt concentrations and kfast is the diffusion limited aggregation rate constant under favorable aggregation conditions.34–38
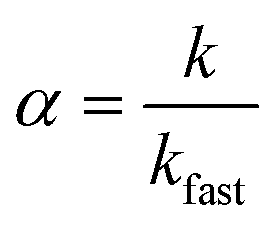
Results and discussion
Synthesis of monodisperse manganese oxide nanocrystals
Manganese oxide nanocrystals with controlled diameters were synthesized through the thermal decomposition of manganese oleate resulting in highly monodisperse particle libraries.21,39 Resulting nanocrystals were stabilized by an amphiphilic organic acid surface coating (oleic acid) and remained well dispersed in various non-polar organic solvents (hexane, toluene, ether, and chloroform). It was observed that the reaction temperature, time, and the ratio of surfactant to metal precursor affected the diameter of nanocrystalline manganese oxide.21 In agreement with past work, we found that the nanocrystalline manganese oxide nanocrystals increased in size from 6.8 ± 0.7 to 12.2 ± 1.2 nm as the reaction temperature increased from 290 to 320 °C (Fig. S1 in the ESI†). The increasing ratio of surfactant to manganese precursor (from 0:5 to 8:5) and longer reaction times (from 30 min to 3 h) also resulted in the larger manganese oxides over 25 nm (Fig. S1 in the ESI†).
In addition to the effect of reaction conditions (temperature, ratio of manganese oleate to oleic acid, and time), the concentration of the starting precursor materials (manganese oleate in 1-octadecene) had a significant effect on the nanocrystal size. Fig. 1 shows transmission electron micrographs of monodisperse manganese oxide nanocrystals from 12.2 ± 1.2 to 27.9 ± 2.8 nm synthesized by varying the concentration of the manganese oleate in 1-octadecene ranging from 0.5 to 1.5 M at 320 °C for 1 h (higher precursor concentrations yielding larger diameter nanocrystals), which has been observed previously for other, similarly sized monodisperse metal oxide nanoparticles.40,41
 | ||
| Fig. 1 TEM micrographs of manganese oxide nanocrystals. The high concentration of manganese oleate (Mn-Ol) increases the diameters of the nanocrystalline manganese oxides. The diameters of manganese oxide nanocrystals are 12.2 ± 1.2 nm (0.5 M of manganese oleate), 18.7 ± 2.8 nm (1.0 M of manganese oleate), and 27.9 ± 2.8 nm (1.5 M of manganese oleate) from (A) to (C). Different concentration of manganese oleate was decomposed in 1-octadecene in the presence of 2 mmol of oleic acid at 320 °C for 1 h. The average diameters of the nanocrystals were analysed by counting over 1000 particles using Image-Pro plus 6 (Fig. S2†). All scale bars are 50 nm. | ||
Synthesized nanocrystalline manganese oxides are shown to have a core- structure of MnO with a thin shell of Mn3O4. As shown in the HR-TEM analysis in Fig. S3 in the ESI,† the lattice fringes of the shell and the core structure were indexed to either (224) and (221) planes of Mn3O4 or (220) and (111) planes of MnO, respectively.26 This is consistent with X-ray diffraction (XRD) patterns of the manganese oxide samples in Fig. 2; all peaks match to MnO (JCPDS card # 07-0230) and Mn3O4 (JCPDS card # 24-0734) with the MnO:Mn3O4 ratio increasing with size as expected for a core–shell structure. That is to say, with increasing particle size, the particle core mass (composed of MnO) increases more than the surface area (covered by Mn3O4). This type of core–shell structure of manganese oxides (MnO@Mn3O4) may be formed through the oxidation Mn(II) on the MnO surface in the presence of oxygen as the reaction cools. Visually, the color of the reaction mixture at 320 °C was grey green (MnO) turning to a deep brown (the formation of Mn3O4) at room temperature and open to the air, as observed by others.21
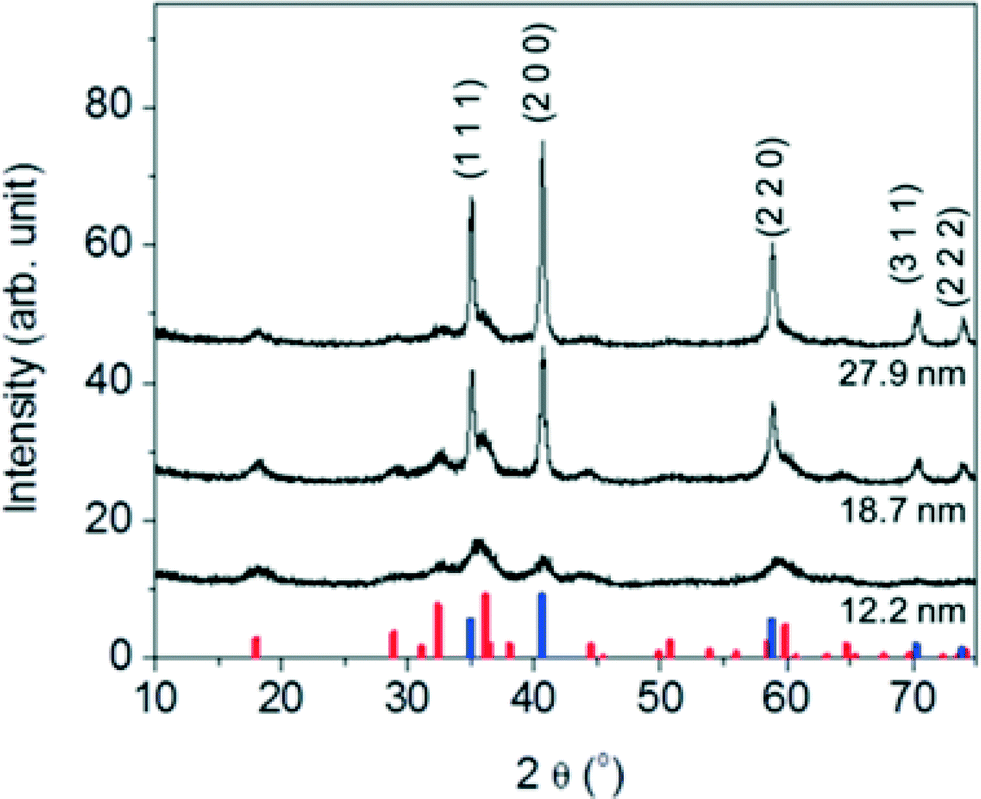 | ||
| Fig. 2 XRD patterns of the nanocrystalline manganese oxides with varying diameters of 12.2 ± 1.2 nm, 18.7 ± 2.8 nm, and 27.9 ± 2.8 nm. The position of the reflections matches to cubic MnO and the tetragonal Mn3O4 phases. The vertical lines at the bottom of the graph corresponds to JCPDS card of MnO (blue, JCPDS card no. 07-0230), and Mn3O4 (red, JCPDS card no. 24-0734). | ||
Phase transfer of monodisperse manganese oxide nanocrystals
To evaluate potential aqueous-based application, nanocrystalline manganese oxides synthesized in organic solvents were phase transferred into water using either by a bilayer strategy, using organic acids (such as, oleyl phosphate, octadecylphosphonic acid, oleic acid, and stearic acid) and/or ligand exchange process using polyethylene glycol (PEG) with different molecular weights. Organic acid bilayer strategies utilize the organic solvent stabilizing oleic acid base (surface) layer (occurring during the synthesis) which can then be tailored with regard to the second, outer layer, which upon self-assembly, interfaces the water phase via hydrophobic head group (here as oleic acid, oleyl phosphate, octadecylphosphonic acid, and stearic acid).31,32 For all materials described, the phase transferred nanocrystals were well dispersed in nano-pure (Milli-Q, 18.2 Ω) water and colloidally stable without any precipitation for over 6 months. The average core diameters and morphologies are identical to those observed for as-synthesized nanocrystals in organic solvents (Fig. 3). In all cases, phase transfer efficiency was over 70%. | ||
| Fig. 3 TEM micrographs of water stable manganese oxide nanocrystals. The diameters of manganese oxide nanocrystals are 12.0 ± 0.9, 18.2 ± 2.2, and 27.5 ± 2.9 nm from (A) to (C). The average diameters of the nanocrystals were analyzed by counting over 1000 particles using Image-Pro plus 6. All scale bars are 50 nm. | ||
Water-stable nanocrystalline manganese oxide samples were characterized by dynamic light scattering (DLS) to measure the hydrodynamic size and zeta potential as it relates to effective surface charge. The hydrodynamic diameters (DH) of the phase transfer agent coated manganese oxide nanocrystals are shown in Fig. 4; DLS measurements revealed that the bilayered structures (using oleyl phosphate, octadecyl phosphonic acid, oleic acid, and stearic acid) and a low molecular weight polymer (poly (ethylene glycol) with 200 molecular weight (PEG 200)) led to thin, compact surface coatings. For example, oleyl phosphate, octadecylphosphonic acid, oleic acid, stearic acid, and PEG 200 applied to nanocrystal cores of 12.2 nm diameter produced materials with hydrodynamic diameters (DH) of 21.7 ± 2.6, 19.2 ± 0.6, 20.4 ± 2.5, 23.6 ± 0.5, and 21.5 ± 2.0 nm, respectively. The larger molecular weight polymers yielded particles with larger hydrodynamic diameters (PEG 1k and 10k coated manganese oxide nanocrystals showed the hydrodynamic diameters of 31.5 ± 2.0, and 46.6 ± 0.7 nm, respectively). The zeta potential of oleyl phosphate, octadecylphosphonic acid, oleic acid stearic acid, poly (ethylene glycol) coated manganese oxide nanocrystals were −46.6 ± 2.3, −43.0 ± 1.9, −33.7 ± 1.9, −32.0 ± 1.1, and −14.7 ± 1.6 mV, respectively, which are in line with expected values for these coatings.42,43
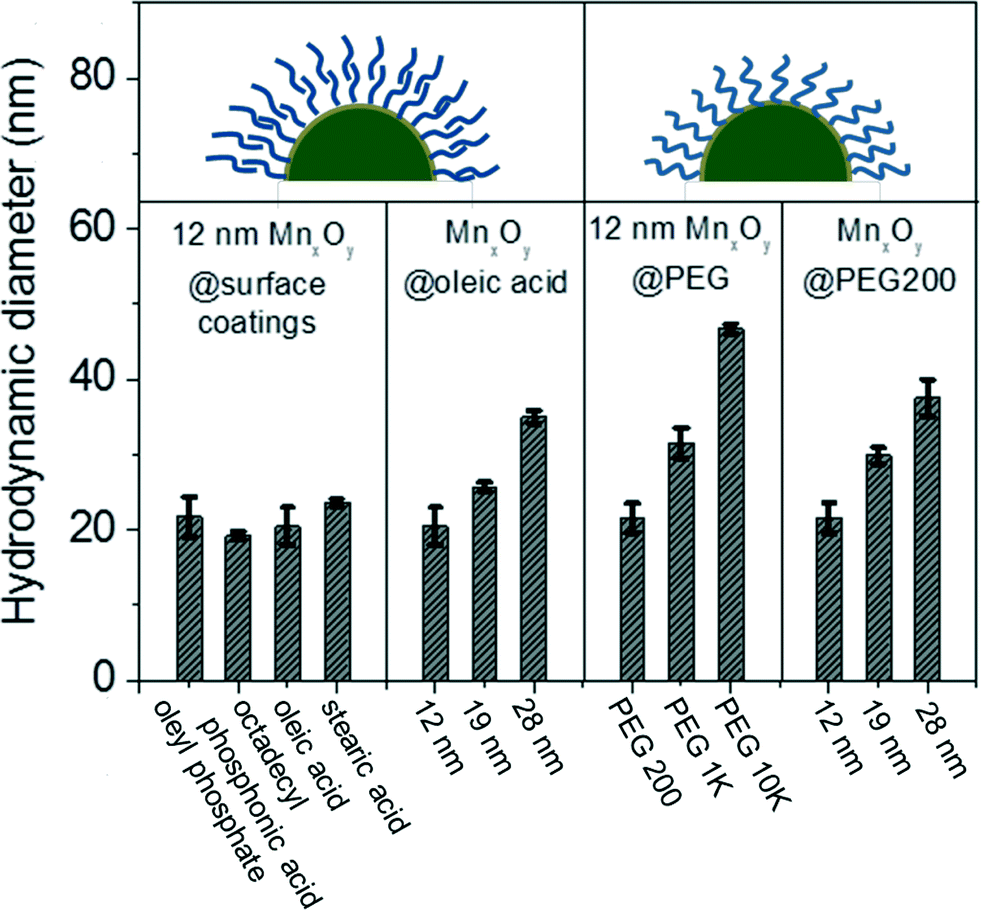 | ||
| Fig. 4 Hydrodynamic diameters of manganese oxide (MnxOy) nanocrystals from the inorganic core (12, 19, and 28 nm) as well as the surface coating (oleyl phosphate, octadecyl phosphonic acid, oleic acid, stearic acid, and poly ethylene glycol). | ||
Once transferred, particle aggregation kinetics were evaluated as a function of environmentally relevant ionic strength and type. Hydrodynamic diameters were monitored by DLS in situ and in real time to determine the critical coagulation concentrations (CCC) of both mono- and di-valent salts (Na+ and Ca2+) for all materials described. As shown in Fig. 5, unsaturated carbon (C9![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C10 double bond) surface coating materials containing phosphonic acid and carboxylic acid bilayered nanocrystals were observed to have higher critical coagulation concentration values compared to both saturated carbon chain analog, bilayered structures and single layered coatings. CCCs for oleyl phosphate- (883.7 mM of NaCl and 19.1 mM of CaCl2) and oleic acid- (810.1 mM of NaCl and 19.2 mM of CaCl2) bilayered nanocrystals were higher than octadecyl phosphonic acid coated (201.5 mM of NaCl and 2.7 mM of CaCl2), stearic acid coated (245.8 mM of NaCl and 5.8 mM of CaCl2), and pegylated nanocrystals (261.4 mM of NaCl and 6.8 mM of CaCl2) (Fig. S8 in the ESI†).
C10 double bond) surface coating materials containing phosphonic acid and carboxylic acid bilayered nanocrystals were observed to have higher critical coagulation concentration values compared to both saturated carbon chain analog, bilayered structures and single layered coatings. CCCs for oleyl phosphate- (883.7 mM of NaCl and 19.1 mM of CaCl2) and oleic acid- (810.1 mM of NaCl and 19.2 mM of CaCl2) bilayered nanocrystals were higher than octadecyl phosphonic acid coated (201.5 mM of NaCl and 2.7 mM of CaCl2), stearic acid coated (245.8 mM of NaCl and 5.8 mM of CaCl2), and pegylated nanocrystals (261.4 mM of NaCl and 6.8 mM of CaCl2) (Fig. S8 in the ESI†).
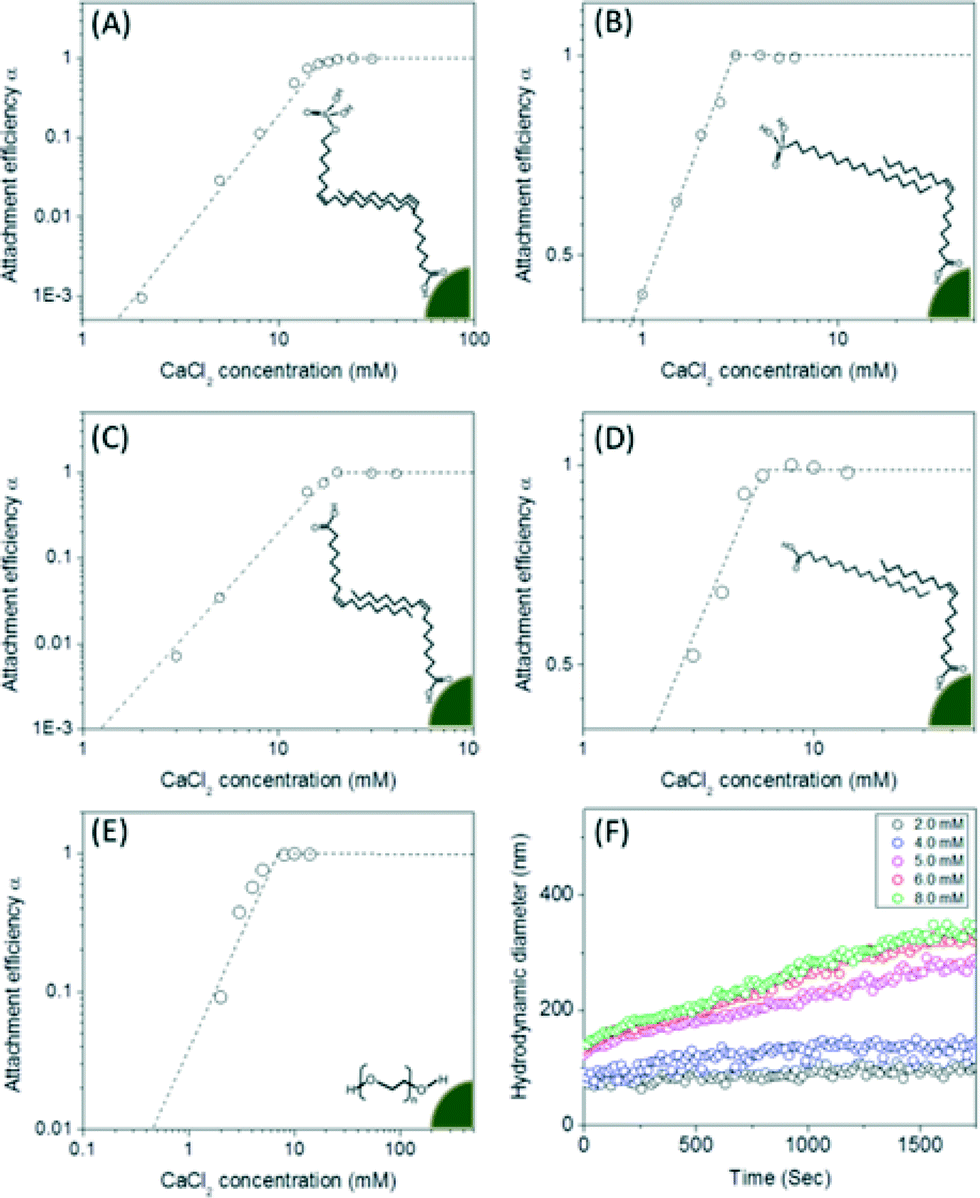 | ||
| Fig. 5 Attachment efficiency (α) as a function of CaCl2 concentrations for both bilayered- (A for oleyl phosphate coated-, B for octadecyl phosphonicacid coated-, C for oleic acid coated-, D for stearic acid coated-manganese oxides) and pegylated-manganese oxide nanocrystals (E) and the aggregation kinetic example of pegylated manganese oxide nanocrystals also as a function of CaCl2 concentration. The attachment efficiency (α) for each nanocrystal sample was calculated by measuring the aggregation rates of the particles as a function of time and salt concentrations (F). The nanocrystal concentration employed was 2.7 × 1014 nanocrystals per L. The depicted images showed the bilayered and single layered surface coating structures. The critical coagulation concentrations were 19.1 mM of CaCl2 for oleyl phosphate coated- (A), 2.7 mM of CaCl2 for octadecylphosphonic acid coated- (B), 19.2 mM of CaCl2 for oleic acid coated- (C), and 5.8 mM of CaCl2 for stearic acid coated- bilayered manganese oxide nanocrystals (D). The critical coagulation concentrations for PEG 200 coated manganese oxide nanocrystals was 6.8 mM of CaCl2 (E). | ||
Uranium sorption and separation using aqueous stabilized manganese oxide nanocrystals
Material libraries were next assessed and compared for their ability to adsorb and thus separate uranium in water. Uranyl sorption isotherms were performed to calculate a maximum adsorption capacity (qmax) and adsorption constant (k) based on the Langmuir isotherm equation, as described in the experimental section at 3 different pH values (pH 5.6, 7.0, and 8.5) for described particle types (size and surface coating combinations).44,45As shown in Fig. 6A, monodisperse nanocrystalline manganese oxides (12.2 nm diameter) demonstrated significantly higher sorption capacities when compared to aggregated commercial available manganese oxides by a factor of over 100 in some cases for oleyl phosphate coated manganese oxides (maximum adsorption capacity was 625.0 mg g−1 (milligram of uranium per gram of manganese)), compared to 5.7 mg g−1 for commercial MnO. Such enhancement is not only a function of more favorable surface chemistry, but also due to the high particle stabilities resulting in the larger effective surface areas when compared to the commercial materials, which due to high surface energies, readily aggregate in water (Fig. 6, S4 and Table S2 in the ESI†). For all surface coatings evaluated, higher sorption values were observed under slightly acidic conditions, here as pH 5.6, compared to neutral (7.0) or slightly basic (pH 8.5) conditions (Fig. 6 and Table S1 in the ESI†), which is an expected trend.44 For example, a maximum uranium adsorption capacity of oleic acid bilayered manganese oxide nanocrystals was 454.55 mg g−1 at pH 5.6, 344.83 mg g−1 at pH 7.0, and 217.39 mg g−1 at pH 8.5 (Fig. 6B). Such higher uranium sorption at slightly acidic condition is hypothesized to be a result of aqueous uranium(VI) speciation (<pH 6, forming UO22+ or UO2(OH)+) which should have a higher electrostatic affinity for negatively charged surfaces, as reported by others.46
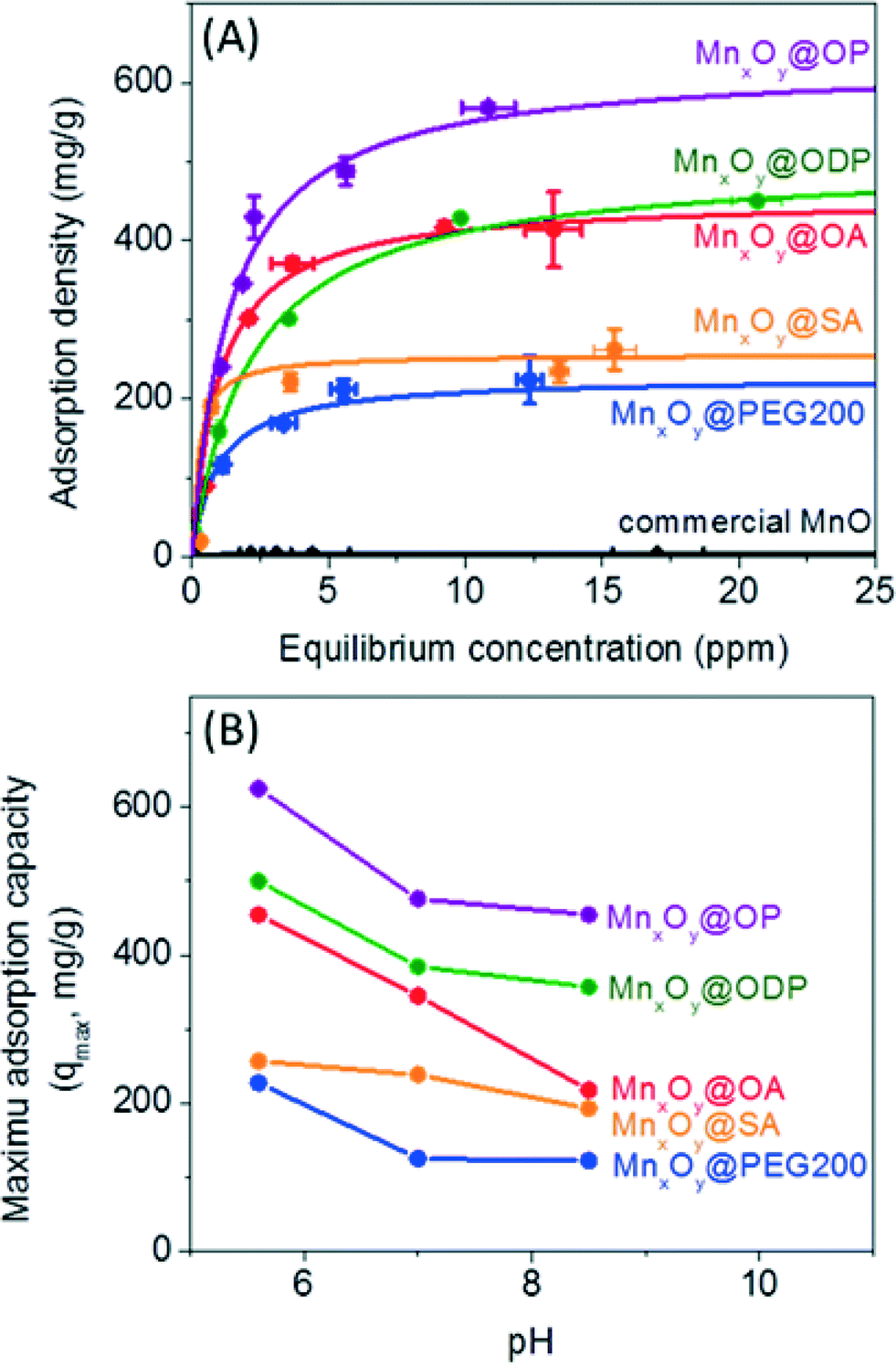 | ||
| Fig. 6 Uranium sorption isotherm on both engineered and commercial manganese oxides. (A) Sorption studies were performed on engineered nanocrystals (oleylphosphate coated- (OP, purple), octadecylphosphonic acid coated- (ODP, green), oleic acid coated- (OA, red), stearic acid coated- (SA, orange), and poly (ethylene glycol) 200 coated- (PEG 200, blue); 0.2 mg of manganese(II)) and commercial MnO (black, 6.7 mg of manganese(II)) at pH 5.6. For these materials, a maximum sorption capacity (qmax) was 47.6, 41.7, 32.3, 18.9, and 16.7 percentage by weight (wt%) per loaded sample (manganese oxide) for oleyl phosphate, octadecylphosphonic acid coated-, oleic acid coated-, stearic acid coated-bilayered manganese oxide nanocrystals, and PEG 200 coated manganese oxide nanocrystals, respectively; commercial manganese oxides showed 0.4 wt%. All solid lines were plotted by Langmuir isotherm equation using the experimental data. (B) Maximum uranium sorption capacities (qmax) for all materials (A) at three different pH (5.6, 7.0, and 8.5). | ||
When normalized for pH and surface coating chemistry effects, both the particle diameter and the surface coating thickness were also observed to play a significant role in uranyl binding capacities. Smaller manganese oxide nanocrystals with thinner, compact surface coating structures demonstrated higher uranium sorption capacities. Fig. S5 in the ESI† shows the particle diameter and surface coating thickness dependent uranium sorption using particle diameters of 12, 19, and 28 nm coated with 3 different surface coating materials (PEG 200, PEG 1k, and PEG 10k). At the same mass concentration, 12 nm manganese oxides (surface area: 2.5 × 1016 nm2), as expected, had the largest uranium sorption capacity due to higher surface area than 19 nm (surface area: 1.6 × 1016 nm2) and 28 nm (surface area: 1.0 × 1016 nm2) materials; at 20 mg L−1 of manganese(II); there are 5.4 × 1013 nanocrystals for 12 nm, 1.4 × 1013 nanocrystals for 19 nm, and 4.3 × 1012 nanocrystals for 28 nm particles. Further, sorption capacity was observed to decrease as a function of surface coatings molecular weight, which scaled well with coating thickness (Fig. 4, S5 and S6 in the ESI†). As shown in Fig. S5 in the ESI,† PEG 200 coated manganese oxide nanocrystals (DH = 21.5 nm) demonstrated the highest maximum sorption capacity of 227.3 mg g−1 compared to 142.9 mg g−1 for PEG 1k and 119.0 mg g−1 for PEG 10k coated manganese oxides. Thinner surface coating materials may allow for more uranyl interfacial surface reactions/binding occurrences.
The surface coating structure and chemistry were observed to play a significant role in sorption. As shown in Fig. 6, bilayered manganese oxide nanocrystals with phosphonate and carboxylate based surface functional groups performed better than single layered PEGylated (HO-PEG-OH) manganese oxide nanocrystals. In one case, a maximum uranium sorption capacity for bilayered nanocrystals (e.g. oleyl phosphate or oleic acid coated manganese oxides) was nearly 3 times higher than the value for the single layered structure, PEG 200 coated materials (Fig. 6, Tables S1 and S2 in the ESI†) for the same core particle at the same pH. High uranium sorption capacities for the bilayered structures is due, in part, to the strong binding of uranium at the functional groups (potentially at either end of the bilayer structure) and the enhanced colloidal stability (the colloidal stability effects are discussed in the next section). For a better understanding of the interaction between uranium and the bilayered stabilized materials, we investigated uranium binding using ATR-FTIR and XPS. As shown in Fig. S7 in the ESI,† upon uranyl sorption, a decrease in intensity and a peak shift at 1226 cm−1 and 1055 cm−1 can be attributed to strong interactions of uranium with phosphonate and carboxyl groups, respectively, as noted by others.47–50 Based on this and in line with literature reports of U affinity for these functional groups under similar water chemistries, we postulate that the binding affinity of the phosphonate group (PO(OH)2) (or carboxyl group (COOH)) to uranium occurs, in part, via electrostatic based mechanism(s) similar to previous reports focused on uranium-ligand binding phenomena.14,15,20,47,50–54
To further evaluate and delineate the mechanism of uranium binding, system redox chemistry was studied using XPS. Redox states for both sorbed uranium and manganese, at the particle surface, were evaluated using oleyl phosphate coated manganese oxide nanocrystals (which demonstrated the highest binding capacity of all material combination studied) before and after uranium sorption. The element’s corresponding oxidation state ratios (uranium (U4f) and manganese (Mn2p)) were calculated by integrating the area under the curves which were fitted by MultiPak V7.0.1 (Table S3 and S4†).55,56 As shown in Fig. 7, 55% of associated uranium(VI) was reduced to uranium(IV) with a corresponding change in manganese oxidation state (ca. 6% decrease in surface located manganese(II) with consequent increase in surface located manganese(IV)) (Table S3 and S4†). Such Mn–U oxidation–reduction coupling likely results, in part, from redox reactions between available uranium species and surface active sites on the nanocrystal surface itself. A complete mechanistic description, including uranyl localization, is currently being delineated via synchrotron based X-ray spectroscopy, via XAFS/XANES analyses, which is beyond the scope of this initial report.
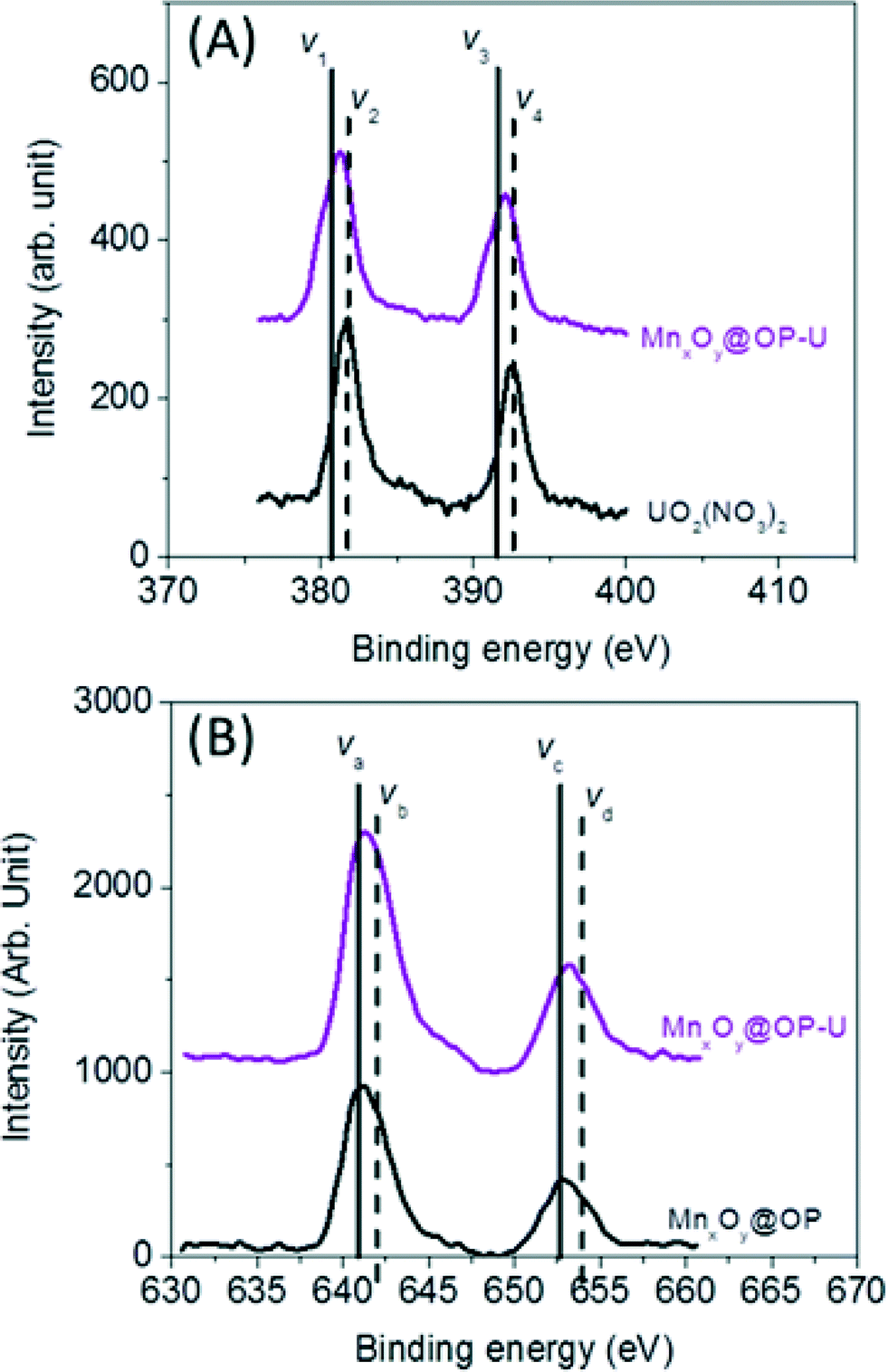 | ||
| Fig. 7 Redox reaction between uranium (U) and manganese (Mn) on the surface of the nanocrystals. (A) The ratio of U(IV) to U(VI) was calculated by (V1 + V3)/(V2 + V4) in the uranium 4f XPS data from the sample before and after the uranium sorption measurement (Table S3†). (B) Manganese 2P XPS data was used to evaluate the ratio of Mn(II)/Mn(III) calculated by (Va + Vc)/(Vb + Vd) from the sample before and after the uranium sorption measurement (Table S4†). | ||
Colloidal stability of single and bilayered nanocrystalline manganese oxides in the presence of mono-/di-valent salt ions
Both oleyl phosphate coated- and oleic acid coated- bilayered structures outperformed the uranium sorption capacities of PEG based coated materials and commercial manganese oxide nanocrystals. Due to relatively large amounts of sorbed uranyl, which could shield functional group interaction(s) with water, we further evaluated the stability of these systems in the presence of elevated uranyl concentrations. Fig. 8 shows that the hydrodynamic diameters of oleyl phosphate and oleic acid bilayered manganese oxide nanocrystals were stable despite increasing uranyl concentrations (from 0.1 to 60 mg L−1) after 24 h. However, octadecylphosphonic acid, stearic acid, and PEG coated manganese oxides showed significant increases in their hydrodynamic diameters (thus losing monomeric stability) as a function of U concentration. Such data are consistent with observed CCC trends for (Na+) and divalent (Ca2+) cations (Fig. 8 and S8 in the ESI†). As discussed for Fig. 5, both oleyl phosphate coated- and oleic acid coated- nanocrystals (bilayered structures) were extremely stable with extended critical coagulation concentration values in the presence of high concentrations of sodium (<880 mM) and calcium (<19 mM) ions; however octadecyl phosphonic acid-, stearic acid-, and PEG 200- coated nanocrystals aggregated at lower critical coagulation concentrations values for sodium (<260 mM) and calcium (<6 mM).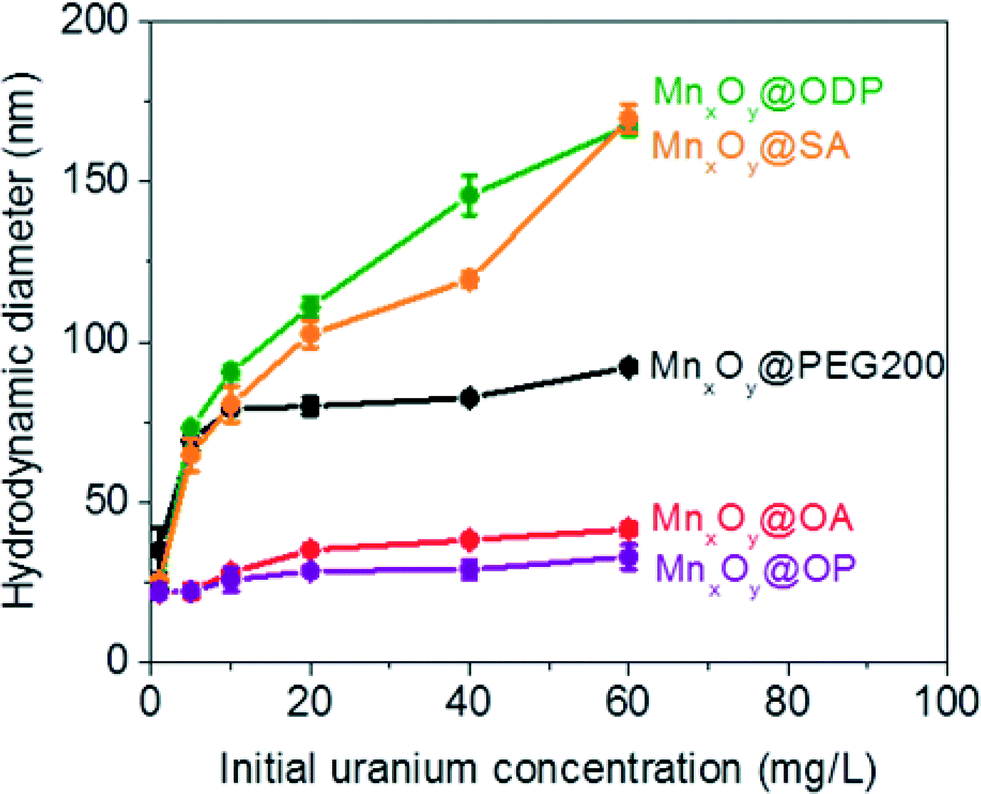 | ||
| Fig. 8 Hydrodynamic diameters of both bilayered- (oleyl phosphate (purple), octadecylphosphonic acid (green), stearic acid (orange), and oleic acid (red) coated) and pegylated- (black) manganese oxide nanocrystals after uranium sorption measurements ranging from 0.1 to 60 mg L−1 of uranium(VI) at pH 5.6 for 24 h. | ||
The enhancement of colloidal stability for the oleyl phosphate and oleic acid bilayered nanocrystals has been reported to result from the increased hydrophobic (sp2 pi bonding environments) interactions between unsaturated carbon chains, resulting in more stable, compact bilayers, by Prakash et al. for oleic acid bilayer stabilized metal oxide nanoparticles.32 It is hypothesized that the kink structure of unsaturated carbon chains (cis forms in both cases here) provides additional layer support via increased Van der Waals interactions (compared to octadecyl phosphonic acid or stearic acid, which are saturated).32 The lower sorption capacities of octadecyl phosphonic acid- and stearic acid- coated bilayered manganese oxide nanocrystals may also be due, in part, to the lower stability in the presence of elevated uranyl (UO22+ or UO2(OH)+) which would decrease the effective surface areas (Fig. 6–8). Highly stable oleyl phosphate and oleic acid coated bilayered structures demonstrated nearly 1.5 times higher uranium sorption capacities than octadecyl phosphonic acid and stearic acid coated bilayered structure samples, respectively.
Conclusions
For the first time, libraries of engineered monodisperse manganese oxide nanocrystals have been systematically synthesized, stabilized in water, characterized and demonstrated as a platform material for advanced uranium, as uranyl, sorption applications. As a function of particle size, surface (layered) structure, and chemistry, these materials are capable of ultra-high uranyl sorption capacities (>50% wt U/wt Mn, with partial U reduction from VI to IV observed) with simultaneous high aqueous stabilities (at environmentally relevant ionic strengths), when optimized via bilayer coating(s) with second (outer-facing) layers consisting of either carboxylic acid or phosphonic acid head groups. Fundamental understanding of the U localization at/in the nanocrystal structures/interfaces as well as a complete system redox evolution are further needed, and on-going, to achieve the next iteration in Mn-based particle design for optimized uranyl sorption and separation application platforms.Acknowledgements
This work is supported by U.S. Army Corps of Engineers (W912HZ-13-2-0009-P00001) and ACS Petroleum Research Fund (52640-DNI10). XRD measurements were made possible by a grant from the U.S. National Science Foundation (EAR-1161543). TEM, DLS, Ultracentrifugation, ICP-OES, and ICP-MS were provided by the Nano Research Facility (NRF) at Washington University in St. Louis, a member of the National Nanotechnology Infrastructure Network (NNIN), which is supported by the National Science Foundation (ECS-0335765). We would like to acknowledge Daniel E. Giammar in his assistance and helpful discussion of uranium adsorption measurement.Notes and references
- P. J. J. Alvarez, V. L. Colvin, J. Lead and V. Stone, ACS Nano, 2009, 3, 1616–1619 CrossRef CAS PubMed.
- R. Bojanowski, L. Radecki and K. Burns, J. Radioanal. Nucl. Chem., 2005, 264, 437–443 CrossRef.
- Z. Chen, Z. Zhuang, Q. Cao, X. Pan, X. Guan and Z. Lin, ACS Appl. Mater. Interfaces, 2013, 6, 1301–1305 Search PubMed.
- A. M. Donia, A. A. Atia, E. M. M. Moussa, A. M. El-Sherif and M. O. A. El-Magied, Hydrometallurgy, 2009, 95, 183–189 CrossRef CAS PubMed.
- M. M. Khin, A. S. Nair, V. J. Babu, R. Murugan and S. Ramakrishna, Energy Environ. Sci., 2012, 5, 8075–8109 CAS.
- H. Q. Sun, G. L. Zhou, S. Z. Liu, H. M. Ang, M. O. Tade and S. B. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6235–6241 CAS.
- Y. S. Wu, F. F. Huang and Y. W. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 1503–1509 CAS.
- J. Yang, H. Zhang, M. Yu, I. Emmanuelawati, J. Zou, Z. Yuan and C. Yu, Adv. Funct. Mater., 2014, 24, 1354–1363 CrossRef CAS PubMed.
- R. A. Crane, M. Dickinson, I. C. Popescu and T. B. Scott, Water Res., 2011, 45, 2931–2942 CrossRef CAS PubMed.
- D. Das, M. K. Sureshkumar, S. Koley, N. Mithal and C. G. S. Pillai, J. Radioanal. Nucl. Chem., 2010, 285, 447–454 CrossRef CAS.
- J. N. Fiedor, W. D. Bostick, R. J. Jarabek and J. Farrell, Environ. Sci. Technol., 1998, 32, 1466–1473 CrossRef CAS.
- L. N. Moyes, R. H. Parkman, J. M. Charnock, D. J. Vaughan, F. R. Livens, C. R. Hughes and A. Braithwaite, Environ. Sci. Technol., 2000, 34, 1062–1068 CrossRef CAS.
- H. Zeng, A. Singh, S. Basak, K. U. Ulrich, M. Sahu, P. Biswas, J. G. Catalano and D. E. Giammar, Environ. Sci. Technol., 2009, 43, 1373–1378 CrossRef CAS.
- Y. F. Yue, R. T. Mayes, J. Kim, P. F. Fulvio, X. G. Sun, C. Tsouris, J. H. Chen, S. Brown and S. Dai, Angew. Chem., Int. Ed., 2013, 52, 13458–13462 CrossRef CAS PubMed.
- M. J. Manos and M. G. Kanatzidis, J. Am. Chem. Soc., 2012, 134, 16441–16446 CrossRef CAS PubMed.
- J. H. Jang, B. A. Dempsey and W. D. Burgos, Water Res., 2008, 42, 2269–2277 CrossRef CAS PubMed.
- D. Renock, M. Mueller, K. Yuan, R. C. Ewing and U. Becker, Geochim. Cosmochim. Acta, 2013, 118, 56–71 CrossRef CAS PubMed.
- D. M. Singer, S. M. Chatman, E. S. Ilton, K. M. Rosso, J. F. Banfield and G. A. Waychunas, Environ. Sci. Technol., 2012, 46, 3821–3830 CrossRef CAS PubMed.
- Z. Wang, S.-W. Lee, J. G. Catalano, J. S. Lezama-Pacheco, J. R. Bargar, B. M. Tebo and D. E. Giammar, Environ. Sci. Technol., 2012, 47, 850–858 CrossRef PubMed.
- Z. Wang, W. Xiong, B. M. Tebo and D. E. Giammar, Environ. Sci. Technol., 2013, 48, 289–298 CrossRef PubMed.
- K. An, M. Park, J. H. Yu, H. B. Na, N. Lee, J. Park, S. H. Choi, I. C. Song, W. K. Moon and T. Hyeon, Eur. J. Inorg. Chem., 2012, 2148–2155 CrossRef CAS PubMed.
- Y. Q. Chang, X. Y. Xu, X. H. Luo, C. P. Chen and D. P. Yu, J. Cryst. Growth, 2004, 264, 232–236 CrossRef CAS PubMed.
- Y. Q. Chang, D. P. Yu, Y. Long, J. Xu, X. H. Luo and R. C. Ye, J. Cryst. Growth, 2005, 279, 88–92 CrossRef CAS PubMed.
- G. H. Lee, S. H. Huh, J. W. Jeong, B. J. Choi, S. H. Kim and H.-C. Ri, J. Am. Chem. Soc., 2002, 124, 12094–12095 CrossRef CAS PubMed.
- M. M. Najafpour, F. Rahimi, M. Amini, S. Nayeri and M. Bagherzadeh, Dalton Trans., 2012, 41, 11026–11031 RSC.
- J. Park, E. Kang, C. J. Bae, J.-G. Park, H.-J. Noh, J.-Y. Kim, J.-H. Park, H. M. Park and T. Hyeon, J. Phys. Chem. B, 2004, 108, 13594–13598 CrossRef CAS.
- J. Rockenberger, E. C. Scher and A. P. Alivisatos, J. Am. Chem. Soc., 1999, 121, 11595–11596 CrossRef CAS.
- G. Salazar-Alvarez, J. Sort, S. Surinach, M. D. Baro and J. Nogues, J. Am. Chem. Soc., 2007, 129, 9102–9108 CrossRef CAS PubMed.
- W. S. Seo, H. H. Jo, K. Lee, B. Kim, S. J. Oh and J. T. Park, Angew. Chem., 2004, 116, 1135–1137 CrossRef PubMed.
- M. Yin and S. O'Brien, J. Am. Chem. Soc., 2003, 125, 10180–10181 CrossRef CAS PubMed.
- D. N. Benoit, H. G. Zhu, M. H. Lilierose, R. A. Verm, N. Ali, A. N. Morrison, J. D. Fortner, C. Ayendano and V. L. Colvin, Anal. Chem., 2012, 84, 9238–9245 CAS.
- A. Prakash, H. G. Zhu, C. J. Jones, D. N. Benoit, A. Z. Ellsworth, E. L. Bryant and V. L. Colvin, ACS Nano, 2009, 3, 2139–2146 CrossRef CAS PubMed.
- E. Vigneau, C. Loisel, M. F. Devaux and P. Cantoni, Powder Technol., 2000, 107, 243–250 CrossRef CAS.
- K. L. Chen and M. Elimelech, Langmuir, 2006, 22, 10994–11001 CrossRef CAS PubMed.
- K. L. Chen, B. A. Smith, W. P. Ball and D. H. Fairbrother, Environ. Chem., 2010, 7, 10–27 CrossRef CAS.
- J. D. Hu, Y. Zevi, X. M. Kou, J. Xiao, X. J. Wang and Y. Jin, Sci. Total Environ., 2010, 408, 3477–3489 CrossRef CAS PubMed.
- E. Illes and E. Tombacz, J. Colloid Interface Sci., 2006, 295, 115–123 CrossRef CAS PubMed.
- D. Jassby and M. Wiesner, Langmuir, 2011, 27, 902–908 CrossRef CAS.
- J. Park, K. J. An, Y. S. Hwang, J. G. Park, H. J. Noh, J. Y. Kim, J. H. Park, N. M. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- S. S. Lee, H. G. Zhu, E. Q. Contreras, A. Prakash, H. L. Puppala and V. L. Colvin, Chem. Mater., 2012, 24, 424–432 CrossRef CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS PubMed.
- S. Ahmed and K. M. Ryan, Chem. Commun., 2009, 6421–6423 RSC.
- S. S. Lee, W. S. Song, M. J. Cho, H. L. Puppala, P. Nguyen, H. G. Zhu, L. Segatori and V. L. Colvin, ACS Nano, 2013, 7, 9693–9703 CrossRef CAS PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- C. J. Werth and M. Reinhard, Environ. Sci. Technol., 1997, 31, 689–696 CrossRef CAS.
- M. Sutton and S. R. Burastero, Chem. Res. Toxicol., 2004, 17, 1468–1480 CrossRef CAS PubMed.
- S. K. Kazy, S. F. D'Souza and P. Sar, J. Hazard. Mater., 2009, 163, 65–72 CrossRef CAS PubMed.
- D. P. J. Pan, A. Senpan, S. D. Caruthers, T. A. Williams, M. J. Scott, P. J. Gaffney, S. A. Wickline and G. M. Lanza, Chem. Commun., 2009, 3234–3236 RSC.
- H. L. Si, H. Z. Wang, H. B. Shen, C. H. Zhou, S. Li, S. Y. Lou, W. W. Xu, Z. L. Du and L. S. Li, CrystEngComm, 2009, 11, 1128–1132 RSC.
- L. Wang, Z. M. Yang, J. H. Gao, K. M. Xu, H. W. Gu, B. Zhang, X. X. Zhang and B. Xu, J. Am. Chem. Soc., 2006, 128, 13358–13359 CrossRef CAS PubMed.
- S. Bachmaf, B. Planer-Friedrich and B. J. Merkel, Geochim. Cosmochim. Acta, 2009, 73, A67 Search PubMed.
- A. Barkleit, H. Foerstendorf, B. Li, A. Rossberg, H. Moll and G. Bernhard, Dalton Trans., 2011, 40, 9868–9876 RSC.
- M. Carboni, C. W. Abney, S. B. Liu and W. B. Lin, Chem. Sci., 2013, 4, 2396–2402 RSC.
- M. R. Romero-Gonzalez, T. Cheng, M. O. Barnett and E. E. Roden, Radiochim. Acta, 2007, 95, 251–259 CrossRef CAS.
- J. W. D. Ng, M. Tang and T. F. Jaramillo, Energy Environ. Sci., 2014, 7, 2017–2024 CAS.
- K. Ramesh, L. Chen, F. Chen, Y. Liu, Z. Wang and Y.-F. Han, Catal. Today, 2008, 131, 477–482 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting information includes detailed descriptions for control over the diameter of manganese oxide nanocrystals, the histograms of the diameter distribution of manganese oxide nanocrystals, HR-TEM image of manganese oxide, uranium sorption isotherm on commercial samples (Fe3O4 and MnO), size and surface coating thickness dependent uranium sorption isotherm on engineered PEGylated manganese oxide nanocrystals, size dependent uranium sorption isotherm on oleic acid bilayered manganese oxide nanocrystals, FT-IR measurement of engineered nanocrystals before and after uranium sorption, attachment efficiencies of both bilayered- and pegylated-manganese oxide nanocrystals, two tables of the maximum uranium sorption capacity and the sorption constant for the materials used in the research, two tables of XPS binding energies of individual peaks of the U4f and Mn2P spectra, and a table of hydrodynamic diameters of the nanocrystal samples designed in the research. See DOI: 10.1039/c5en00010f |
| This journal is © The Royal Society of Chemistry 2015 |