
Impact of Pahokee Peat humic acid and buffer identity on goethite aggregation and reactivity†
Amanda M.
Vindedahl
a,
William A.
Arnold
*b and
R.
Lee Penn
*a
aDepartment of Chemistry, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455-0431, USA. E-mail: rleepenn@umn.edu
bDepartment of Civil, Environmental, and Geo- Engineering, University of Minnesota, 500 Pillsbury Dr. SE, Minneapolis, MN 55455-0116, USA. E-mail: arnol032@umn.edu
First published on 25th August 2015
Abstract
Natural organic matter (NOM) has been shown to strongly influence both reactions occurring at the solid–liquid interface and particle aggregation. Ferrous iron bound to iron oxides, such as goethite, can serve as a reductant towards environmental contaminants. Little is known, however, about how NOM affects reactivity and aggregation of iron oxide nanoparticles in aqueous conditions. Here, the rate of 4-chloronitrobenzene (4-ClNB) reduction is used to assess the reactivity of Fe(II) on goethite, and this reactivity is tracked as a function of increasing NOM concentration, using either 3-(N-morpholino)propanesulfonic acid (MOPS) or bicarbonate buffer at pH 7. Pahokee Peat humic acid (PPHA), extracted from an agricultural peat soil, was selected to simulate the NOM present in groundwater. Generally speaking, the 4-ClNB degradation rate decreases with increasing PPHA concentration. Characterization of the goethite nanoparticles after repeated cycles of 4-ClNB degradation demonstrated that oxidative crystal growth occurred mainly along the goethite c-axis, reaction rates progressively slowed with each cycle, and no new phases precipitated. Finally, bicarbonate buffer, a model for groundwater, dramatically affects both aggregation and reaction rates, with reaction rates an order of magnitude slower and aggregates substantially larger than observed in suspensions prepared using MOPS.
Nano impactNatural organic matter (NOM) is ubiquitous in natural waters, and these complicated and heterogeneous molecules are known to interact with mineral surfaces. These molecules are important players in nanoparticle and contaminant fate and transport. The influence of Pahokee Peat humic acid on goethite nanoparticle reactivity and aggregation in two different buffer systems is investigated. These data provide a novel reference for goethite behavior during reduction reactions in the presence of a representative natural organic matter in environmentally relevant systems. |
Introduction
Some highly oxidized contaminants, such as pesticides and industrial chemicals, are transformed to less harmful chemicals by reductive pathways. Two forms of ferrous iron, structural iron within a mineral and surface-bound Fe(II), serve as effective reductants towards a broad range of such highly oxidized contaminants. Reductive degradation reactions involving the oxidation of surface-bound Fe(II), which binds due to complexation of Fe(II) with mineral surface hydroxyl moieties, are more rapid than with dissolved Fe(II).1–10 Commonly studied contaminants include metal cations,11–13 halogenated organic compounds,1–6,14–16 and nitroaromatic compounds.17–22The reactivity of mineral bound, redox active species is strongly sensitive to solution conditions. The presence and concentration of Fe(II),2,5,23 suspension pH,2,5,24,25 buffer identity,26 and extent of adsorption of other species1,7,8,23,26–28 impact the reactivity of mineral bound Fe(II). In addition, the physical and chemical properties (e.g. mineral identity29,30 and size31,32) of the mineral play important roles in reactivity. Recently, reduced reactivity has been directly linked to mineral particle aggregation. For example, work by Vikesland et al. demonstrated that aggregation of magnetite particles increased with increasing ionic strength, which was accompanied by decreased rates of reductive degradation of carbon tetrachloride.31 They hypothesized that the total reactive surface area decreased upon aggregation, causing the rates of degradation to slow.31 The detailed arrangement of crystallites in an aggregate is also an important consideration. Aggregation that involves direct contact between the most (or least) reactive surfaces could result in substantial loss (or no change) in overall reactivity. Furthermore, reactivity likely drops substantially with increasing density of aggregates, because transport of reactive species to crystal surfaces residing at the interior of the aggregate would be inhibited.
Natural organic matter (NOM) consists of macromolecules formed from the decomposition of biomaterial, and the amount present within an environmental system varies from one system to the next, as well as spatially or temporally within a single system. NOM has been shown to play complex roles in the reductive degradation of environmental contaminants in aqueous systems. The presence of NOM may dramatically increase rates of contaminant degradation by serving as an electron transfer mediator because it undergoes oxidation-reduction cycles.33 In contrast, the presence of NOM may dramatically decrease reaction rates by several mechanisms, such as by adsorbing onto the surface of the iron-bearing mineral34–38 and blocking surface sites from taking up Fe(II).23 In addition, rates decrease when NOM prevents interaction between the contaminant and the adsorbed Fe(II) or when NOM oxidizes or complexes either aqueous or adsorbed Fe(II).23
Recent work has demonstrated that NOM causes changes in iron oxide nanoparticle aggregation39,40 as well as aggregation of engineered nanoparticles.41–46 For example, the addition of Suwannee River fulvic acid to aqueous suspensions of titanium dioxide nanoparticles (pH 8) decreased aggregate size.47 Furthermore, the impact of NOM on aggregation state depends on concentration. At low concentrations (ca. 2–10 mg L−1) of Suwannee River humic acid, large aggregates of titanium dioxide46 and iron oxide39 were observed, but disaggregation was observed at high concentrations. From a mechanistic point of view, NOM could result in increased reaction rates by increasing accessible surface area or by serving as an electron shuttle between surface-bound species and dissolved species or could result in decreased reaction rates by blocking reactive surface sites or decreasing accessible surface area.
Here, we present results tracking goethite aggregation and rates of 4-chloronitrobenzene (4-ClNB) reduction by surface-bound Fe(II) as a function of the concentration of Pahokee Peat humic acid (PPHA) in aqueous suspension at circumneutral pH. PPHA was selected because it was collected from an agricultural peat soil and thus serves as a surrogate for the NOM present in groundwater. For facile comparison to previous work,48 synthetic goethite nanoparticles and 4-ClNB were selected as the model iron oxide and contaminant species, respectively. Goethite (α-FeOOH) is a highly abundant iron oxide due to its high degree of thermal stability.49 4-ClNB was selected as a model contaminant due to its previous use as a probe compound19,48,50–54 and because it represents relevant classes of environmental contaminants (explosives and pesticides).55 Evolving goethite nanoparticle reactivity was also quantified via sequential degradation reactions of 4-ClNB.
Recent studies have demonstrated that buffers cause a release of Fe(II) from the mineral surface and alter the aggregation state of iron oxide nanoparticles.26,48 Good's buffers are hypothesized to complex with surface Fe(II) via the six-membered ring.26,56 Thus, reactivity was quantified in batch reactors prepared using either 3-(N-morpholino)propanesulfonic acid (MOPS), a Good's buffer widely employed in studies involving Fe(II)-iron oxide systems,48 or sodium bicarbonate (NaHCO3) to better simulate groundwater conditions.
Finally, a major goal is to directly link aggregation state with observed reaction kinetics. Goethite aggregation state was characterized using two primary methods: dynamic light scattering (DLS) and cryogenic-transmission electron microscopy (cryo-TEM). DLS measures the intensity of scattered light from suspended objects, enabling dynamic assessment of aggregation state. Cryo-TEM enables the direct imaging of objects suspended in solution, which are collected at any point during a reaction and thus serve as snapshots of the aggregation state at any time. DLS is semi-quantitative with a short data collection time and has numerous and well documented limitations.57,58 Cryo-TEM is quantitative in the sense that objects are measured from calibrated images, but it has the problem of very limited sampling. Thus, we have employed both DLS and cryo-TEM as complementary methods to characterize the evolving aggregation state before, during, and after reaction.
Experimental
Nanoparticle synthesis
Goethite nanoparticles were synthesized according to Anschutz and Penn.59 Post-synthesis, the particles were left in suspension with Milli-Q water, adjusted to pH 4 with 4 M HNO3 (Mallinkrodt Chemicals, 68-70%, 0.41 mM HNO3) to inhibit particle aggregation, and left under continuous stirring. The amount of goethite nanoparticles by mass per unit volume was determined by drying different volumes of suspension and determining the mass of each sample until constant mass was maintained for 24 hours. Average goethite particle dimensions of 89 ± 35 nm by 10 ± 4 nm were measured via transmission electron microscopy (TEM) because nanoparticle aggregation effects were not of concern in this sample. A surface area of 117 m2 g−1 was measured via nitrogen adsorption and desorption (affected by nanoparticle aggregation effects) and 262 m2 g−1 was calculated using TEM measurements (based on single crystal dimensions). X-Ray diffraction (XRD) results, in combination with Rietveld Refinement analysis, demonstrate the material is 99.7% w/w goethite and 0.3% w/w hematite (ESI;† Fig. S1).Batch 4-ClNB reactors
Batch reactors were prepared in duplicate in an anaerobic chamber (Coy Laboratory Products, 5% H2/95% N2), using 121 mL serum bottles, which were sealed with PTFE-lined rubber septa. The reactors contained 39 mg of goethite nanoparticles (mass loading of 0.325 g L−1, surface area of 38.0 m2 L−1) and 25 mM MOPS (3-(N-morpholino)propanesulfonic acid, Sigma-Aldrich, ≥99.5%, pH 7, adjusted with 10 M NaOH) or 10 mM NaHCO3 buffer (Sigma-Aldrich, pH 7, adjusted with 1 M H2SO4 (final concentration 600 μM)). The MOPS buffer concentration was selected to yield maximum goethite reactivity48 while the NaHCO3 buffer concentration was selected to match the ionic strength of a well-characterized groundwater sample containing significant levels of dissolved iron.60 An acidified 1.0 M FeCl2 (Fisher Scientific, 101.0%) solution was prepared in the glove bag by adding 5 mL of a 1.1 M FeCl2 stock solution to 0.5 mL of a 1.0 M HCl (BDH Aristar, 36.5–38%) solution. The acidified solution was added to the batch reactors to yield a final concentration of 1.0 mM Fe(II) (to ensure excess). Pahokee Peat humic acid (PPHA, International Humic Substances Society, IHSS, 1S103H) stock solution was prepared by suspending the as-received dry sample in Milli-Q water. PPHA stock solution was added to the serum bottle to achieve organic carbon (OCPPHA, determined based on elemental composition reported by IHSS) concentrations ranging from 1 to 50 parts per million OC (representative of a wide range of natural water systems).23,61 The suspension was allowed to equilibrate for 21 hours while rotating on an IKA RO 15 rotator along its longitudinal axis (at 22 °C).A methanolic stock solution of 4-chloronitrobenzene (4-ClNB) was added via automatic pipette to the reactor to yield a final concentration of 100 μM, and buffer was then added so that no headspace was present in the reactor (pH values of the suspension were approximately 6.8 before and after 4-ClNB degradation). Samples were removed from the reactor at predetermined time intervals over the course of 50 minutes using a syringe and filtered using PALL Life Sciences Acrodisc® 13 mm Syringe Filters with a 0.2 μm nylon membrane. Thus, headspace was present within the reactor throughout the reaction. Septa were replaced between sampling times when NaHCO3 buffer was used to minimize CO2 loss into the glovebag atmosphere. The 4-ClNB concentration was quantified by an Agilent 1100 Series system equipped with an ultraviolet detector. Sample (20 μL) was injected onto a Zorbax SB-C18 column (4.6 mm × 150 mm, 5 μm). The 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 acetonitrile:ammonium acetate (1 g L−1) pH 7 mobile phase was operated at a flow rate of 0.7 mL min−1 for a total separation time of 7 minutes. The absorbance at 254 nm was monitored.
:30 acetonitrile:ammonium acetate (1 g L−1) pH 7 mobile phase was operated at a flow rate of 0.7 mL min−1 for a total separation time of 7 minutes. The absorbance at 254 nm was monitored.
Sequential spike reactors
Two sets of five identical batch reactors were prepared as above, with one set suspended in MOPS and the other set in NaHCO3 buffer, with 10 ppm OC PPHA. Samples for 4-ClNB monitoring were removed from the first reactor only. Upon complete degradation of the parent compound, a sample was analyzed by the ferrozine assay to determine the concentration of aqueous Fe(II). Solids from the first reactor were collected for analysis by TEM and XRD as detailed below.Based on the measured Fe(II) concentration, an aliquot of the Fe(II) stock solution was added to the remaining four reactors to yield an Fe(II) concentration of 1.0 mM Fe(II), and the reactors were allowed to equilibrate. Degradation reactions were reinitiated in the remaining reactors with the addition of 4-ClNB and the entire process was repeated, sacrificing one reactor per cycle. Solids were collected from each reactor sacrificed, yielding goethite nanoparticles that had been exposed to 1, 2, 3, 4, or 5 sequential spikes of Fe(II) and 4-ClNB.
Ferrozine assay
A 5 mg mL−1 ferrozine (3-(2-pyridyl)-5,6-diphenyl-1,2,4-triazine-p,p'-disulfonic acid disodium salt, Acros) stock solution was prepared by dissolving the solid in Milli-Q. A total of three samples were prepared with 0.400 mL of stock ferrozine, 3.400 mL of buffer, and 0.200 mL of filtered solution were combined in a polystyrene cuvette inside the anaerobic chamber. The absorbance of each assay solution was quantified at 562 nm by an Agilent 8453 UV-Visible spectrometer three times. Final Fe(II) concentrations were determined using a five point calibration curve plotted using the absorbance determined for standard solutions of known concentrations prepared the same day. Reported values were calculated by averaging the three average Fe(II) concentration from each assay sample.TOC analysis
Total organic carbon (TOC) was analyzed by a Shimadzu OCT-L 8-Port Sampler paired with a Shimadzu TOC-L Total Organic Carbon Analyzer. Supernatant from centrifuged suspensions containing goethite, Fe(II), and NOM were compared to solutions of NOM to calculate percent OC adsorbed onto goethite in each buffer. Samples were acidified to pH 2–3 with 1 M HCl, sparged with high purity air (80 mL min−1) for 1.5 minutes to remove inorganic carbon prior to analysis. Measurements were compared to a calibration curve comprised of potassium hydrogen phthalate (Sigma Aldrich, >99.95%) standards generated from a stock solution (1000 ppm OC).Nanoparticle characterization
Zeta potential and electrophoretic mobility were measured using the same instrument with the ZetaPALS Zeta Potential Analyzer option. The zeta potential probe was inserted into the sample-containing cuvette inside the anaerobic chamber and immediately measured in an attempt to prevent Fe(II) oxidation. Operational parameters consisted of 5 runs of 10 cycles applying the Smoluchowski zeta potential model. Water was selected as the solvent at a pH of 7.00 and 25 °C. The particles were modeled to be 100.0 nm in size and 0.325 mg mL−1 mass loading.
Results and discussion
Reactor preparation conditions
To ensure reproducible and comparable datasets, several experiments testing the effects of the experimental procedures were performed. Neither the order of Fe(II) and PPHA addition nor equilibration time between additions affected the goethite nanoparticle aggregate size or the rate at which 4-ClNB degraded. DLS-based goethite aggregate size and 4-ClNB degradation rate constant in 25 mM MOPS with varying equilibration times are presented in Table S1.† Both the average aggregate size and pseudo-first order rate constants of 4-ClNB degradation were statistically indistinguishable (see ESI† for t-test results). These data support the results of Colón et al.,23 where changing the order of Fe(II) and Suwannee River humic acid addition did not result in statistically significant differences in the initial rate of p-cyanonitrobenzene degradation by goethite nanoparticles suspended in 4-morpholineethanesulfonic acid (a Good's buffer).Variation in PPHA concentration
Generally speaking, the rate of 4-ClNB degradation decreased with increasing PPHA concentration. Fig. 1 shows the pseudo-first order rate constant for 4-ClNB degradation and the average DLS-based aggregate size versus total OC added as PPHA, and the variation in the DLS measurements is shown in Fig. S2.† Control experiments containing (i) only goethite, (ii) only Fe(II), (iii) goethite/Fe(II), (iv) Fe(II)/2 ppm PPHA, (v) Fe(II)/10 ppm PPHA, and (vi) Fe(II)/50 ppm PPHA were conducted (results detailed in Table S4†) with no observed 4-ClNB degradation except in case (iii). No detectable 4-ClNB degradation was observed in the reactors prepared without Fe(II) and goethite. Furthermore, there was no evidence of adsorption of 4-ClNB onto goethite, and XRD analysis showed no formation of siderite (data not shown). In all reactors, the pH decreased slightly after 21 hours of equilibration (pH = 6.8) and remained constant before and after 4-ClNB degradation.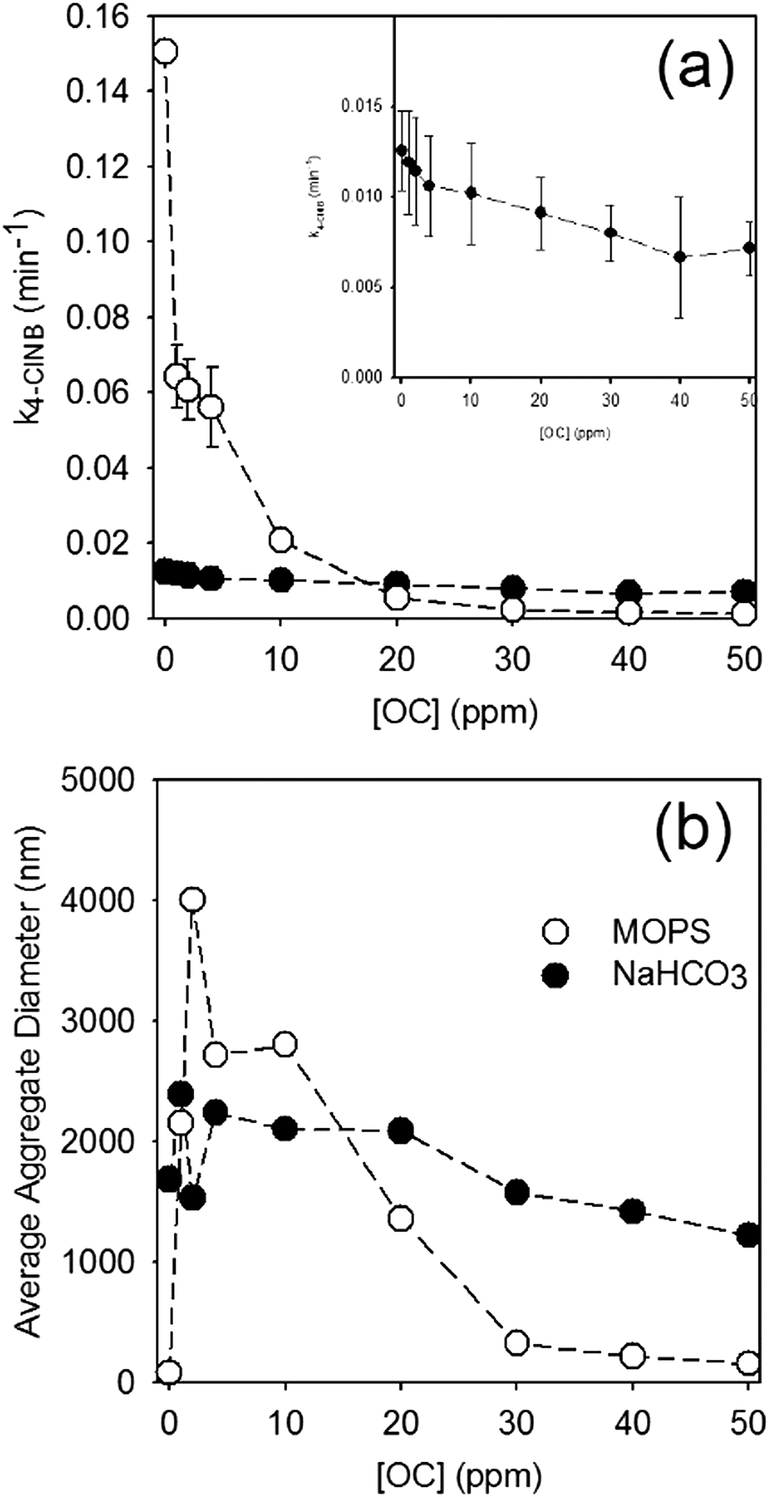 | ||
| Fig. 1 (a) Rate of 4-ClNB degradation (initial concentration 100 μM) and (b) average nanoparticle aggregate size for 1 mM Fe(II) and 0.325 g L−1 goethite suspended in 25 mM MOPS (open circles) and 10 mM NaHCO3 (closed circles) as a function of PPHA concentration, expressed as ppm organic carbon. In all plots, dashed lines are provided to help guide the eye. Inset in (a) displays an expanded view of the NaHCO3 data. All error bars in (a) represent the 95% confidence interval from regression of the rate constant. All DLS measurements are reported in the ESI,† Fig. S2. | ||
The use of bicarbonate to maintain pH dramatically affects both aggregation and reaction rates, with observed reaction rates an order of magnitude slower and aggregates substantially larger than observed in suspensions prepared using MOPS. Not only are the reaction rates substantially faster in reactors prepared using MOPS, they are also far more sensitive to PPHA concentration, with a dramatic decrease in rate even at the lowest concentration of PPHA. In NaHCO3, which more closely simulates natural aquatic systems, the reaction rates are comparatively slow, but the general trend of decreasing rate with increasing concentration of PPHA were still observed (p-value = 5.2 × 10−5).
In terms of aggregation state, addition of PPHA to goethite suspensions prepared with MOPS results in a dramatic increase in aggregation followed by a decrease in aggregation at concentrations higher than 10 ppm OC. At high OC concentrations, therefore, PPHA blocks reactive surface sites from reacting with 4-ClNB. It is difficult to determine, however, whether the decreased 4-ClNB degradation rate at concentrations lower than 10 ppm OC is due to site blocking, aggregation, or – most likely – both. In contrast, goethite suspensions prepared with NaHCO3 were highly aggregated and no statistically significant changes in aggregation state could be detected by DLS. The steady decrease in reactivity with increasing PPHA likely indicates that site blocking does play a role in the observed reduction in reactivity. Total organic carbon measurements (Table S5†) demonstrate that more PPHA adsorbed onto the goethite surface when the concentration was increased from 10 to 30 ppm OC in NaHCO3 (corresponding data was not obtainable in MOPS due to signal interference by buffer molecules). Zeta potential measurements, which provide a measure of nanoparticle surface charge, of goethite in aqueous solutions of Fe(II) prepared in NaHCO3 were much less positive than with MOPS (Table S5†), which explains why larger aggregates were observed in bicarbonate buffer. Similarly, the electrophoretic mobility of the particles in NaHCO3 is lower than in MOPS. Upon addition of PPHA, the zeta potential (and electrophoretic mobility) values in both buffers dropped substantially, to nearly the same value, which suggests more similar surface chemistries dominated by association of PPHA with the particles.
A potential cause of the loss in reactivity in bicarbonate buffer is that less Fe(II) adsorbs onto the surface. However, results using the ferrozine assay demonstrated that the amount of adsorbed Fe(II) in 10 mM NaHCO3 was comparable to the amount of adsorbed Fe(II) in 25 mM MOPS up to 20 ppm OCPPHA concentrations, with potentially lower adsorbed Fe(II) levels at 30–50 ppm OCPPHA (Fig. S3†). Samples containing only buffer, Fe(II), and NOM (open circles) indicate any loss of Fe(II) due to either complexation with NOM or oxidation to Fe(III) by NOM (Fig. S3†). Theoretical calculations of complexed Fe(II) by PPHA in a similar manner to Royer et al.63 were in good agreement with the values observed via the ferrozine assay. This suggests that ferrozine cannot extract Fe(II) from NOM complexes over the timescale of these measurements. The difference in Fe(II) concentration between these samples and those containing goethite (closed circles) are indicative of the amount of Fe(II) adsorbed onto the mineral surface. While some loss of Fe(II) is due to complexation with or oxidation by PPHA, these processes do not appear to hinder Fe(II) adsorption by goethite, for an excess of aqueous Fe(II) is available in all samples. Although total adsorbed Fe(II) may be similar between both buffer systems, it is hypothesized that in the NaHCO3 system, less Fe(II) is adsorbed on the most reactive goethite surface which yields an overall decrease in the observed 4-ClNB rate constants, although we cannot rule lower amounts of Fe(II) adsorption at high OC levels also playing a role.
Like MOPS,26 NaHCO3 can interact with the goethite surface, which could alter the availability of reactive sites by inhibiting Fe(II) adsorption. Carbonate species adsorb and form bidentate inner-sphere complexes with oxygen atoms on the goethite mineral surface (≡(FeO)2CO).64–68 The high surface charge of goethite suspended in MOPS (in the absence of PPHA) is consistent with the very small aggregate size detected by DLS, and lower surface charge for goethite suspended in NaHCO3 (in the absence of PPHA) is consistent with a higher degree of aggregation. We conclude that NaHCO3 significantly interacts with the mineral surface, causing the decrease in 4-ClNB degradation compared with MOPS in the absence of PPHA. This dramatic difference means that interpretation of data collected from simulations using MOPS, or other Good's buffers, may not be useful in interpreting what is happening in a natural water environment.
Cryo-TEM was used to obtain in situ images of goethite nanoparticle aggregates to reveal further details about aggregation state (Fig. 2). PPHA has low contrast in TEM, resulting in a cloudy appearance of nanoparticle aggregates in some of the images. The aggregates observed via cryo-TEM support both the average size and variances in size observed via DLS. For example, goethite aggregate sizes with 50 ppm OCPPHA in 25 mM MOPS were consistently small both in the cryo-TEM images and across every measurement by DLS. Conversely, in 10 mM NaHCO3, nanoparticle aggregates displayed a wide range of sizes, from a few 100 nm to a few 1000 nm in length, via both DLS and cryo-TEM measurements. Neither the presence of 4-ClNB nor its degradation altered the extent of nanoparticle aggregation. The average aggregate size was consistently large or small across pre- and post- 4-ClNB samples for the same buffer. Thus, the initial nanoparticle aggregation state remains constant throughout the degradation process and is unaffected by 4-ClNB degradation. This is in direct contrast to the results of previous work,48 where the average aggregate size increased significantly upon addition of 4-ClNB. This discrepancy may to be due to differences in nanoparticle storage after synthesis. Previous work involved the use of dried goethite nanoparticles, whereas never-dried nanoparticles were used in this study.
 | ||
| Fig. 2 Cryo-TEM images of 0.325 g L−1 goethite and 1 mM Fe(II) at 0 ppm (top row), 2 ppm (middle row), and 50 ppm (bottom row) OCPPHA in 25 mM MOPS (first two columns) and 10 mM NaHCO3 (right two columns) one hour before and after 4-ClNB addition (initial concentration 100 μM) as indicated within the figure. All scale bars represent 500 nm. | ||
Determination of reactive surface
Previous work examining the reductive degradation of 4-ClNB by Fe(II) on goethite in MOPS demonstrated that (021) goethite was the most reactive surface, yielding oxidative crystal growth along the c-axis, and that reaction rates decreased as a function of reaction progress.19 Oxygen atoms of the same coordination, however, are present on other mineral surfaces, albeit in lower concentrations.10 It is possible, therefore, that a different surface is the most reactive in the presence of NOM. This was tested in the sequential spike batch reactors prepared with MOPS or NaHCO3 buffers and 10 ppm OCPPHA with the rate of 4-ClNB degradation quantified and mineral solids collected for characterization using dry TEM.The rate of reaction slows by nearly an order of magnitude after five injections, which is consistent with previous work.19Fig. 3 shows the pseudo-first order rate constant for reduction of 4-ClNB with each injection of Fe(II) and 4-ClNB. These data clearly demonstrate a consistent decrease in the rate of 4-ClNB with each injection, despite replenishing both the total Fe(II) and 4-ClNB to the levels used for the first injection. No new mineral phases were detected by XRD, which suggests that goethite nanoparticles grew by oxidative growth.
 | ||
| Fig. 3 Rate of 4-ClNB degradation by 0.325 g L−1 goethite and 1.0 mM Fe(II) in the presence of 10 ppm OCPPHA in 25 mM MOPS (open circles) and 10 mM NaHCO3 (closed circles). Error bars represent the 95% confidence interval of the regression of the pseudo-first order rate equation. | ||
Results quantifying the change in nanocrystal dimensions as a function of reaction progress demonstrate that the (021) goethite surface remains the most reactive in the presence of PPHA, regardless of the buffer employed. The length and width of several hundred nanoparticles per sample were measured from calibrated TEM images, and histograms depicting the frequency of each measurement for the stock nanoparticles (initial) and nanoparticles collected after five injections of Fe(II) and 4-ClNB (final) are displayed in Fig. 4 (histograms of injections one and three, in addition to the results of the F-tests, t-tests, and XRD analysis are reported in the ESI†). In both buffer systems, the average lengths after five 4-ClNB degradations are statistically different than the nanoparticles of the original stock suspension. While the average widths are also statistically different than that of the stock suspension, the histograms suggest that the distributions are not dissimilar. Fewer than the ideal number of nanoparticles (500) were measured due to the high degree of aggregation resulting in the inability to determine exact boundaries between particles. This qualification notwithstanding, the data suggest that the (021) surfaces of goethite remain the most reactive surface in the presence of PPHA. This conclusion is supported in Fig. 5, where the predicted length, based on mass balance under the assumption that the nanoparticles only grow in the length dimension, is compared to the actual, measured, length (calculation details are provided in the ESI†). We hypothesize, however, that once the degradation is inhibited at the (021) surface, growth on the other facets becomes detectable. Fig. 4d indicates that detectable growth in the width dimension may have occurred. This is the first indication of goethite growth in this dimension and future experiments could include more injections of 4-ClNB such that growth in the width dimension is statistically detectable by calibrated TEM images.
 | ||
| Fig. 4 Particle length (l) and width (w) distributions for goethite nanoparticles before (initial) and after (final) five reactions of 4-ClNB in 10 mM NaHCO3 (a, b) and 25 mM MOPS (c, d) in the presence of 10 ppm OCPPHA. The average values of length and width and the standard deviation are indicated in the legend. | ||
 | ||
| Fig. 5 Comparison of the actual, measured, lengths and the predicted lengths calculated using mass balance of 1, 3, and 5 reactions of 0.325 g L−1 goethite, 1.0 mM Fe(II), and 100 μM 4-ClNB in pH 7 25 mM MOPS (open circles) and 10 mM NaHCO3 (closed circles) in the presence of 10 ppm OCPPHA. A 1:1 line is provided to model complete agreement between the observed and calculated nanoparticle lengths. | ||
Conclusions
The rate of 4-ClNB degradation generally decreased with increasing PPHA concentration. Furthermore, results demonstrate that commonly used organic buffers, such as Good's buffers, yield substantially different results than when bicarbonate, which is commonly found in groundwater, buffer is used. Thus, caution should be exercised in the use of Good's buffers when attempting to model environmental conditions. The reduction in reactivity is most pronounced in the Good's buffer. At high OCPPHA concentrations, PPHA blocks reactive surface sites from reaction with 4-ClNB, but does not appear to affect the sorption of Fe(II) to the surface. Repeated cycles of 4-ClNB degradation demonstrated definitive oxidative crystal growth parallel to the c-axis, slowed reactions with successive exposure, and no precipitation of new mineral phases. There is evidence of crystal growth in the particle width dimension, which, if present, is the first known observation of oxidative growth on that goethite crystal face. Regardless, neither the use of NaHCO3 (instead of MOPS) nor the addition of PPHA altered which goethite face is the most reactive.Acknowledgements
The authors would like to thank the University of Minnesota (Interdisciplinary Doctoral Fellowship, Graduate School) and the National Science Foundation (ECS-1012193) for financial support. Parts of this work were carried out in the Characterization Facility, University of Minnesota, a member of the NSF-funded Materials Research Facilities Network (http://www.mrfn.org) via the MRSEC program. In addition, we would like to recognize Benjamin Wilson who performed BET analysis and Jeanette Tensfeldt who conducted zeta potential experiments. We also thank the reviewers for their constructive comments.References
- R. A. Maithreepala and R.-A. Doong, Environ. Sci. Technol., 2004, 38, 6676–6684 CrossRef CAS PubMed.
- K. Pecher, S. B. Haderlein and R. P. Schwarzenbach, Environ. Sci. Technol., 2002, 36, 1734–1741 CrossRef CAS PubMed.
- M. Elsner, S. B. Haderlein, T. Kellerhals, S. Luzi, L. Zwank, W. Angst and R. P. Schwarzenbach, Environ. Sci. Technol., 2004, 38, 2058–2066 CrossRef CAS PubMed.
- C. L. Chun, R. M. Hozalski and W. A. Arnold, Environ. Sci. Technol., 2005, 39, 8525–8532 CrossRef CAS PubMed.
- J. E. Amonette, D. J. Workman, D. W. Kennedy, J. S. Fruchter and Y. A. Gorby, Environ. Sci. Technol., 2000, 34, 4606–4613 CrossRef CAS.
- T. P. Klupinski, Y.-P. Chin and S. J. Traina, Environ. Sci. Technol., 2004, 38, 4353–4360 CrossRef CAS PubMed.
- E. J. O'Loughlin, K. M. Kemner and D. R. Burris, Environ. Sci. Technol., 2003, 37, 2905–2912 CrossRef.
- R. A. Maithreepala and R.-A. Doong, Environ. Sci. Technol., 2004, 38, 260–268 CrossRef CAS PubMed.
- A. G. B. Williams and M. M. Scherer, Environ. Sci. Technol., 2004, 38, 4782–4790 CrossRef CAS PubMed.
- V. Barrón and J. Torrent, J. Colloid Interface Sci., 1996, 177, 407–410 CrossRef.
- B.-H. Jeon, B. A. Dempsey, W. D. Burgos, M. O. Barnett and E. E. Roden, Environ. Sci. Technol., 2005, 39, 5642–5649 CrossRef CAS PubMed.
- K. Amstaetter, T. Borch, P. Larese-Casanova and A. Kappler, Environ. Sci. Technol., 2010, 44, 102–108 CrossRef CAS PubMed.
- S. R. Kanel, D. Nepal, B. Manning and H. Choi, J. Nanopart. Res., 2007, 9, 725–735 CrossRef CAS.
- X. Liang, R. P. Philp and E. C. Butler, Chemosphere, 2009, 75, 63–69 CrossRef CAS PubMed.
- H. Shao and E. C. Butler, Environ. Sci. Technol., 2009, 43, 1896–1901 CrossRef CAS PubMed.
- V. Sarathy, A. J. Salter, J. T. Nurmi, G. O. B. Johnson, R. L. Johnson and P. G. Tratnyek, Environ. Sci. Technol., 2010, 44, 787–793 CrossRef CAS PubMed.
- D. Colón, E. J. Weber, J. L. Anderson, P. Winget and L. A. Suarez, Environ. Sci. Technol., 2006, 40, 4449–4454 CrossRef.
- R. Matta, K. Hanna, T. Kone and S. Chiron, Chem. Eng. J., 2008, 144, 453–458 CrossRef CAS.
- C. L. Chun, R. L. Penn and W. A. Arnold, Environ. Sci. Technol., 2006, 40, 3299–3304 CrossRef CAS PubMed.
- T. B. Hofstetter, C. G. Heijman, S. B. Haderlein, C. Holliger and R. P. Schwarzenbach, Environ. Sci. Technol., 1999, 33, 1479–1487 CrossRef CAS.
- K. B. Gregory, P. Larese-Casanova, G. F. Parkin and M. M. Scherer, Environ. Sci. Technol., 2004, 38, 1408–1414 CrossRef CAS PubMed.
- P. G. Tratnyek and D. L. Macalady, J. Agric. Food Chem., 1989, 37, 248–254 CrossRef CAS.
- D. Colón, E. J. Weber and J. L. Anderson, Environ. Sci. Technol., 2008, 42, 6538–6543 CrossRef.
- K. M. Danielsen and K. F. Hayes, Environ. Sci. Technol., 2004, 38, 4745–4752 CrossRef CAS PubMed.
- T. P. Klupinski and Y.-P. Chin, Environ. Sci. Technol., 2003, 37, 1311–1318 CrossRef CAS.
- A. Buchholz, C. Laskov and S. B. Haderlein, Environ. Sci. Technol., 2011, 45, 3355–3360 CrossRef CAS PubMed.
- J. A. Hakala, Y.-P. Chin and E. J. Weber, Environ. Sci. Technol., 2007, 41, 7337–7342 CrossRef CAS PubMed.
- J. A. Hakala and Y.-P. Chin, J. Agric. Food Chem., 2010, 58, 12840–12846 CrossRef CAS PubMed.
- M. Elsner, R. P. Schwarzenbach and S. B. Haderlein, Environ. Sci. Technol., 2004, 38, 799–807 CrossRef CAS PubMed.
- P. J. Vikesland and R. L. Valentine, Environ. Sci. Technol., 2002, 36, 512–519 CrossRef CAS PubMed.
- P. J. Vikesland, A. M. Heathcock, R. L. Rebodos and K. E. Makus, Environ. Sci. Technol., 2007, 41, 5277–5283 CrossRef CAS PubMed.
- J. J. Erbs, B. Gilbert and R. L. Penn, J. Phys. Chem. C, 2008, 112, 12127–12133 CrossRef CAS.
- D. L. Macalady and K. Walton-Day, in Aquatic Redox Chemistry, American Chemical Society, 2011, vol. 1071, pp. 85–111 Search PubMed.
- M. Meier, K. Namjesnik-Dejanovic, P. A. Maurice, Y.-P. Chin and G. R. Aiken, Chem. Geol., 1999, 157, 275–284 CrossRef CAS.
- L. Weng, W. H. Van Riemsdijk, L. K. Koopal and T. Hiemstra, Environ. Sci. Technol., 2006, 40, 7494–7500 CrossRef CAS PubMed.
- B. Gu, J. Schmitt, Z. Chen, L. Liang and J. F. McCarthy, Environ. Sci. Technol., 1994, 28, 38–46 CrossRef CAS PubMed.
- C. R. Evanko and D. A. Dzombak, Environ. Sci. Technol., 1998, 32, 2846–2855 CrossRef CAS.
- T. Saito, L. K. Koopal, W. H. V. Riemsdijk, S. Nagasaki and S. Tanaka, Langmuir, 2004, 20, 689–701 CrossRef CAS PubMed.
- M. Baalousha, Sci. Total Environ., 2009, 407, 2093–2101 CrossRef CAS PubMed.
- K. L. Chen, S. E. Mylon and M. Elimelech, Environ. Sci. Technol., 2006, 40, 1516–1523 CrossRef CAS PubMed.
- X. Liu, M. Wazne, Y. Han, C. Christodoulatos and K. L. Jasinkiewicz, J. Colloid Interface Sci., 2010, 348, 101–107 CrossRef CAS PubMed.
- A. P. Gondikas, E. K. Jang and H. Hsu-Kim, J. Colloid Interface Sci., 2010, 347, 167–171 CrossRef CAS PubMed.
- X. Liu, M. Wazne, T. Chou, R. Xiao and S. Xu, Water Res., 2011, 45, 105–112 CrossRef CAS PubMed.
- K. L. Chen and M. Elimelech, J. Colloid Interface Sci., 2007, 309, 126–134 CrossRef CAS PubMed.
- O. Furman, S. Usenko and B. L. T. Lau, Environ. Sci. Technol., 2013, 47, 1349–1356 CAS.
- F. Loosli, P. Le Coustumer and S. Stoll, Water Res., 2013, 47, 6052–6063 CrossRef CAS PubMed.
- R. F. Domingos, N. Tufenkji and K. J. Wilkinson, Environ. Sci. Technol., 2009, 43, 1282–1286 CrossRef CAS PubMed.
- A. M. Stemig, T. A. Do, V. M. Yuwono, W. A. Arnold and R. L. Penn, Environ. Sci.: Nano, 2014, 1, 478–487 RSC.
- R. M. Cornell and U. Schwertmann, The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses, 2nd edn, Wiley-VCH, 2003 Search PubMed.
- A. Hartenbach, T. B. Hofstetter, M. Berg, J. Bolotin and R. P. Schwarzenbach, Environ. Sci. Technol., 2006, 40, 7710–7716 CrossRef CAS PubMed.
- J. Klausen, S. P. Tröber, S. B. Haderlein and R. P. Schwarzenbach, Environ. Sci. Technol., 1995, 29, 2396–2404 CrossRef CAS PubMed.
- D. Naka, D. Kim and T. J. Strathmann, Environ. Sci. Technol., 2006, 40, 3006–3012 CrossRef CAS PubMed.
- A. Neumann, T. B. Hofstetter, M. Lüssi, O. A. Cirpka, S. Petit and R. P. Schwarzenbach, Environ. Sci. Technol., 2008, 42, 8381–8387 CrossRef CAS PubMed.
- C. A. Schultz and T. J. Grundl, Environ. Sci. Technol., 2000, 34, 3641–3648 CrossRef CAS.
- R. P. Schwarzenbach, P. M. Gschwend and D. M. Imboden, Environmental Organic Chemistry, 2nd edn, John Wiley & Sons, Inc., Hoboken, NJ, 2003 Search PubMed.
- M. Ostermeier, C. Limberg and B. Ziemer, Z. Anorg. Allg. Chem., 2006, 632, 1287–1292 CrossRef CAS.
- W. Tscharnuter, in Encyclopedia of Analytical Chemistry, John Wiley & Sons, Ltd, 2006 Search PubMed.
- E. Tomaszewska, K. Soliwoda, K. Kadziola, B. Tkacz-Szczesna, G. Celichowski, M. Cichomski, W. Szmaja and J. Grobelny, J. Nanomater., 2013, 2013, 10 Search PubMed.
- A. J. Anschutz and R. L. Penn, Geochem. Trans., 2005, 6, 60–66 CrossRef CAS.
- J. D. Hem, U.S. Geological Survey Water-Supply Paper 2254 Search PubMed.
- T. Zeng, K. L. Ziegelgruber, Y.-P. Chin and W. A. Arnold, Environ. Sci. Technol., 2011, 45, 6814–6822 CrossRef CAS PubMed.
- W. S. Rasband, ImageJ, U.S. National Institutes of Health, Bethesda, Maryland, USA, 1997 Search PubMed.
- R. A. Royer, W. D. Burgos, A. S. Fisher, R. F. Unz and B. A. Dempsey, Environ. Sci. Technol., 2002, 36, 1939–1946 CrossRef CAS PubMed.
- A. van Geen, A. P. Robertson and J. O. Leckie, Geochim. Cosmochim. Acta, 1994, 58, 2073–2086 CrossRef CAS.
- M. Villalobos and J. O. Leckie, Geochim. Cosmochim. Acta, 2000, 64, 3787–3802 CrossRef CAS.
- M. Villalobos and J. O. Leckie, J. Colloid Interface Sci., 2001, 235, 15–32 CrossRef CAS PubMed.
- T. Hiemstra, R. Rahnemaie and W. H. van Riemsdijk, J. Colloid Interface Sci., 2004, 278, 282–290 CrossRef CAS PubMed.
- R. Rahnemaie, T. Hiemstra and W. H. van Riemsdijk, J. Colloid Interface Sci., 2007, 315, 415–425 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5en00141b |
| This journal is © The Royal Society of Chemistry 2015 |