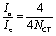DOI:
10.1039/C5RA09252C
(Paper)
RSC Adv., 2015,
5, 60570-60580
Poly(ethylene glycol-co-1,4-cyclohexanedimethanol terephthalate) random copolymers: effect of copolymer composition and microstructure on the thermal properties and crystallization behavior
Received
18th May 2015
, Accepted 6th July 2015
First published on 8th July 2015
Abstract
A series of poly(ethylene glycol-co-1,4-cyclohexanedimethanol terephthalate) (PETG) random copolymers were synthesized and characterized using 1H and 13C nuclear magnetic resonance, infrared spectroscopy and viscometry. Differential scanning calorimetry, wide-angle X-ray diffraction, and thermogravimetric analysis were used to probe the effects of copolymer composition and microstructure on the thermal properties, crystallization behavior, and thermal stability of the PETG copolymers, respectively. The mechanical properties were evaluated by tensile testing and dynamic mechanical measurements. The partial replacement in the polymerization feed of ethylene glycol by 1,4-cyclohexanedimethanol led to statistically random PETG copolymers with adjustable compositions and suitable molecular weights, which were thermally stable above 380 °C. The number-average sequence length of ethylene glycol terephthalate decreased with the increasing 1,4-cyclohexanedimethanol terephthalate (CT) content. In contrast, the number-average sequence length of CT increased gradually with the increasing CT content. The crystalline structure of the PETG copolymers changed from a PET-type lattice to a PCT-type lattice at a lower CT content. The crystallinity decreased at first, and then increased remarkably with the increasing CT content. It was interesting to notice that the rigid structure of the CT unit controlled the crystallization. The incorporation of CT in the PET chain, significantly altered the thermal transitions of the polyester. The glass transition temperature increased linearly with the increasing CT content. The melting temperature of the segments in crystalline domains strongly depended on the corresponding average sequence length. An increase in the average sequence length resulted in a higher melting temperature and an increase in the melting enthalpy.
1 Introduction
Aliphatic aromatic polyesters are a class of thermoplastic polyesters with unique properties, such as good mechanical properties, good chemical resistance, excellent surface appearance, and stable electrical insulation properties.1 As one of the most famous aliphatic aromatic polyesters, semicrystalline poly(ethylene terephthalate) (PET) has broad applications and is usually used in the form of fibers or films.2–6 The properties of PET are subject to the degree of crystallization.7 However, at the glass transition temperature (Tg) (∼70 °C) the mobility of the polymer chains in amorphous region increases considerably, resulting in the decrease of stiffness.8 Hence, PET is not suitable for applications in high temperature environments.
Increasing the Tg of polyesters, PET in particular, is currently an open front in the injection-moulding area. PET with higher Tg displays greater dimensional stability in thermal processing and better barrier properties in packing.9 Streamline hotfilling bottle process requiring materials with a Tg above filling temperature is a good example, which drives the development of the innovative PET derivatives.10
Copolymerization has been and continues to be the most appealing approach addressed to modify the properties of aromatic polyesters.11 Cyclic monomers, which tend to restrict the chain mobility, are able to render polyesters with enhanced Tg.12 Cycloaliphatic diols, such as 1,4-cyclohexanedimethanol (CHDM) can impart rigidity to the polyester chain. Poly(ethylene glycol-co-1,4-cyclohexanedimethanol terephthalate) (PETG), a modified PET, is prepared by partially replacing the ethylene glycol groups of PET with the CHDM groups. The crystallization rate and crystallinity of the prepared PETG copolymers are decreased, which can be attributed to the reduced molecular regularity by the introduction of CHDM into PET molecular chains.13
In this study, PET and a series of PETG random copolymers with different compositions were synthesized via bulk polycondensation. The copolymer composition and microstructure of the PETG copolymers were determined by 1H and 13C nuclear magnetic resonance (NMR) spectroscopy. Attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) was used to analyze the chemical structure of the PETG copolymers. Their thermal properties, crystallization behavior, and thermal stability were investigated using differential scanning calorimetry (DSC), wide-angle X-ray diffraction (WAXD), and thermogravimetric analysis (TGA), respectively. The tensile testing and dynamical mechanical thermal analysis (DMTA) were used to evaluate their mechanical properties. The aim of this study was to investigate the effects of the copolymer composition and the microstructure on the thermal properties, crystallization behavior, and thermal stability of the PETG copolymers.
2 Experimental
2.1 Materials
Terephthalic acid (TPA) (99%), ethylene glycol (EG) (99%), and 1,4-cyclohexanedimethanol (CHDM) (99%) were purchased from Yangzi Petrochemical Engineering Co., Ltd, Shanghai Lingfeng Chemical Reagent Co., Ltd, and SK Chemicals Co., Ltd, respectively. The solvents used for purification and characterization, such as methanol, phenol, and 1,1,2,2-tetrachloroethane, were analytical grade and used as received. The catalysts (antimony acetate, cobalt acetate, and organic tin) were friendly supplied by Jiangsu Jinghong New Material Technology Co., Ltd. The antioxidants (Irganox 1010 and Irgafos 168) were purchased from Ciba Specialty Chemicals Co., Ltd.
2.2 Synthesis
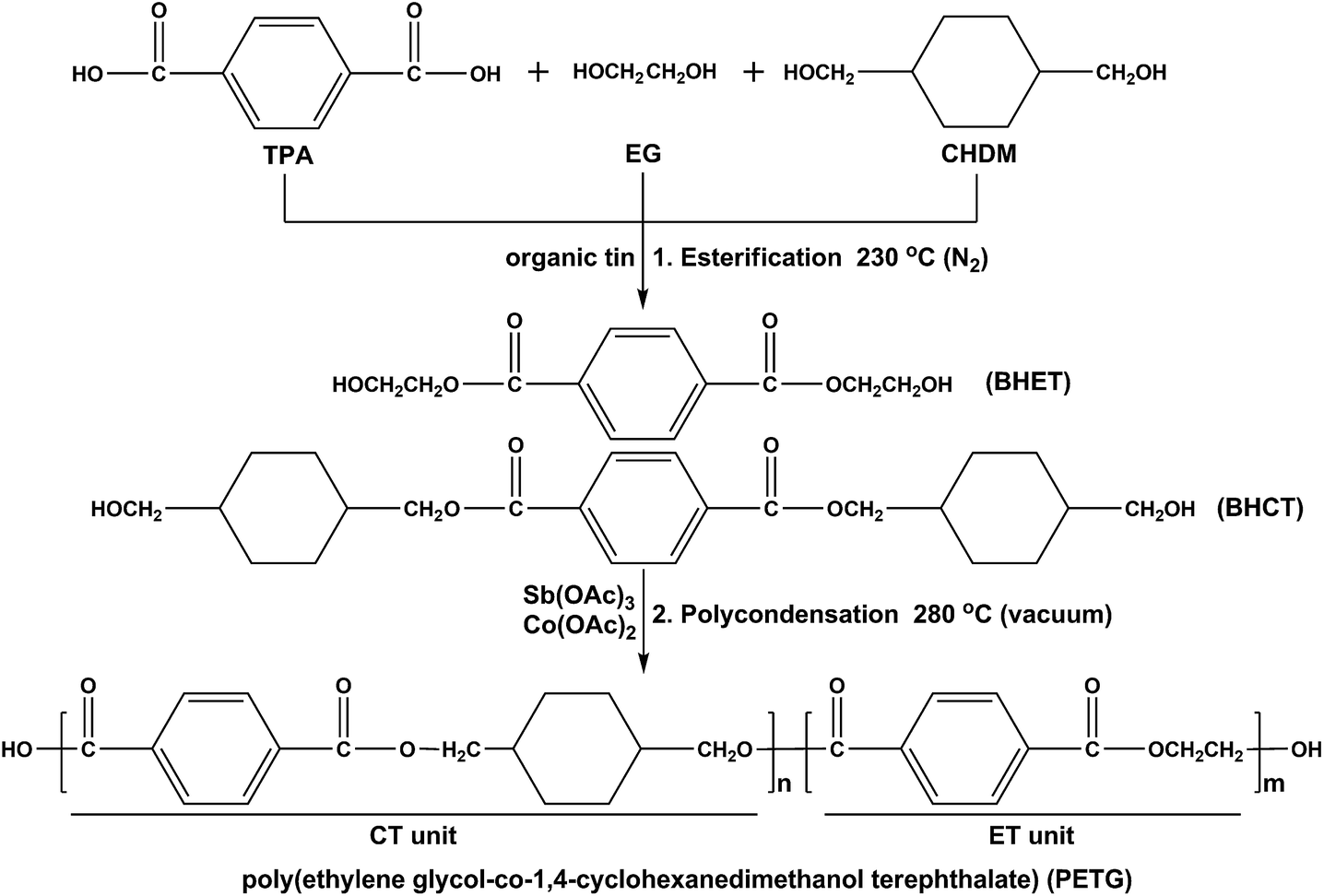
The PETG copolymers and PET used in this study were prepared from TPA, EG, and CHDM using organic tin, antimony acetate and cobalt acetate as catalysts. The two-step polymerization was performed on a laboratory-scale polymerization reactor in the molten state. The first step was the esterification reaction of TPA, EG, and CHDM with catalytic organic tin. The second step was the polycondensation reaction using antimony acetate and cobalt acetate as catalysts. The monomers, catalysts, and antioxidants were premixed in an autoclave reactor, and the mixture was gradually heated under constant stirring. The esterification reaction was carried out around 230 °C under nitrogen atmosphere, and the reaction was controlled by the amount of the distilled water. The polycondensation of bis(hydroxyethyl)terephthalate (BHET) and bis(hydroxymethylcyclohexane)terephthalate (BHCT) was carried out around 280 °C under high-vacuum condition. At the end of the reaction, the products in the melt were quenched into a water bath and followed by drying in a vacuum oven at room temperature for 48 h. The monomer composition in the feed was varied from 0, 15, 30, 50, to 70 mol% of CHDM. The resulting polymers were purified by methanol. The reactions involved in these syntheses are formulated in Scheme 1.
 |
| | Scheme 1 Synthesis of the PETG copolymers. | |
2.3 Characterization
The intrinsic viscosity [η] measurements were carried out in a mixed solvent of phenol/1,1,2,2-tetrachloroethane (1/1, w/w) at 25 ± 0.1 °C using an Ubbelohde viscometer.14 1H NMR spectroscopy was used for determining the copolymer composition. 13C NMR spectroscopy was used for determining the dyad sequence distribution in the copolymers. 1H and 13C NMR spectra were recorded on a Bruker DRX 500 operating at 500 and 125 MHz, respectively. About 12 and 100 mg of sample dissolved in 0.5 mL deuterated trifluoroacetic acid (TFA-d1) for 1H and 13C NMR spectra, respectively. On determination of the dyad sequence distribution, the relative peak intensities for EE, EC, CE and CC dyads are deconvoluted, and their peak areas are considered to be corresponding dyad quantities.
ATR-FTIR spectroscopy was performed on a FTIR spectrometer (Thermo Fisher Nicolet IS5, America) in the attenuated total reflectance mode. The spectra were scanned from 4000 to 600 cm−1 with a resolution of 4 cm−1.
2.4 WAXD measurements
The WAXD experiments were performed using a WAXD diffractometer (Shimadzu 6000, Japan). The operated voltage and current were 40 kV and 30 mA, respectively. The samples were annealed at 150 °C for 1 h prior to the measurement. The samples were scanned from 2θ = 10° to 40° at a speed of 5° min−1. The lamellar thickness (L) was calculated using the Scherrer's formula:15| |
L = Kλ/β![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ cosθ
| (1) |
where K is a structure factor, taken as 1.0; λ is the wavelength of the monochromatic X-ray beam [nm] (λ = 0.154 nm for CuKα radiation15); β is the full width at half maximum for the diffraction peak [rad]; and θ is the Bragg angle [°].
2.5 DSC measurements
A differential scanning calorimeter (TA Q20, USA) was applied to investigate the thermal properties of the PETG copolymers and PET. Temperature and heat flow were calibrated using a high purity indium standard (156.6 °C and 28.45 J g−1). All samples were dried in a vacuum oven at 60 °C for 12 h prior to the test. The measurement was performed using about 10 mg sample sealed in an aluminum pan under the nitrogen atmosphere with a purge flow of 50 mL min−1. The sample was heated to 300 °C at 40 °C min−1 and kept isothermally for 1 min to erase the previous thermal history. Then, the sample was quenched to the amorphous state. Thereafter, the sample was heated to 300 °C at 10 °C min−1 to clearly detect the glass transition temperature (Tg). Finally, a second cool scan and a third heat scan were performed at 10 °C min−1 to obtain the thermal properties.
The crystallinity (Xc) of sample was calculated as follows:16
where Δ
Hm is the melting enthalpy, and Δ
H*m is the melting enthalpy of 100% crystalline PET, which is equal to 130 J g
−1.
17
2.6 TGA measurements
The thermal stability of the PETG copolymers and PET were evaluated by TGA. The thermal degradation process was recorded using a TGA thermal analyzer (TA Q50, USA). For each experiment, about 20 mg of the sample was used and a nitrogen flow rate of 50 mL min−1 was adopted. The samples, under a nitrogen protective atmosphere, were heated from 40 to 800 °C at a rate of 20 °C min−1.
2.7 Tensile and shrinkage properties measurements
Samples for tensile testing were prepared with a thickness of 0.5 mm by compression molding. Subsequently, substantially amorphous samples were obtained by minimizing crystallization, using iced water bath, and followed by drying in a vacuum oven at room temperature for 48 h. The samples were cut into dumbbell shape with a width of 4 mm while the distance between the testing marks was 20 mm. The Young's modulus, yield stress, and elongation at break were measured at a stretching rate of 5 mm min−1 on a universal testing machine (CMT 5254, SANS Co. China) at room temperature.
The monoaxial shrinkage of the PETG copolymers and PET was performed on rectangular samples (50 × 10 mm2) cut from amorphous, isotropic films with a thickness of about 0.5 mm. The tests were conducted on a universal testing machine (CMT 4254, SANS Co. China) equipped with a temperature-controlled thermal cabinet. The sample was stretched to a stretch ratio of 3 × 1 for 5 min at 90 °C. Then, the sample was fixed under this strain and cooled to 25 °C for 10 min to measure the original length (L0). Finally, the sample was placed into the thermal cabinet again at 90 °C for 5 min without any stress and the final length (Lf) was recorded. The shrinkage at 90 °C was calculated by the following equation:18
| | |
Shrinkage = (L0 − Lf)/L0 × 100%
| (3) |
2.8 DMTA measurements
Samples for dynamic mechanical measurements were obtained by compression molding. Subsequently, substantially amorphous samples were obtained by minimizing crystallization, using iced water bath, and followed by drying in a vacuum oven at room temperature for 48 h. Samples were cut into a 25 × 6 × 1 mm3 rectangular shape. Dynamic mechanical measurements were performed with a dynamical mechanical thermal analyzer (Anton Paar MCR 302, Austria), at a frequency of 1 Hz and a heating rate of 3 °C min−1, over a temperature range from 30 to 160 °C.
3 Results and discussion
3.1 NMR analysis
The 1H NMR spectra of the PETG copolymers and PET and the assignment of each peak are shown in Fig. 1. The equation for determining the copolymer composition from respective peak area is given as follow:| |
 | (4) |
where a, b, c, d, e, and f represent the areas of corresponding peaks in Fig. 1, and X and Y denote the mole fractions of ethylene glycol terephthalate (ET) and 1,4-cyclohexanedimethanol terephthalate (CT) units, respectively. When the copolymer compositions as calculated by eqn (4) are compared with the feed compositions, it is revealed that the copolymer compositions are nearly equal to the feed compositions (results are shown in Table 1).
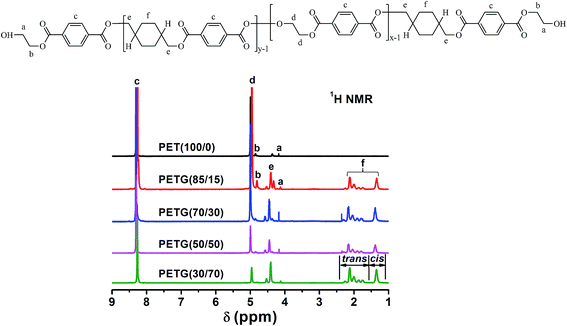 |
| | Fig. 1 1H NMR spectra of the PETG copolymers and PET and the assignment of each peak. | |
Table 1 Compositions, number-average molecular weights, intrinsic viscosities, and viscosity average molecular weights of the PETG copolymers and PETa
| Copolymer |
Feed composition (mol%) |
Copolymer compositionb (mol%) |
CHDMb (mol%) |
NET |
NCT |
![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) nb (g mol−1) nb (g mol−1) |
[η]c (dL g−1) |
vd (g mol−1) |
| ET/CT |
ET/CT |
trans/cis |
| NET, NCT: total repeating units of ET and CT, respectively. Measured by 1H NMR. Measured in a mixed solvent of phenol and 1,1,2,2-tetrachoroethane (1/1, w/w) using an Ubbelhode viscometer at 25 °C. Viscosity average molecular weights were estimated from the [η] using the Mark–Houwink equation. |
| PET(100/0) |
100/0 |
100/0 |
0/0 |
74.5 |
0 |
1.4 × 104 |
0.69 |
2.6 × 104 |
| PETG(85/15) |
85/15 |
84.4/15.6 |
66.7/33.3 |
64.5 |
13.3 |
1.6 × 104 |
0.79 |
3.1 × 104 |
| PETG(70/30) |
70/30 |
70.9/29.1 |
70.4/29.6 |
45.1 |
21.3 |
1.4 × 104 |
0.70 |
2.6 × 104 |
| PETG(50/50) |
50/50 |
51.6/48.4 |
69.9/30.1 |
30.8 |
27.3 |
1.3 × 104 |
0.67 |
2.5 × 104 |
| PETG(30/70) |
30/70 |
31.6/68.4 |
68.0/32.0 |
16.9 |
30.8 |
1.2 × 104 |
0.75 |
2.9 × 104 |
In the 1H NMR spectra, since peak b (δ = 4.85 ppm) is slightly overlapped with peak d (δ = 5.00 ppm), peak a (δ = 4.18 ppm) was used as a chain end proton for polymer structure estimation.19 The total repeating units of ET (NET) and CT (NCT) in copolymers can be calculated by the integration ratio of chain end to corresponding repeating units by the 1H NMR spectra, using the following equations:
| |
 | (5) |
| |
 | (6) |
where
Ia,
Id, and
Ie are the integration values for the corresponding peak at positions a, d, and e, and
NET and
NCT are the total repeating units of ET and CT, respectively. The total number-average molecular weights of the whole polymer (
n) can thus be calculated by
n/Dalton = 192 ×
NET + 274 ×
NCT (where 192 and 274 are the molecular weights of the repeating units of ET and CT), with the results listed in
Table 1. The estimated number-average molecular weights are in the range of 1.2 × 10
4–1.6 × 10
4 g mol
−1.
For the copolymers synthesized, viscosity average molecular weights (v) were estimated from the measured intrinsic viscosities ([η]) using the Mark–Houwink equation with constants α and k which were determined previously by Ward20 for PET homopolymer:
| | |
[η] = 2.75 × 10−4 0.77v
| (7) |
The results are listed in Table 1. The estimated viscosity average molecular weights are in the range of 2.5 × 104–3.1 × 104 g mol−1, indicating that the resulting polymers have relatively high molecular weight, which are able to form films. In fact, eqn (7) was found for PET homopolymer rather than PETG copolymers. The Mark–Houwink constants are not known for other copolymer compositions, therefore values of v are used for relative comparison.
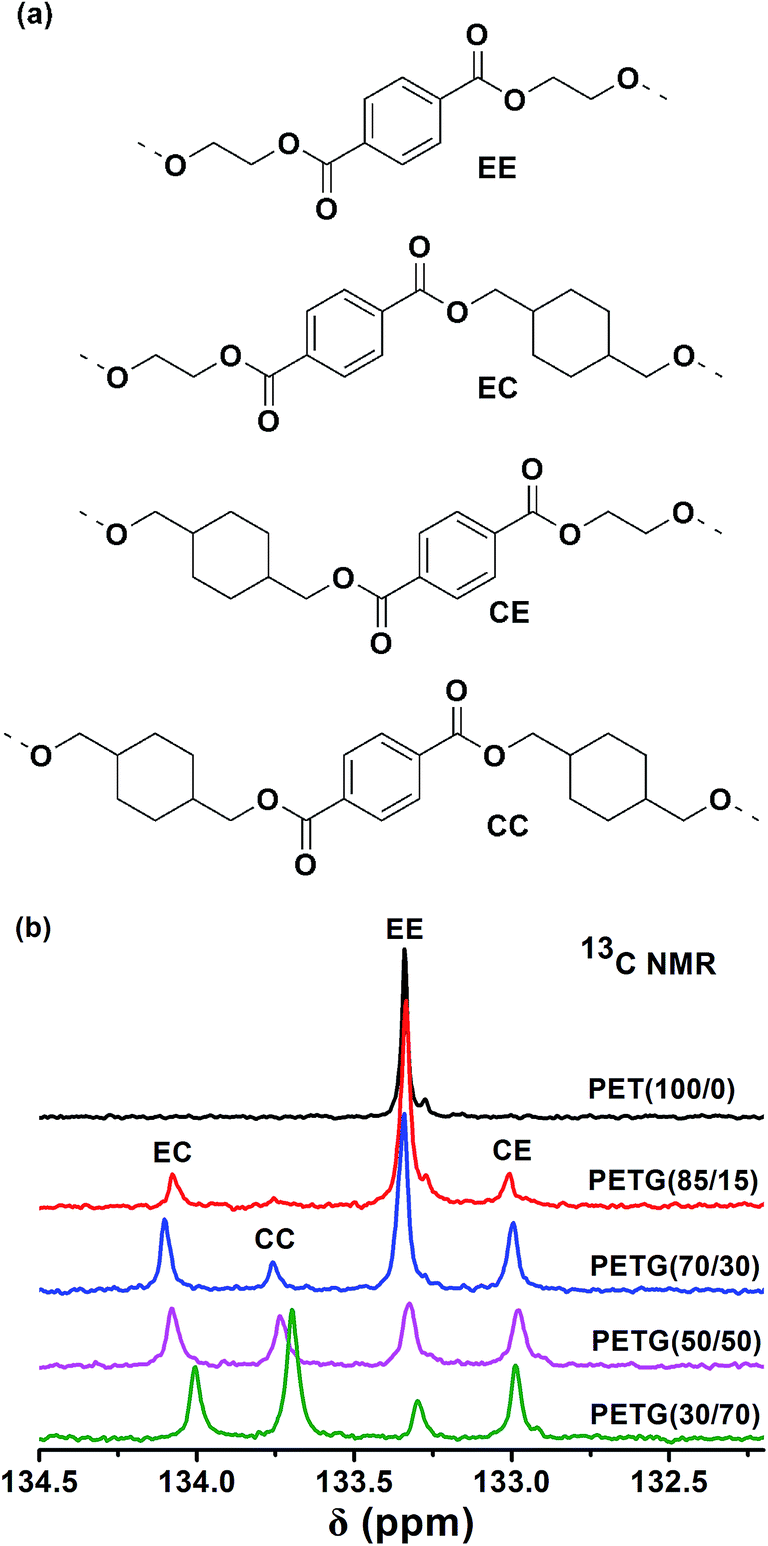
The microstructure of the PETG copolymers is analyzed using 13C NMR spectroscopy. The quaternary aromatic carbon resonances appear to be sensitive to dyad sequence effects.21 The signals due to these carbons appear to split giving a total of four peaks since four types of dyads (EE, EC, CE and CC) are possible. The four different dyads possible in the copolymer repeat units are depicted in Fig. 2a. The 132.5–134.5 ppm region containing the resonance signals produced by such carbons is shown in Fig. 2b. The peaks arising from EE, EC, CE and CC dyads appear well resolved as to be quantified by integration with reliability. The relative concentrations of the four dyads are determined from deconvoluted areas of the four signals.
 |
| | Fig. 2 13C NMR spectra: (a) the four possible dyads occurring in the PETG copolymers, (b) the non-protonated aromatic carbon region in the 13C NMR spectra of the PETG copolymers and PET. | |
In Fig. 3, the dyad contents are plotted against the copolymer compositions. The dashed lines represent the theoretically calculated contents for statistically random copolymers, in which the dyad sequence distribution obeys the Bernoullian statistics model:22
where
Nij and
Xi denote the mole fractions of the
ij dyad sequence and
i component in the copolymer chains, respectively. Since the fractions of the dyad sequence determined experimentally are in good agreement with the dyad sequence distributions calculated by the Bernoullian statistics. It is suggested that the PETG samples synthesized in this study are statistically random copolymers.
 |
| | Fig. 3 Plot of dyad contents vs. copolymer compositions. The dashed lines represent the calculated distribution based on the Bernouillian statistics. | |
According to Yamadera and Murano,23 if four kinds of signals due to homolinks and heterolinks are observed in the NMR spectroscopy of the copolymer, then the average sequence length and the degree of randomness of the copolymer can be further determined. The number-average sequence lengths (n) of the ET and CT homogeneous sequences, as well as the degree of randomness (R), are estimated for each PETG copolymers by using the following equations:24
| | |
nET = (NEE + 1/2(NEC + NCE))/1/2(NEC + NCE)
| (11) |
| | |
nCT = (NCC + 1/2(NEC + NCE))/1/2(NEC + NCE)
| (12) |
| | |
R = (1/nET) + (1/nCT)
| (13) |
For random copolymers, R is unity. The value of R equal to zero indicates a mixture of homopolymers, while a value of 2 indicates an alternating distribution.23 Results from these calculations are summarized in Table 2, indicating that the sequence distribution in the PETG copolymers is essentially random in the entire range of the compositions as the values of R are very close to unity. The number-average sequence length nET decreases with the increasing CT unit content. In contrast, nCT increases gradually with the increasing CT unit content. The concentration of the esterification product BHCT increases with the increasing content of monomer CHDM, which leads to an increase of the probability that BHCT reacts with BHCT during the polycondensation reaction. This will increase the number-average sequence length of the CT homogeneous sequences. On the contrary, it will leads to the decreasing nET.
Table 2 Composition and microstructure of the PETG copolymersa
| Copolymer |
Dyad contentb (%) |
Sequence lengths |
R |
| NEE |
NCE |
NCC |
nET |
nCT |
| n: number-average sequence lengths. R: degree of randomness. Measured by 13C NMR. |
| PETG(85/15) |
74.67 |
23.18 |
2.15 |
7.44 |
1.19 |
0.97 |
| PETG(70/30) |
53.83 |
39.66 |
6.51 |
3.71 |
1.33 |
1.02 |
| PETG(50/50) |
29.30 |
50.60 |
20.10 |
2.16 |
1.79 |
1.02 |
| PETG(30/70) |
10.62 |
45.50 |
43.88 |
1.47 |
2.93 |
1.02 |
3.2 ATR-FTIR analysis
The chemical structure of the PETG copolymers was analyzed by ATR-FTIR spectroscopy taking PET as the reference in Fig. 4. The spectra were baseline corrected and normalized at 1712 cm−1, the major absorbance peak representing the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of ester groups.1 The peaks at 2968 and 2907 cm−1 are attributed to the asymmetric and symmetric aliphatic C–H stretching vibrations in PET.25,26 The C–H symmetrical and asymmetrical stretching due to existence of methylene groups in the PETG copolymers can be observed between 2924 and 2852 cm−1.27 The C(O)–O stretching peak, CH2 bending peak, and C–H stretching peak of cyclohexylene ring are observed at 1260, 1451, and 958 cm−1, respectively, in the PETG copolymers with various CT content.28–31 The C–H stretching peak of cyclohexylene ring in the PETG copolymers increases with the increasing CT content. This result is accordance with the copolymer composition measured by 1H NMR.
O of ester groups.1 The peaks at 2968 and 2907 cm−1 are attributed to the asymmetric and symmetric aliphatic C–H stretching vibrations in PET.25,26 The C–H symmetrical and asymmetrical stretching due to existence of methylene groups in the PETG copolymers can be observed between 2924 and 2852 cm−1.27 The C(O)–O stretching peak, CH2 bending peak, and C–H stretching peak of cyclohexylene ring are observed at 1260, 1451, and 958 cm−1, respectively, in the PETG copolymers with various CT content.28–31 The C–H stretching peak of cyclohexylene ring in the PETG copolymers increases with the increasing CT content. This result is accordance with the copolymer composition measured by 1H NMR.
 |
| | Fig. 4 ATR-FTIR spectra of the PETG copolymers and PET. | |
3.3 WAXD analysis
The crystallization behavior of the PETG copolymers and PET was probed by WAXD. The diffraction patterns of the as-obtained samples exhibit a typical semicrystalline polymer behavior. The diffraction patterns of the samples with the composition close to the 50:50 is not well resolved. Hence, all the samples were annealed at 150 °C for 1 h prior to the further WAXD measurement. After annealing, the diffraction peaks were well resolved. Fakirov and co-workers investigated this phenomena and suggested that the emergence of the well resolved diffraction peaks might be ascribed to the crystallization-induced sequential reordering in the copolymers during the annealing process.32 Fig. 5 shows the WAXD patterns of the PETG copolymers and PET annealed at 150 °C for 1 h. The corresponding WAXD data are represented in Table 3. A large amorphous hump appears in PETG(70/30), implying that PETG(70/30) is mainly amorphous. However, the other four samples present a series of sharp diffraction peaks related to their crystalline structures. The crystalline structures of PET and poly(1,4-cyclohexylene dimethylene terephthalate) (PCT) are reported to be triclinic.33–35 The three main peaks observed are assigned to (010), (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10), and (100) planes of PET, at 2θ values of about 17.68°, 22.78°, and 26.24°. PETG(85/15) shows a similar pattern to PET, indicating that PETG(85/15) crystallized in the PET lattice. PCT shows three main diffraction peaks at 2θ = 16.63°, 19.10°, and 23.41°, indexing as (010), (10), and (100) planes.21 The diffraction peaks shown in the patterns of PETG(50/50) and PETG(30/70) are at the same position as that of PCT, revealing that PETG(50/50) and PETG(30/70) crystallized in the PCT lattice. The WAXD patterns (Fig. 5) can be divided into two groups according to the CT content in the copolymer: the PET-type crystal and the PCT-type crystal. Under 15 mol% CT content, the PET-type crystalline structure is in dominance, whereas above 50 mol% CT the PCT-type crystalline structure develops. This result indicates that the crystalline structure of the PETG copolymers changes from PET-type lattice to PCT-type lattice at a lower CT content. These findings suggest that the rigid structure of CT unit controls the crystallization.
10), and (100) planes of PET, at 2θ values of about 17.68°, 22.78°, and 26.24°. PETG(85/15) shows a similar pattern to PET, indicating that PETG(85/15) crystallized in the PET lattice. PCT shows three main diffraction peaks at 2θ = 16.63°, 19.10°, and 23.41°, indexing as (010), (10), and (100) planes.21 The diffraction peaks shown in the patterns of PETG(50/50) and PETG(30/70) are at the same position as that of PCT, revealing that PETG(50/50) and PETG(30/70) crystallized in the PCT lattice. The WAXD patterns (Fig. 5) can be divided into two groups according to the CT content in the copolymer: the PET-type crystal and the PCT-type crystal. Under 15 mol% CT content, the PET-type crystalline structure is in dominance, whereas above 50 mol% CT the PCT-type crystalline structure develops. This result indicates that the crystalline structure of the PETG copolymers changes from PET-type lattice to PCT-type lattice at a lower CT content. These findings suggest that the rigid structure of CT unit controls the crystallization.
 |
| | Fig. 5 WAXD patterns of the PETG copolymers and PET annealed at 150 °C for 1 h. | |
Table 3 WAXD data of the PETG copolymers and PET annealed at 150 °C for 1 ha
| Samples |
Crystal plane |
2θ (°) |
d (Å) |
β (°) |
L (Å) |
| θ: Bragg angle, d: interplanar distance, L: lamellar thickness. The WAXD data is only presented to the samples which have measurable diffraction peaks. |
| PET(100/0) |
01 |
16.56 |
5.35 |
1.10 |
81.06 |
| 010 |
17.68 |
5.01 |
1.36 |
65.66 |
| 11 |
21.80 |
4.07 |
1.32 |
68.07 |
| 10 |
22.78 |
3.90 |
— |
— |
| 100 |
26.24 |
3.39 |
2.29 |
39.56 |
| PETG(85/15) |
01 |
16.44 |
5.39 |
1.16 |
76.85 |
| 010 |
17.66 |
5.02 |
— |
— |
| 11 |
21.60 |
4.11 |
— |
— |
| 10 |
22.68 |
3.92 |
— |
— |
| 100 |
26.08 |
3.41 |
2.60 |
34.84 |
| PETG (50/50) |
011 |
15.12 |
5.85 |
1.22 |
72.96 |
| 010 |
17.06 |
5.19 |
1.67 |
53.43 |
| 10 |
19.42 |
4.57 |
— |
— |
| 100 |
23.72 |
3.75 |
1.98 |
45.54 |
| 11 |
26.10 |
3.41 |
1.66 |
54.56 |
| PETG (30/70) |
011 |
15.18 |
5.83 |
1.15 |
77.40 |
| 010 |
16.91 |
5.24 |
1.07 |
83.37 |
| 10 |
19.10 |
4.64 |
— |
— |
| 100 |
23.40 |
3.80 |
1.75 |
51.49 |
| 11 |
25.82 |
3.45 |
1.32 |
68.58 |
As shown in Table 3, the lamellar thicknesses of the crystal planes of PET are larger than that of PETG(85/15) at the same crystallization condition, which implies that the crystallization rate of PET is superior to that of PETG(85/15). As for the PETG copolymers crystallized in the PCT lattice, the lamellar thicknesses of PETG(30/70) in the crystal planes are larger than that of PETG(50/50), indicating that PETG(30/70) has a faster crystallization rate than that of PETG(50/50). These results reveal that the crystallization rate of the PETG copolymers decreases at first and then increases remarkably with the increasing CT content.
3.4 DSC analysis
The DSC cooling traces and heating traces of the PETG copolymers and PET at 10 °C min−1 are depicted in Fig. 6. The detailed data, crystallization temperature (Tc), cold crystallization temperature (Tcc), melting temperature (Tm), and the corresponding enthalpies (ΔHc, ΔHcc, and ΔHm), are listed in Table 4. As shown in Fig. 6a, a crystallization peak is observed in both PET(100/0) and PETG(30/70). The crystallization peak occurs due to the melt crystallization of the samples. However, the crystallization peak is not found in PETG(85/15), PETG(70/30), and PETG(50/50), which suggests that these three copolymers could not melt crystallization under this condition. These results indicate that the crystallization rate of PET(100/0) and PETG(30/70) is faster than these three copolymers. As regards the DSC heating traces (Fig. 6b), a cold crystallization peak and a melting peak are observed in both PETG(85/15) and PETG(50/50). The cold crystallization peak occurs due to the cold crystallization of the samples during the heating process. The melting peak is attributed to the crystals formed from the cold crystallization. Moreover, just one melting peak is observed in PET(100/0) and PETG(30/70), indicating that these two copolymers have completed the crystallization during the cooling process. However, neither the cold crystallization peak nor the melting peak is found in PETG(70/30), which suggests that PETG(70/30) is completely amorphous.
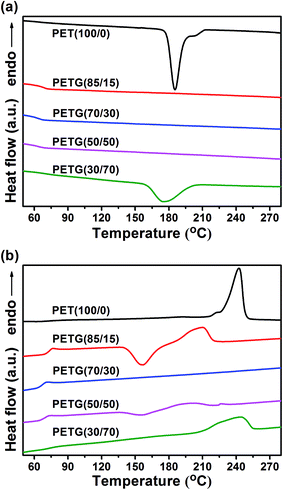 |
| | Fig. 6 DSC traces of the PETG copolymers and PET at 10 °C min−1: (a) cooling traces, (b) heating traces. | |
Table 4 Thermal properties of the PETG copolymers and PET obtained from DSCa
| Sample Codes |
Sequence lengthsb |
Cooling |
Heating |
| nET |
nCT |
Tc (°C) |
ΔHc (J g−1) |
Tcc (°C) |
ΔHcc (J g−1) |
Tm (°C) |
ΔHm (J g−1) |
Xc (%) |
| Crystallization (Tc), cold crystallization (Tcc) and melting (Tm) temperatures, as well as their enthalpies (ΔHc, ΔHcc, and ΔHm) measured by DSC with the cooling/heating rate of 10 °C min−1. Calculated using the data from 13C NMR. |
| PET(100/0) |
|
|
185.4 |
43.8 |
— |
— |
242.5 |
45.1 |
34.7 |
| PETG(85/15) |
7.44 |
1.19 |
— |
— |
157.1 |
22.9 |
210.1 |
22.9 |
17.6 |
| PETG(70/30) |
3.71 |
1.33 |
— |
— |
— |
— |
— |
— |
— |
| PETG(50/50) |
2.16 |
1.79 |
— |
— |
156.5 |
5.1 |
197.1 |
9.6 |
7.4 |
| PETG(30/70) |
1.47 |
2.93 |
174.7 |
27.4 |
— |
— |
243.6 |
25.5 |
19.6 |
The incorporation of CT in the PET chain, significantly altered the thermal transitions of the polyester. The CT unit is a stiff and bulky structure, which destroys the regularity of the PET chain. As shown in Table 4, although PET is semicrystalline with a melting temperature of around 242.8 °C, PETG(70/30) appears to be essentially amorphous, which is in agreement with the WAXD analysis. In fact, obvious endothermic peaks characteristic of melting are observed in PETG(85/15), PETG(50/50), and PETG(30/70). Also, their melting temperatures are lower than that of PET, and meanwhile involved with much smaller associated enthalpies. The crystallinity of the PETG copolymers decreases at first, and then increases remarkably with the increasing CT content. These results clearly indicate that there is a strong effect exerted by the CT units on the crystallinity. According to the molecular features of CT, both chain packing and crystallization rates appeared to be hampered by copolymerization, which will be discussed in the following part.
The Tg of the PETG copolymers could be clearly detected in the heating DSC traces of samples previously quenched from the melt (Fig. 7). A cold crystallization peak is observed in PET(100/0), PETG(85/15), and PETG(30/70). The cold crystallization peak occurs due to the cold crystallization of the quenched samples during the heating process. However, the cold crystallization peak is not found in PETG(70/30) and PETG(50/50), which suggests that these two copolymers could not crystallize under this condition. These results reveal that the crystallization ability of PET(100/0), PETG(85/15), and PETG(30/70) is stronger than these two copolymers.
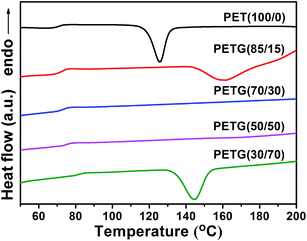 |
| | Fig. 7 DSC traces of the PETG copolymers and PET recorded at heating from quenched samples for Tg observation. | |
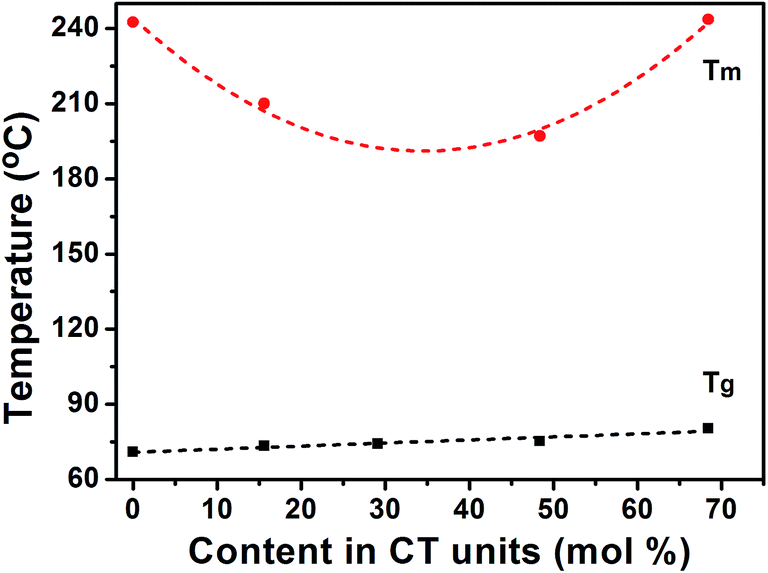
The variation of melting and glass transition temperatures as the function of composition is plotted in Fig. 8. The Tg of the PETG copolymers increases linearly with the increasing CT content, which also confirmed the fact that the copolymers synthesized in this study have a random distribution of ET and CT units. Furthermore, the CT structure is able to render stiffness and viscosity to the polymer chain, and therefore, it will reduce the free volume as well. It is interesting to note that Tg plays an important role in controlling the crystallization. Moreover, the copolymer composition of around 30 mol% CT showing the minimum melting temperature corresponds to a critical composition where the crystal transition between PET-type and PCT-type crystal lattices occurs. Therefore, it is reasonable to believe that the PET-type crystal develops when CT content is not above than 30 mol%, while the PCT-type crystal develops when CT content is above 30 mol%. This result is in accordance with the WAXD analysis.
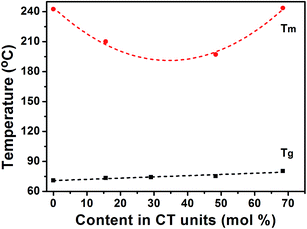 |
| | Fig. 8 Dependence of glass transition and melting temperatures on the copolymer composition. | |
Research indicated that the melting temperature of the segments in crystal region strongly depends on the corresponding average sequence length.36 According to the WAXD analysis, PET(100/0) and PETG(85/15) crystallize in the PET lattice, while PETG(50/50) and PETG(30/70) crystallize in the PCT lattice. The 13C NMR results reveal that the number-average sequence length nET decreases with the increasing CT unit content. On the contrary, nCT increases gradually with the increasing CT unit content. As shown in Table 4, the melting temperature and the melting enthalpy of the copolymers crystallized in PET lattice, such as PET(100/0) and PETG(85/15), decrease significantly with the decreasing average sequence length nET. As for the copolymers crystallized in PCT lattice, such as PETG(50/50) and PETG(30/70), an increase in the average sequence length nCT results in the higher melting temperature and the increase of the melting enthalpy.
3.5 TGA analysis
In order to investigate the thermal stability of the PETG copolymers, TGA was conducted under a nitrogen atmosphere using PET as the reference. Fig. 9 shows the TGA thermograms of the PETG copolymers and PET. The detailed data, including the initial degradation temperature at 5 wt% loss of the original weight (T5%), the temperature of the maximum degradation rate (Td,max),37 and the weight percentage of the residue at 800 °C, are summarized in Table 5.
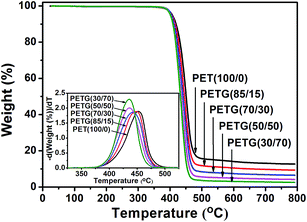 |
| | Fig. 9 TGA thermograms of the PETG copolymers and PET at a heating rate of 20 °C min−1 in nitrogen. | |
Table 5 TGA data of the PETG copolymers and PET in nitrogena
| Samples |
T5% (°C) |
Td,max (°C) |
Residue (wt%) |
| T5%: the initial decomposition temperature at 5 wt% loss of the original weight. Td,max: the temperature at the maximum degradation rate. |
| PET(100/0) |
407.5 |
451.5 |
12.7 |
| PETG(85/15) |
405.7 |
449.1 |
9.4 |
| PETG(70/30) |
404.6 |
442.2 |
6.5 |
| PETG(50/50) |
401.5 |
435.9 |
4.3 |
| PETG(30/70) |
401.2 |
435.7 |
2.6 |
The PETG samples appear as materials with high thermal stability. Under nitrogen flow the degradation process begins above 380 °C and occurs in a single step up to a complete weight loss, as shown in Fig. 9. The temperatures of the initial degradation (T5%), reported in Table 5, indicate that all samples have a very similar behavior. Moreover, the PETG copolymers show a decreased Td,max from 449.1 °C of PETG(85/15) to 435.7 °C of PETG(30/70). A close inspection of data reveals that although the thermal stability of these copolymers decreased slightly with the increasing CT content, it remains close to that of PET. The residue of PET is 12.7 wt% and the residue of the PETG copolymers ranges from 9.4 to 2.6 wt%. PETG with the more cyclohexylene ring contents exhibit less residue at high temperature, which might result from the reduced carbonization due to the presence of cyclohexylene ring.
3.6 Tensile and shrinkage properties
A preliminary evaluation of the mechanical properties of the PETG copolymers has been carried out by tensile testing. The stress–strain curves of the PETG copolymers and PET at room temperature are shown in Fig. 10. The detailed data, including Young's modulus, yield stress, and elongation at break, are presented in Table 6. At room temperature, the films undergo yielding followed by plastic deformation region and ultimate failure of the films. The Young's modulus and yield stress of the PETG copolymers decreases with the increasing CT content. The elongation at break of PETG(85/15) and PETG(50/50) is relatively low, whereas a high deformation is reached for PETG(70/30) and PETG(30/70).
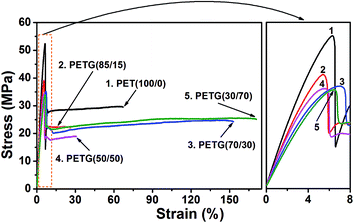 |
| | Fig. 10 Stress–strain curves of the PETG copolymers and PET at room temperature. | |
Table 6 Tensile properties of the PETG copolymers and PET at room temperature
| Samples |
Young's modulus (MPa) |
Yield stress (MPa) |
Elongation at break (%) |
| PET(100/0) |
1085.5 ± 21.8 |
51.3 ± 1.0 |
55 ± 10.2 |
| PETG(85/15) |
927.8 ± 39.7 |
37.3 ± 1.7 |
27 ± 2.5 |
| PETG(70/30) |
839.3 ± 45.7 |
36.4 ± 2.0 |
151 ± 8.6 |
| PETG(50/50) |
875.2 ± 10.0 |
34.8 ± 2.1 |
21 ± 7.9 |
| PETG(30/70) |
712.2 ± 32.3 |
33.1 ± 1.2 |
190 ± 34.1 |
The shrinkages of the PETG copolymers with different CHDM content (0%, 15%, 30%, 50%, and 70%), stretched into 3 × 1 at 90 °C, tested at 90 °C, are 58%, 62%, 63%, 62%, and 48%, respectively. The testing temperature is higher than the Tg values detected by DSC. PETG(70/30) has a higher shrinkage than other copolymers. Presumably, this occurs due to the weakest crystallization ability of PETG(70/30) among these copolymers and it is essentially amorphous according to the DSC analysis. The shrinkages of the PETG copolymers and PET are more than 40% when the testing temperature is higher than Tg. This result indicates that both the PETG copolymers and PET could be potentially used as heat-shrinkable materials with the operation temperature at 90 °C.
3.7 DMTA analysis
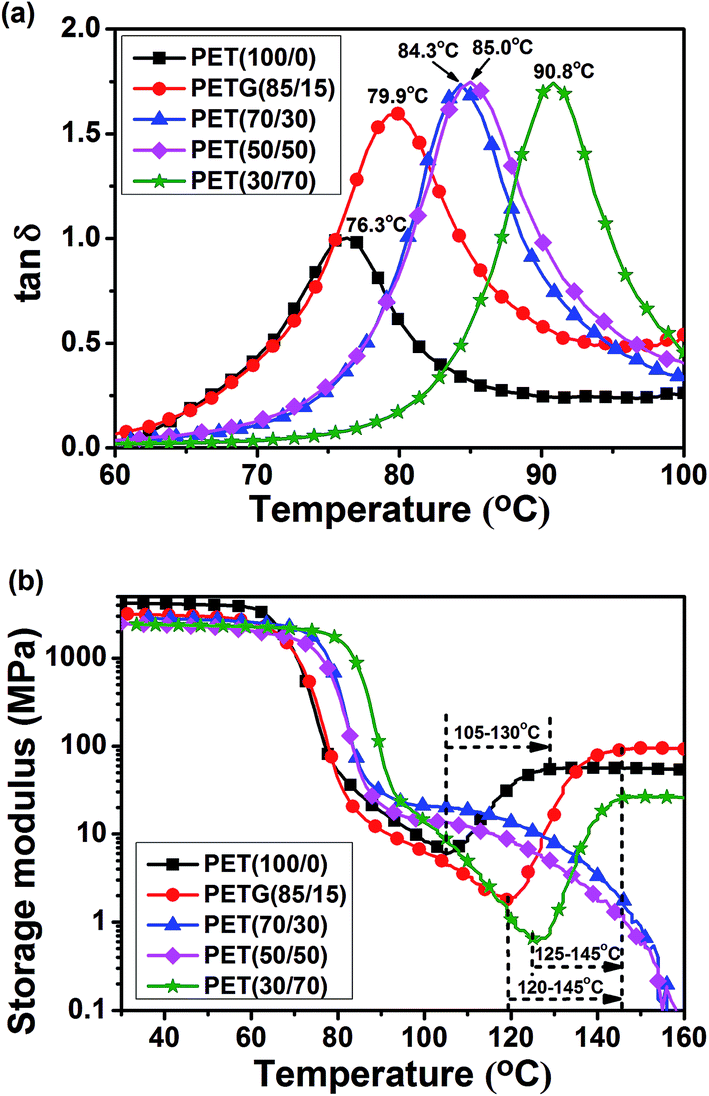
Fig. 11 shows the DMTA spectra of the PETG copolymers and PET. In the temperature range from 60–100 °C the samples exhibit one peak of tanδ, denoted as α in order of increasing temperature. As regards the assignment of the observed relaxation, the peak is confidently assigned to the glass-to-rubber transition. Indeed, there is a good correspondence between the Tg values detected by DSC and the Tα values determined by DMTA. The value of Tα increases with the increasing CT content, which indicates that the copolymer with higher CT content is suitable for applications in higher temperature environment. Moreover, from Fig. 11a, it is evident that the intensity of the α peak of the PETG copolymers is higher than that of PET. This might be ascribed to the cyclic structure (cyclohexylene ring) of CHDM, which can absorb energy through the interconversion of chair and boat conformations.38
 |
| | Fig. 11 DMTA spectra of the PETG copolymers and PET: (a) tanδ, (b) storage modulus. | |
As regards the storage modulus, at relatively low temperature it remains basically unchanged with increasing temperature, whereas, in correspondence with the glass relaxation, it shows a steep decrease of about two orders of magnitude. From the DMTA spectra of PET(100/0), PETG(85/15), and PETG(30/70), an activated dispersion region corresponding to the glass relaxation can be observed, followed by a slight pre-crystallization stiffening. The latter is presumably related to the density modulation preceding the formation of a crystalline phase, which was already observed by Imai et al.39 The rather steep increase of the elastic modulus observed in the range 105–130 °C for PET(100/0), 120–145 °C for PETG(85/15), and 125–145 °C for PETG(30/70) must be ascribed to the growth of crystalline domains. It is evident that PET(100/0), PETG(85/15), and PETG(30/70) cold crystallize in the DMTA measurement process. Moreover, a slight pre-crystallization stiffening is not observed in the spectra of PETG(70/30) and PETG(50/50), indicating that these two copolymers could not crystallize under the DMTA measurement condition. These results suggest that the crystallization ability of PET(100/0), PETG(85/15), and PETG(30/70) is stronger than that of PETG(70/30) and PETG(50/50). These results are in accordance with the DSC analysis (Fig. 7).
4 Conclusions
A series of statistically random PETG copolymers with adjustable compositions and suitable molecular weights were synthesized in this study. Their copolymer composition and microstructure were determined by 1H NMR and 13C NMR spectroscopy. The effects of the copolymer composition and the microstructure on the thermal properties, crystallization behavior, and thermal stability of the PETG copolymers were systematically investigated. Their copolymer compositions were nearly equal to the feed compositions. The number-average sequence length nET decreased with the increasing CT content. On the contrary, nCT increased gradually with increasing CT content. The crystalline structure of the PETG copolymers changed from PET-type lattice to PCT-type lattice at a lower CT content. The crystallinity of the PETG copolymers decreased at first, and then increased remarkably with the increasing CT content. It was interesting to notice that the rigid structure of CT controlled the crystallization. The incorporation of CT in the PET chain, significantly altered the thermal transitions of the polyester. The Tg of the PETG copolymers increased linearly with the increasing CT content, which also confirmed the fact that the copolymers synthesized in this study had a random distribution of ET and CT units. The melting temperature of the segments in crystal region strongly depended on the corresponding average sequence length. An increase in the average sequence length resulted in a higher melting temperature and an increase in the melting enthalpy.
Acknowledgements
This work was supported by the Innovation Foundation for Graduate Students of Jiangsu Province (KYLX_0744) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). The authors would like also to express their appreciation to Mr Hao Wang and Mr Mingfu Chen for their help in the preparation of the manuscript.
References
- I. N. Strain, Q. Wu, A. M. Pourrahimi, M. S. Hedenqvist, R. T. Olsson and R. L. Andersson, J. Mater. Chem. A, 2015, 3, 1632–1640 CAS
 .
. - G. Xu, S. Qin, J. Yu, Y. Huang, M. Zhang and W. Ruan, RSC Adv., 2015, 5, 29924–29930 RSC .
- Y. Rao, J. Greener, C. A. Avila-Orta, B. S. Hsiao and T. N. Blanton, Polymer, 2008, 49, 2507–2514 CrossRef CAS PubMed .
- M. R. Patel, J. V. Patel and V. K. Sinha, Polym. Degrad. Stab., 2005, 90, 111–115 CrossRef CAS PubMed .
- X. K. Jing, X. S. Wang, D. M. Guo, Y. Zhang, F. Y. Zhai, X. L. Wang, L. Chen and Y. Z. Wang, J. Mater. Chem. A, 2013, 1, 9264–9272 CAS .
- Y. Jahani, M. Ghetmiri and M. R. Vaseghi, RSC Adv., 2015, 5, 21620–21628 RSC .
- S. Fakirov, E. W. Fischer, R. Hoffmann and G. F. Schmidt, Polymer, 1977, 18, 1121–1129 CrossRef CAS .
- J. M. Huang, P. P. Chu and F. C. Chang, Polymer, 2000, 41, 1741–1748 CrossRef CAS .
- T. Higashioji, T. Tsunekawa and B. Bhushan, Tribol. Int., 2003, 36, 437–445 CrossRef CAS .
- G. W. McNeely and A. J. Woodward, US pat., 5 250 333, 1993 .
- D. P. R. Kint and S. Muñoz-Guerra, Polym. Int., 2003, 52, 321–336 CrossRef CAS PubMed .
- C. Japu, A. Martínez de Ilarduya, A. Alla and S. Muñoz-Guerra, Polymer, 2014, 55, 2294–2304 CrossRef CAS PubMed .
- Y. Tsai, C. H. Fan, C. Y. Hung and F. J. Tsai, J. Appl. Polym. Sci., 2008, 109, 2598–2604 CrossRef CAS PubMed .
- J. Yang, W. Li, A. Yu, P. Xi, X. A. Huang and S. Li, J. Appl. Polym. Sci., 2009, 111, 2751–2760 CrossRef CAS PubMed .
- A. Bigi, E. Boanini, C. Capuccini, M. Fini, I. N. Mihailescu, C. Ristoscu, F. Sima and P. Torricelli, Biomaterials, 2009, 30, 6168–6177 CrossRef CAS PubMed .
- N. Vasanthan, N. J. Manne and A. Krishnama, Ind. Eng. Chem. Res., 2013, 52, 17920–17926 CrossRef CAS .
- R. J. Müller, H. Schrader, J. Profe, K. Dresler and W. D. Deckwer, Macromol. Rapid Commun., 2005, 26, 1400–1405 CrossRef PubMed .
- W. K. Shih, Polym. Eng. Sci., 1994, 34, 1121–1128 CAS .
- W. Huang, Y. Wan, J. Chen, Q. Xu, X. Li, X. Yang, Y. Li and Y. Tu, Polym. Chem., 2014, 5, 945–954 RSC .
- I. Ward, Nature, 1957, 180, 141–142 CrossRef CAS PubMed .
- T. E. Sandhya, C. Ramesh and S. Sivaram, Macromolecules, 2007, 40, 6906–6915 CrossRef CAS .
- Y. G. Jeong, W. H. Jo and S. C. Lee, Macromolecules, 2000, 33, 9705–9711 CrossRef CAS .
- R. Yamadera and M. Murano, J. Polym. Sci., Part A: Polym. Chem., 1967, 5, 2259–2268 CrossRef CAS PubMed .
- C. Japu, A. Martinez de
Ilarduya, A. Alla, M. G. Garcia-Martin, J. A. Galbis and S. Muñoz-Guerra, Polym. Chem., 2013, 4, 3524–3536 RSC .
- R. H. Guo, S. Q. Jiang, C. W. M. Yuen and M. C. F. Ng, J. Mater. Sci.: Mater. Electron., 2009, 20, 735–740 CrossRef CAS .
- O. Prasad, L. Sinha, N. Misra, V. Narayan, N. Kumar and J. Pathak, J. Mol. Struct.: THEOCHEM, 2010, 940, 82–86 CrossRef CAS PubMed .
- R. Jayakumar, M. Rajkumar, R. Nagendran and S. Nanjundan, J. Appl. Polym. Sci., 2002, 85, 1194–1206 CrossRef CAS PubMed .
- A. Bessadok, S. Roudesli, S. Marais, N. Follain and L. Lebrun, Composites, Part A, 2009, 40, 184–195 CrossRef PubMed .
- S. S. Umare, A. S. Chandure and R. A. Pandey, Polym. Degrad. Stab., 2007, 92, 464–479 CrossRef CAS PubMed .
- H. Nakamura, H. Sakai, S. Aoshima and M. Abe, J. Oleo Sci., 2002, 51, 781–787 CrossRef CAS .
- S. W. Lee, W. Huh, Y. S. Hong and K. M. Lee, Korea Polym. J., 2000, 8, 261–267 Search PubMed .
- Z. Denchev, M. Sarkissova, H. J. Radusch, T. Luepke and S. Fakirov, Macromol. Chem. Phys., 1998, 199, 215–221 CrossRef CAS .
- Y. Kitano, Y. Kinoshita and T. Ashida, Polymer, 1995, 36, 1947–1955 CrossRef CAS .
- R. P. Daubeny and C. Bunn, Proc. R. Soc. London, Ser. A, 1954, 226, 531–542 CrossRef CAS .
- C. Boye, J. Polym. Sci., 1961, 55, 275–284 CrossRef CAS PubMed .
- A. A. Deschamps, D. W. Grijpma and J. Feijen, Polymer, 2001, 42, 9335–9345 CrossRef CAS .
- Y. Zhang, L. Chen, J. J. Zhao, H. B. Chen, M. X. He, Y. P. Ni, J. Q. Zhai, X. L. Wang and Y. Z. Wang, Polym. Chem., 2014, 5, 1982–1991 RSC .
- H. Ni, J. L. Daum, P. R. Thiltgen, M. D. Soucek, W. J. Simonsick, W. Zhong and A. D. Skaja, Prog. Org. Coat., 2002, 45, 49–58 CrossRef CAS .
- M. Imai, K. Kaji and T. Kanaya, Macromolecules, 1994, 27, 7103–7108 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.