
Water-soluble TRIS-terminated PAMAM dendrimers: microwave-assisted synthesis, characterization and Cu(II) intradendrimer complexes
Ali Serol Ertürk*a,
Mustafa Ulvi Gürbüzb,
Metin Tülüb and
Abdürrezzak Emin Bozdoğanb
aDepartment of Metallurgy and Material Science Engineering, Adıyaman University, 02040, Adıyaman, Turkey. E-mail: aserturk@adiyaman.edu; traserturk@gmail.com; Fax: +90 416 223 3809; Tel: +90 416 223 3800/2760
bDepartment of Chemistry, Yıldız Technical University, 34210, Istanbul, Turkey
First published on 2nd July 2015
Abstract
This study is the first report describing the microwave-assisted synthesis (MAS) of poly(amido amine) (PAMAM) dendrimers with TRIS surface functional groups (PAMAM–TRIS). Six PAMAM–TRIS dendrimers were synthesized using both newly developed conventional and microwave methods. Five of them are novel. Three different cores, one polymeric Jeffamine® T-403 and two monomeric, ethylenediamine and diethylenetriamine, were used in the syntheses. All the reactions were monitored by attenuated total reflectance (ATR). It was observed that microwave reactions proceeded 3.5 to 4.0 times faster than conventional reactions. Therefore, a fast, easy and one-pot MAS of six different water-soluble PAMAM–TRIS dendrimers was accomplished with high (90–96%) yields in short (110–140 min) reaction times and under mild reaction conditions, using methanol as solvent. The other ester terminated half generation precursor PAMAM (PAMAM–OCH3) dendrimers used for the synthesis of the PAMAM–TRIS dendrimers were obtained by utilizing conventional and microwave methods together. For the purification of all the PAMAM dendrimers, a liquid phase polymer-based retention (LPR) technique was used. The PAMAM–TRIS dendrimers were characterized by 1H NMR, 13C NMR, ATR (IR), EA, potentiometric and spectroscopic titrations. Furthermore, Cu(II)–PAMAM–TRIS dendrimer complexes were prepared and characterized by UV-Vis spectroscopy. The synthesized PAMAM–TRIS dendrimers can be considered as new drug carrier systems and should find use in widespread application fields, especially in future pharmaceutical and catalytic studies but also in other fields.
Introduction
Poly(amido amine) (PAMAM) dendrimers are the class of most widely studied starburst macromolecules. They are distinguishable from linear polymers by their unusual chemical and physical properties.1 These properties can be altered depending on the construction units, which consist of the core, repeating branches and surface groups. While the segment of PAMAM dendrimers comprising the core and repeating branch units is called the internal cavity, the segment, including the surface groups is called the outer periphery. Control of these segments allows PAMAM dendrimers to be used as target materials in a wide range of applications including catalysis,2,3 antibacterial agents,4–6 gene therapy,7–9 and drug-delivery.10,11Aqueous solubility of PAMAM dendrimers is very important. For example, water-soluble PAMAM dendrimers can be a host for small hydrophobic acidic guest molecules within their internal cavities. Several amides and tertiary amine groups are present in these cavities. These groups can entrap small molecules such as drugs in the cavity of the dendrimer by hydrogen bonding, electrostatic interaction, or both. Thus, these cavities can play the role of a container for various hydrophobic drugs. On the other hand, the outer periphery with hydrophilic groups can provide PAMAM dendrimers with appropriate water solubility.12 As a result, they can be used as potential drug carriers and delivery systems in physiological environments. Moreover, PAMAM dendrimers are used as templates to control size, stability, and solubility of nanoparticles in the range of 1 nm to 4–5 nm.2 Crooks et al.2 showed dendrimers to be good hosts for metal nanoparticles. These well-defined nanoparticles have uniform structures. Therefore, they can take part in catalytic reactions, resist agglomeration, and be selective to control encapsulation of small substrate molecules.13,14 Furthermore, a stable host–guest interaction is desired for both intradendrimer metal complexes, and drug–drug carrier conjugates. In particular, the synthesis of water-soluble derivatives of drugs with different chemical formulations is generally tried while developing efficient drug-delivery systems. However, even small structural changes to improve water solubility can often lead to a dramatic decrease in the efficacy of drug. For this reason, drug carrier systems can be helpful to increase the water solubility of drugs by encapsulation since the therapeutic efficacy and the ability of the drug to access the target sites are increased.
Modifying the structure of PAMAM dendrimers is important to reach the desired water solubility. TRIS is the abbreviation of the common known organic compound, tris(hydroxymethyl)aminomethane, with the formula (HOCH2)3CNH2. It is widely used in biochemistry, molecular biology, and is highly water-soluble. Ester-terminated PAMAM dendrimers (PAMAM–OCH3) are generally water insoluble. Dendrimers are known to be designable polymers and can be used as building blocks. Surface modification of water insoluble PAMAM–OCH3 dendrimers with TRIS makes them water-soluble. Not only are TRIS-terminated PAMAM dendrimers (PAMAM–TRIS) water-soluble drug-delivery agents, but they are also synthetic precursors for dendrimer-encapsulated metal nanoparticles. However, preparation of these PAMAM dendrimers requires three to four days under conventional heating using DMSO as solvent.12,15 So far, a little attention has been paid to develop a fast, easy and green synthesis of the PAMAM–TRIS dendrimers. Therefore, alternative approaches are necessary.
Microwave-assisted synthesis (MAS) of PAMAM dendrimers is an attractive subject field on the agenda and a challenging research topic. A few studies have been conducted up to now on the fast, facile and efficient synthesis of PAMAM dendrimers by using hyphenated synthesis and advanced purification techniques such as liquid phase polymer retention (LPR) or membrane filtration (MF) together.16,17 When reaction times, chemical wastage of solvents, reaction conditions, and environmental effects are taken into consideration, MAS can shorten reaction times,18 convert hours to minutes, enhance reaction rate, prevent side product formation and give higher yields when compared with conventional methods. In addition, it can stop wastage of solvents, present neat and sustainable reaction conditions and protocols,19 and assist in reducing global warming. Hence, MAS is a helpful technique and can be a good alternative for the synthesis of water-soluble PAMAM–TRIS dendrimers.
This paper presents a fast, efficient and one-pot synthesis of six PAMAM–TRIS dendrimers, five of which are novel, from PAMAM–OCH3 dendrimer precursors using newly developed conventional and microwave-assisted methods. By using these methods, starting from two different monomeric cores, ethylenediamine (E), diethylenetriamine (D), and one polymeric core, Jeffamine® T-403 (P), MAS of third and fourth-generation PAMAM–TRIS dendrimers was performed. The synthesized PAMAM–TRIS dendrimers were characterized by 1H NMR, 13C NMR, ATR (IR), EA and UV-Vis spectroscopy in addition to potentiometric and spectroscopic titrations. Finally, Cu(II)–PAMAM–TRIS dendrimer complexes were prepared and structural defects in the internal cavities of the PAMAM–TRIS dendrimers were investigated to show the purity and monodispersity of them by spectroscopic Cu(II) titrations.
Results and discussion
Synthesis of PAMAM–TRIS dendrimers
The terminal groups of PAMAM dendrimers provide them with unique properties. Amine-terminated PAMAM dendrimers (PAMAM–NH2) are water-soluble and mostly present in protonated conformation at the pH media of the living cells’ physiological functions.20 Thus, when they come in contact with negatively charged cells, hemolysis of cells occurs. This phenomenon has a negative effect on the future development of clinical applications of amine terminated dendrimers.21On the other hand, drug delivery and toxicity studies on surface modified derivatives of PAMAM dendrimers revealed that carboxyl terminated PAMAM dendrimers (PAMAM–COOH) are less toxic, and their negative charge on the periphery prohibits them from binding or interacting with the negatively charged surface of the cells.22,23 Ideally, an interaction between a drug carrier or delivery system and a cell is expected. Thus, we decided to synthesize a series of water-soluble TRIS-terminated water-soluble PAMAM dendrimers (Scheme 1). Syntheses were performed by both conventional (Method A) and microwave-assisted (Method B) methods. As a result, we have developed a new microwave-assisted method for the surface modification of PAMAM dendrimers with TRIS functional groups to improve their water solubility.
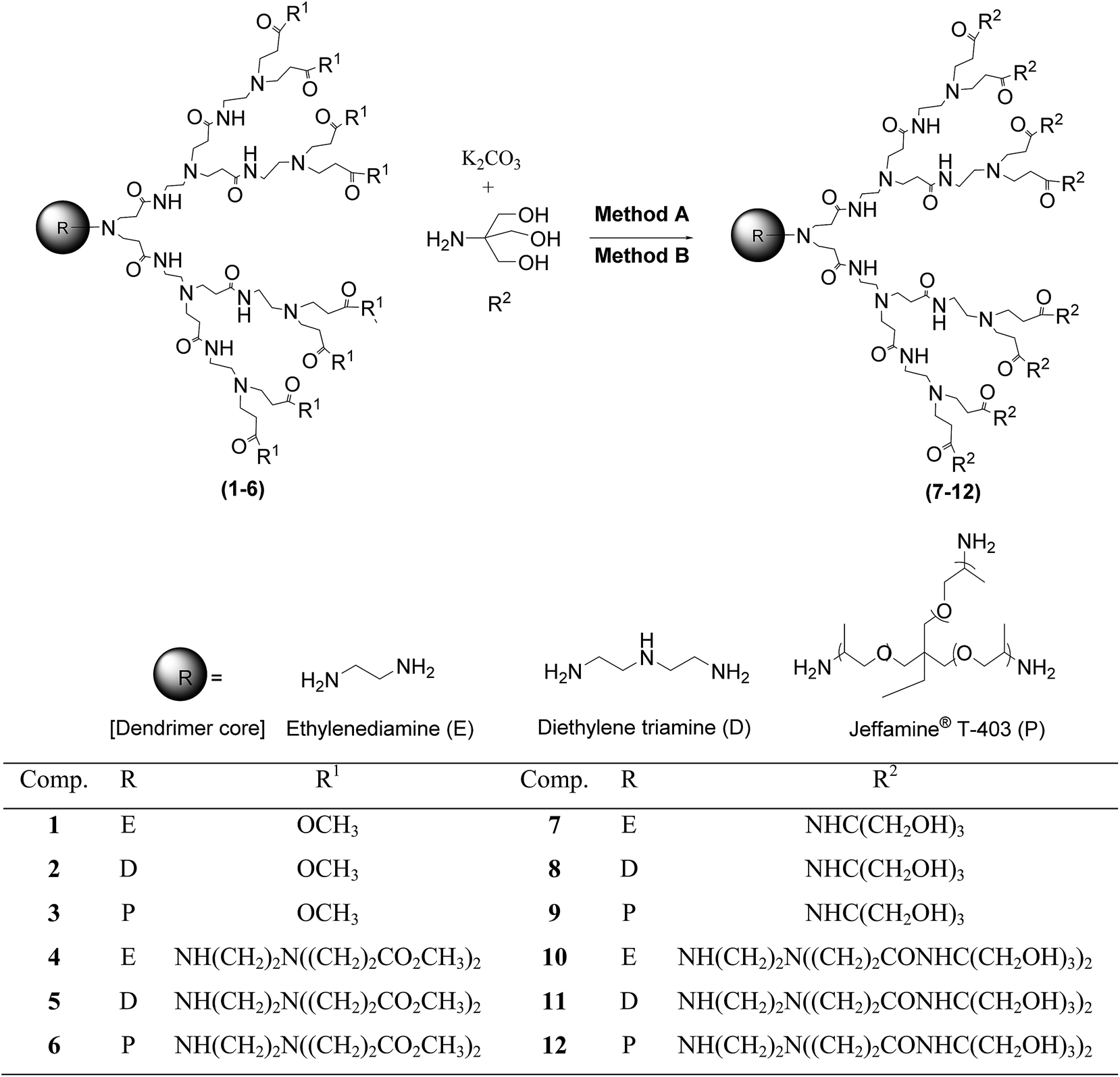 | ||
| Scheme 1 Synthesis of PAMAM–TRIS dendrimers (Cn.TRIS) (7–12) from PAMAM–OCH3 dendrimers (Cn.5) (1–6). | ||
As can be seen from Scheme 1, six different PAMAM–OCH3 dendrimer precursors (1–6) were used in the synthesis of PAMAM–TRIS dendrimers (7–12). PAMAM–OCH3 dendrimers (1–6) were synthesized according to our recently developed microwave-assisted divergent synthesis method.17 For the surface modification, three different types of dendrimer core, ethylenediamine (E), diethylenetriamine (D), and Jeffamine T-403 (P), were used. Therefore, a series of water-soluble PAMAM–TRIS dendrimers (7–12) possessing various numbers of terminal hydroxyl groups and properties were obtained. The coded generations and the physico-chemical properties of these PAMAM dendrimers are summarized in Table 1.
| Dendrimer | Generation | Molecular formula | MW (g mol−1) | Number of tertiary amines (3N) | Number of terminal esters (OCH3) | Number of terminal hydroxyls (OH) |
|---|---|---|---|---|---|---|
| a The theoretical characteristic data including the masses, the number of tertiary amines,24 terminal esters and hydroxyl groups (three folds of the number of terminal ester groups) of dendrimers were calculated according to the literature20,24 by using Scheme 1. | ||||||
| 1 | E2.5 | C126H224N26O44 | 2808 | 14 | 16 | — |
| 2 | D2.5 | C159H283N33O55 | 3537 | 18 | 20 | — |
| 3 | P2.5 | C207H377N39O71 | 4562 | 21 | 24 | — |
| 4 | E3.5 | C270H480N58O92 | 6012 | 30 | 32 | — |
| 5 | D3.5 | C339H603N73O115 | 7543 | 38 | 40 | — |
| 6 | P3.5 | C423H761N87O143 | 9368 | 45 | 48 | — |
| 7 | E3.TRIS | C174H336N42O76 | 4234 | 14 | — | 48 |
| 8 | D3.TRIS | C199H383N53O95 | 5230 | 18 | — | 60 |
| 9 | P3.TRIS | C279H545N63O119 | 6700 | 21 | — | 72 |
| 10 | E4.TRIS | C366H704N90O156 | 8865 | 30 | — | 96 |
| 11 | D4.TRIS | C459H883N113O195 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 109 109 |
38 | — | 120 |
| 12 | P4.TRIS | C567H1097N135O239 | 13647 |
45 | — | 144 |
When TRIS is attached to PAMAM–OCH3 dendrimers, the number of resulting terminal hydroxyl groups is increased three fold (Table 1). Thus, PAMAM–TRIS dendrimers could gain very high water solubility and can be used as drug carrier systems in many applications.12 Newkome et al.15,25 reported the synthesis of PAMAM–TRIS dendrimers from commercially available ethylenediamine cored dendrimers previously using conventional synthesis methods. By applying the same procedure, Beezer et al.12 synthesized three water-soluble PAMAM–TRIS dendrimers from PAMAM–OCH3 dendrimers. However, no other alternative conventional or microwave-assisted methods have been reported for the fast, one-pot synthesis of water-soluble TRIS surface modified PAMAM dendrimers up to now.
We herein, as a new approach, show the synthesis of third (C3.TRIS) and fourth (C4.TRIS) generation PAMAM–TRIS dendrimers with E, D and P cores using a MAS method. Monitoring the reaction conditions, deciding the amount of solvents, and where to stop the reactions were determined with a similar approach used in our recent study.17 In ATR monitoring, the disappearance of the ester 1730 cm−1 peak, and formation of ∼3260 cm−1 broad hydroxyl peak, and ∼1635 and ∼1554 cm−1 amide I and amide II peaks were used as the indicators of the completion of reactions (Fig. 1). In Table 2, MAS conditions for the TRIS surface modification of PAMAM–OCH3 dendrimers using Method B (1–6) are presented. First of all, before obtaining the optimum conditions reported in Table 2, a series of trial experiments with different power (watt), time (min), and various molar ratios of PAMAM–OCH3 dendrimer precursors to TRIS or K2CO3 were performed. Afterwards, the best reaction conditions were determined as shown in Table 2. Furthermore, the ideal molar ratios of TRIS and K2CO3 to PAMAM–OCH3 dendrimers (1–6) were determined to be 1.20 and 1.50 times molar equivalence of the number of terminal ester groups of PAMAM–OCH3 dendrimers (1–6), respectively. Finally, the syntheses of PAMAM–TRIS products (7–12) were performed within 110–140 min when compared to three12 and four days15,25 in conventional methods (Table 2).
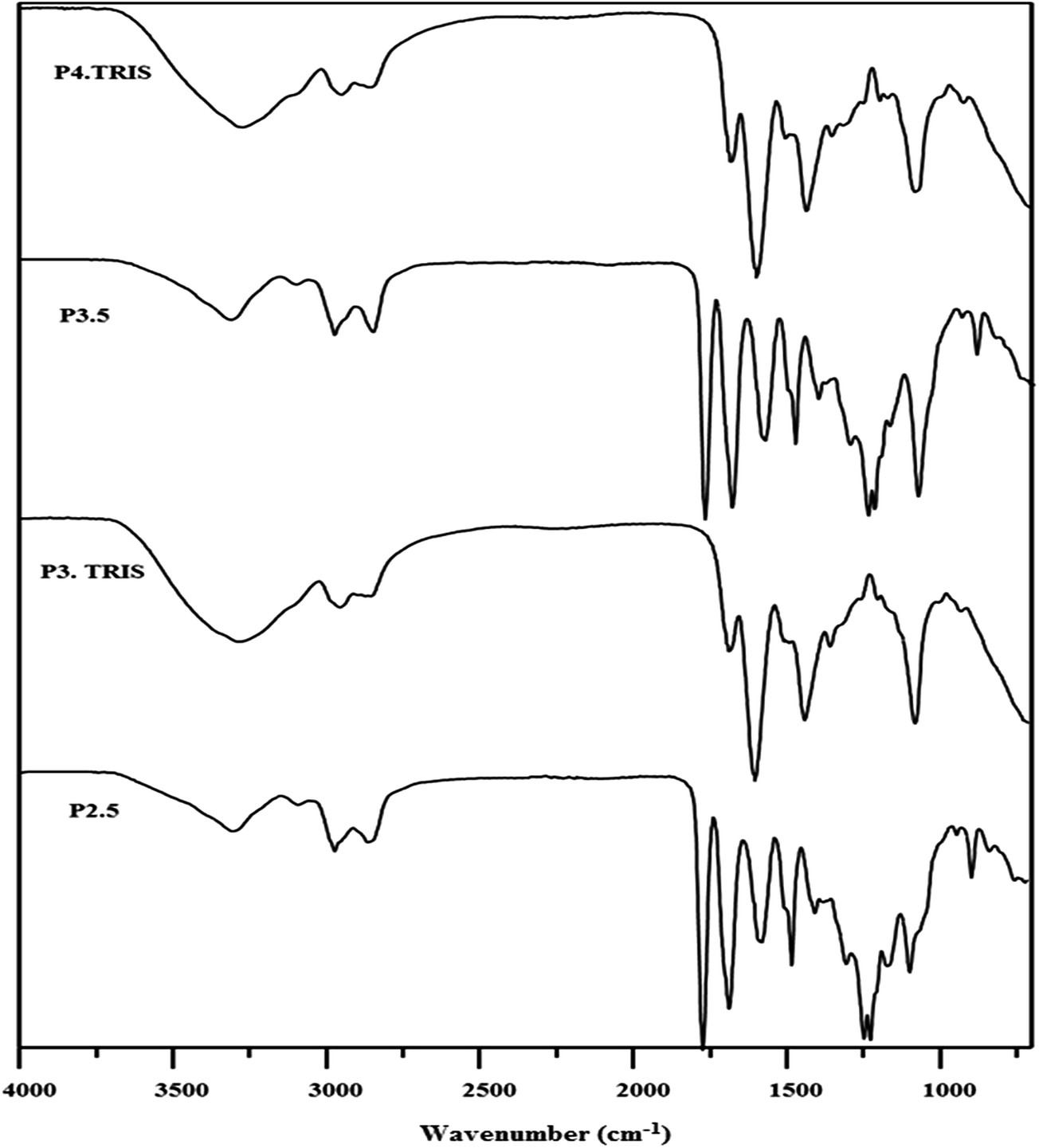 | ||
| Fig. 1 A representative ATR (IR) spectra of pure ester (Cn.5) and TRIS-terminated (Cn.TRIS) for Jeffamine® T-403 cored PAMAM dendrimers. | ||
| Product | Precursor | Precursor g (mmol) | TRIS g (mmol) | K2CO3 g (mmol) | MeOH (mL) | MW (watt) | Time (min) | Yielda (%) |
|---|---|---|---|---|---|---|---|---|
| a Isolated percent yield after LPR purification. | ||||||||
| 7 | 1 | 1.72 (0.61) | 1.43 (11.77) | 2.03 (14.71) | 10 | 200 | 120 | 96 |
| 8 | 2 | 0.98 (0.27) | 0.80 (6.64) | 1.15 (8.29) | 15 | 200 | 110 | 95 |
| 9 | 3 | 0.68 (0.15) | 0.53 (4.33) | 0.75 (5.42) | 10 | 200 | 120 | 91 |
| 10 | 4 | 0.99 (0.16) | 0.76 (6.29) | 1.08 (7.85) | 10 | 200 | 140 | 93 |
| 11 | 5 | 1.12 (0.14) | 0.81 (6.70) | 1.16 (8.40) | 13 | 200 | 125 | 94 |
| 12 | 6 | 0.84 (0.09) | 0.62 (5.16) | 0.89 (6.46) | 10 | 200 | 135 | 93 |
The synthesized PAMAM–TRIS dendrimers (7–12) could be easily characterized via 1H NMR and 13C NMR. All the expected signals are at the correct intensity and position (Fig. 2 and 3). The resonances from the methyl ester of 6 at 3.59 ppm are no longer visible and confirm the complete conversion of the ester functional groups to TRIS groups with a new singlet at 3.63 ppm (Fig. 2). This singlet resulting from the resonances of the new methylene protons adjacent to the terminal hydroxyl groups also indicates the full conversion to 12. Moreover, the 13C NMR spectrum of 12 shows the right number of carbon signals. The signals at 181.09 from exterior amides, and 175.5, 174.65 ppm from interior amides correspond to TRIS, and the interior amides prove the complete conversion of 6 to 12 (Fig. 3). Likewise to the 1H NMR, the strong resonance corresponding to the methyl groups of the terminal methyl group at 172.43 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 51.15 (COOCH3) ppm in the 13C NMR is no longer present (Fig. 3). This is also an indication of good purity. In the 13C NMR of TRIS terminated conversions, the formation of 56.38 (NHCR3), 63.6 ppm (CH2OH) bands indicated the full formation of TRIS-terminated PAMAM dendrimers. Therefore, 1H NMR and 13C NMR spectroscopy evaluations prove good purity.
O), 51.15 (COOCH3) ppm in the 13C NMR is no longer present (Fig. 3). This is also an indication of good purity. In the 13C NMR of TRIS terminated conversions, the formation of 56.38 (NHCR3), 63.6 ppm (CH2OH) bands indicated the full formation of TRIS-terminated PAMAM dendrimers. Therefore, 1H NMR and 13C NMR spectroscopy evaluations prove good purity.
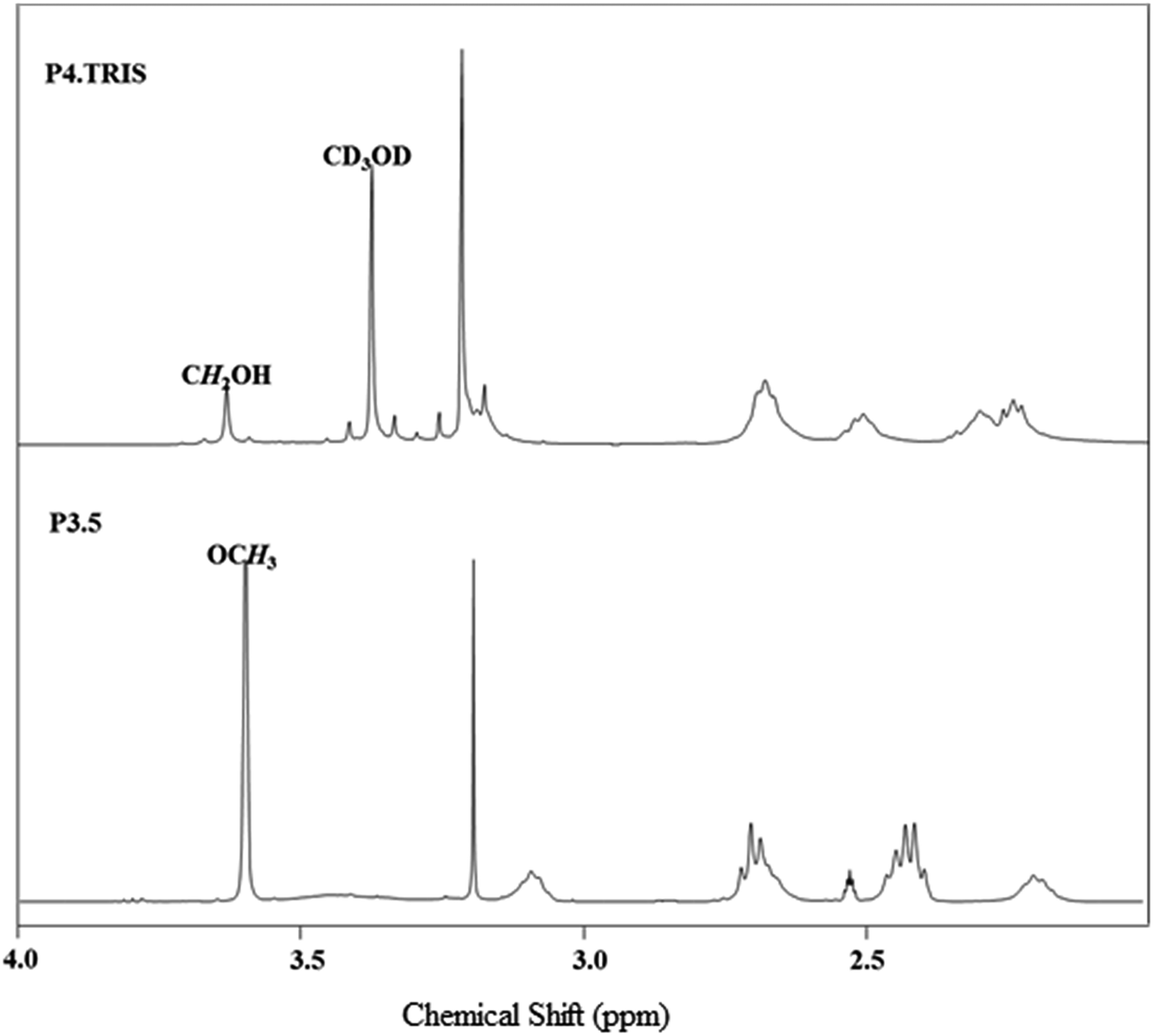 | ||
| Fig. 2 1H NMR spectrum monitoring of the conversion of 6 (bottom, in DMSO-d6) to 12 (top, in CD3OD). | ||
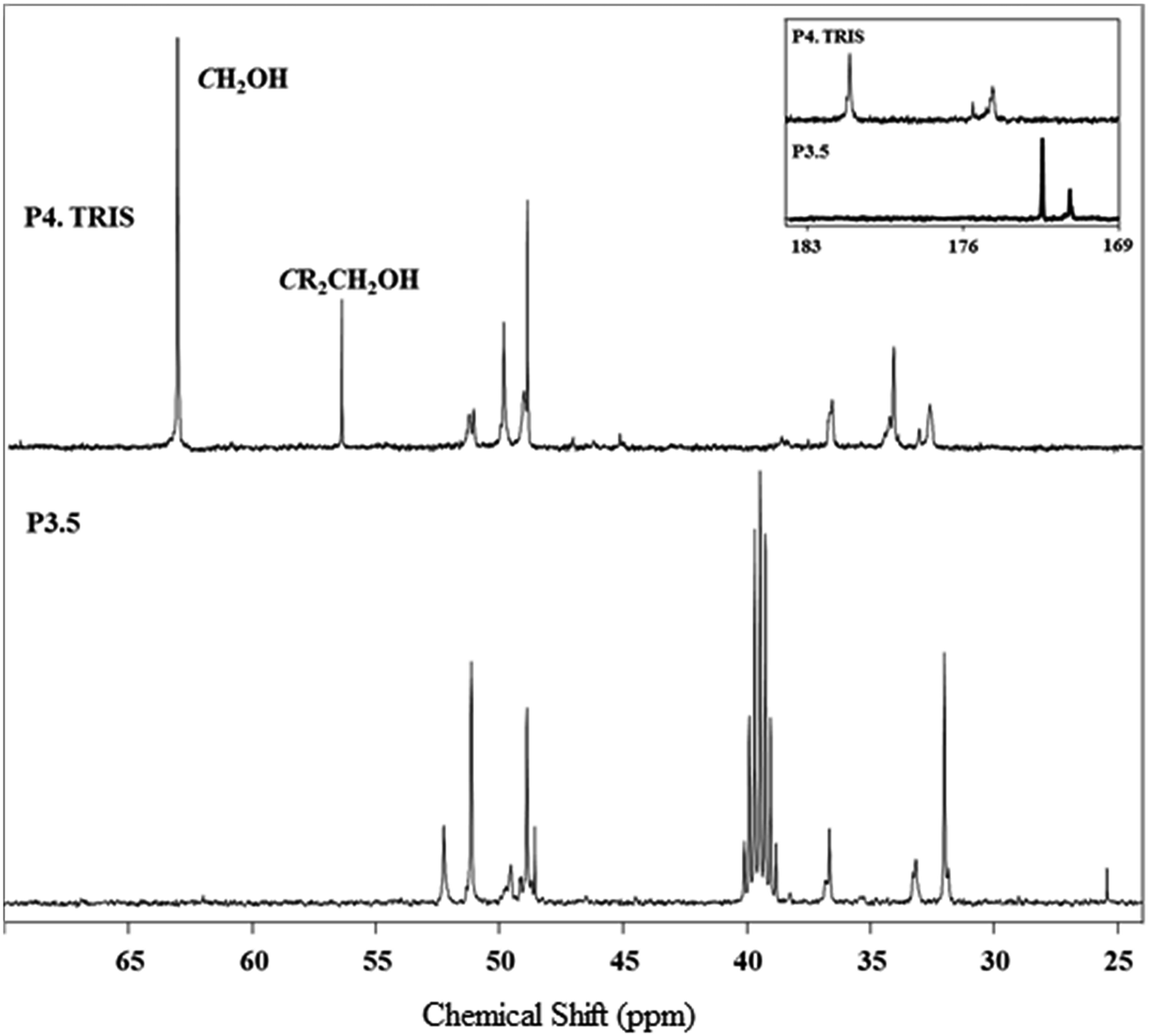 | ||
| Fig. 3 13C NMR spectrum monitoring of the conversion of 6 (bottom, in DMSO-d6) to 12 (top, in CD3OD). | ||
Comparison of microwave-assisted method (Method B) with conventional method (Method A)
Surface modification of 3 with TRIS to synthesize 9 was selected as a model representative reaction for the conversion of PAMAM–OCH3 dendrimers to water-soluble PAMAM dendrimers with terminal hydrophilic hydroxyls. This conversion reaction was compared by Method A (preheated oil bath) and Method B (CEM discovery Labmade-Open Vessel mode). Conversions were determined with ATR (IR) monitoring of the disappearance of ester 1730 cm−1 and formation of ∼1635 cm−1 and ∼1554 cm−1 amide I and amide II peaks, and ∼3260 cm−1 broad hydroxyl peaks by taking identical aliquots at specific time intervals (Fig. 4). The conversions achieved by Method B were after 30 min (91%), 60 min (93%), 120 min (100%) while they were 60 min (56%), 120 min (61%), 180 min (80%), 240 min (92%), 300 min (97%), 360 min (99%) and 420 min (100%) by Method A (Fig. 5). Comparison of the observed conversion values revealed that Method B is 3 to 4 times faster than Method A when compared under same reaction conditions (Table 3). Microwave reactions were performed under a series of different power and time trial experiments by refluxing at 70–90 °C bulk temperature using methanol as solvent. The optimum conditions were summarized comparative to conventional synthesis in Table 3. As can be seen from Table 3, syntheses of PAMAM–TRIS dendrimers (7–12) were accomplished not only in shorter reaction times but also with higher product yields of 91–96%. Compounds 8–12 are novel water-soluble PAMAM–TRIS dendrimers with hydroxyl end groups. We have increased the product yield of compound 7 from 54% (96 h),12 83% (72 h)26 to 96% (120 min). In fact, we have observed that the reaction time required for 7 under conventional methods is shortened by up to 36–38 times. It is also important to state in terms of green chemistry that this is the first report where methanol is the solvent (Table 2).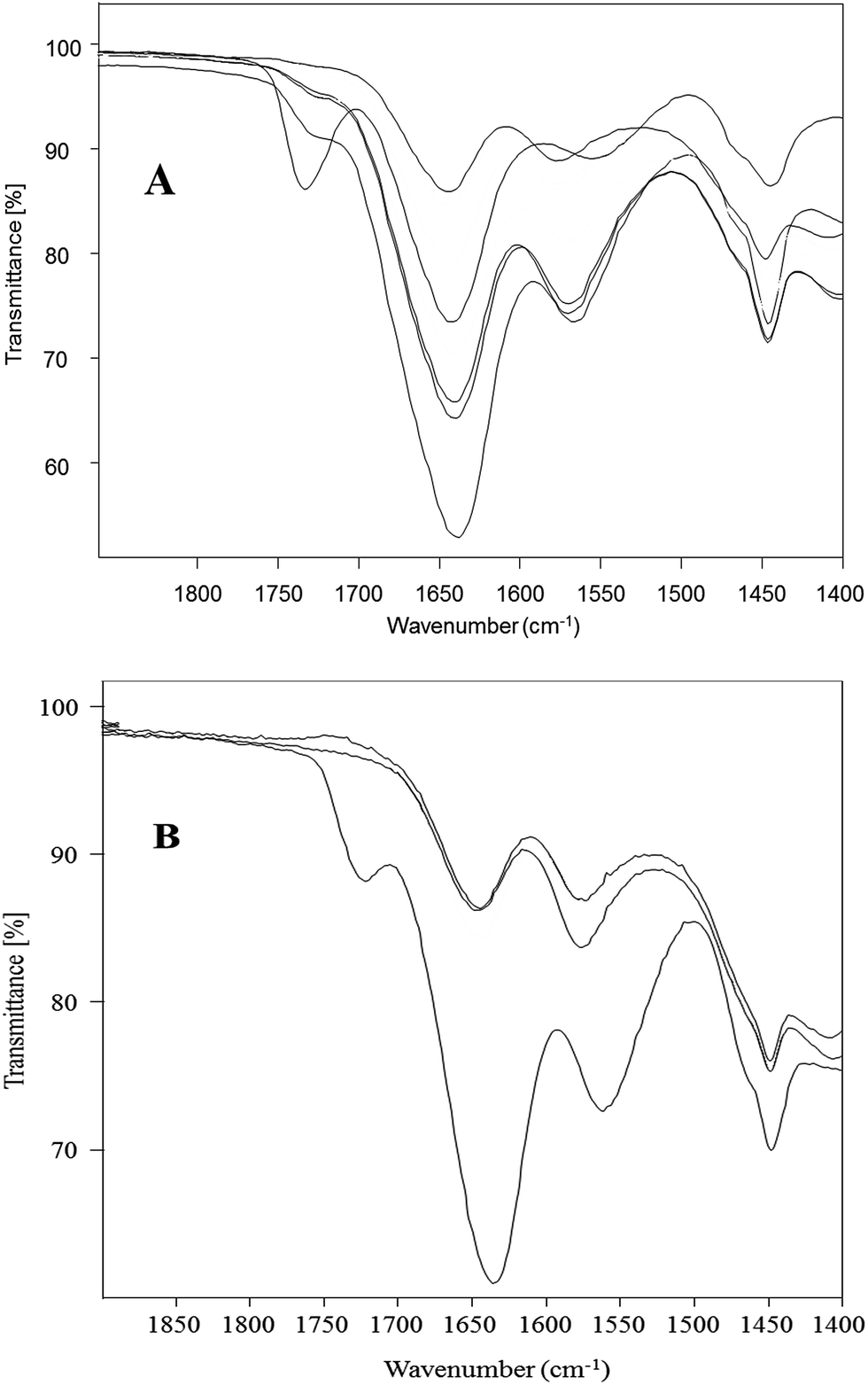 | ||
| Fig. 4 ATR (IR) monitored conversion spectrum from 3 to 9. (A) Method A conditions: oil bath, reflux, 7.5 h. 3 to TRIS and K2CO3 molar ratio 1:1.20 and 1:1.50 respectively (B) Method B conditions: open vessel, 200 W, reflux, 120 min, 3 to TRIS and K2CO3 molar ratio 1:1.20 and 1:1.50 respectively. | ||
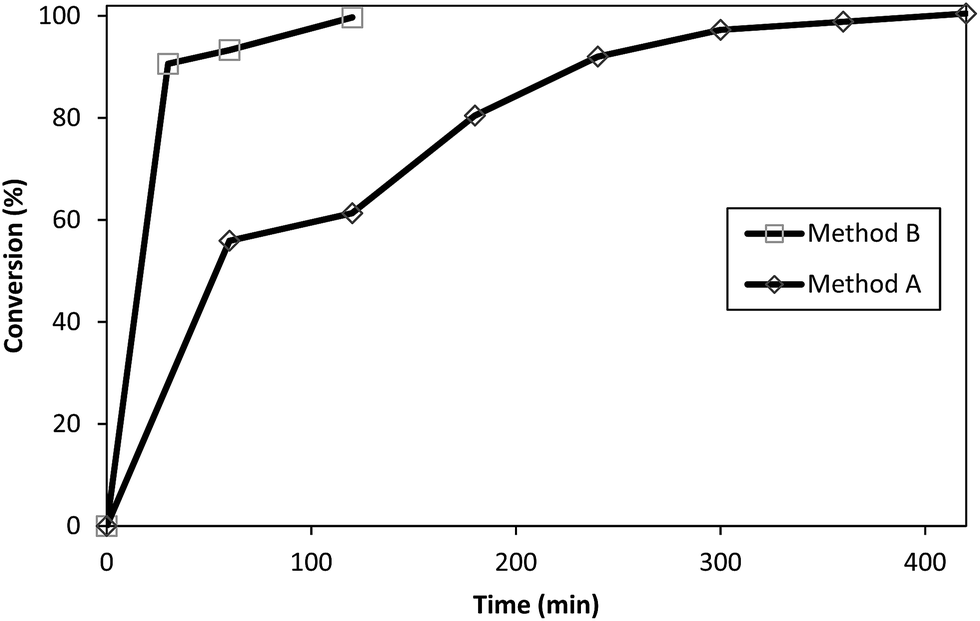 | ||
| Fig. 5 Conversion of 3 to 9. Reaction time: 420 min for Method A in oil bath, and 120 min for Method B. MW open vessel mode at 200 W (conversion (%) was calculated from the ATR (IR) spectra in Fig. 4). | ||
| Core (Cn.5) | Product (Cn.TRIS) | Method A (conv.) | Method B (MW) | Literature | |||
|---|---|---|---|---|---|---|---|
| Time (h) | Yield (%) | Time (min) | Yield (%) | Time (days) | Yield (%) | ||
| a Data taken from ref. 26. Reaction was carried out at 40 °C in DMSO under conventional conditions.b Data taken from ref. 12. Reaction was carried out at 50 °C in DMSO under conventional conditions. | |||||||
| 1 | 7 | 7.5 | 95 | 120 | 96 | 4a, 3b | 83a, 54b |
| 2 | 8 | 7.5 | 95 | 110 | 95 | — | — |
| 3 | 9 | 7.5 | 90 | 120 | 91 | — | — |
| 4 | 10 | 8 | 93 | 140 | 93 | — | — |
| 5 | 11 | 8.5 | 94 | 125 | 94 | — | — |
| 6 | 12 | 8 | 92 | 135 | 93 | — | — |
Potentiometric titrations of PAMAM–TRIS dendrimers
All potentiometric titrations of PAMAM–TRIS dendrimers were carried out by using a TitroLine® 7000 autotitrator with a ultra precise IoLine glass electrode with an iodine/iodide reference system. Titrisoft® 2.73 software was used to manage the titration processes over a personal computer. Secure mode was used to collect pH data reducing the pH drift error to a minimum on the pH reading error of 0.002 and volume reading error of 0.001 mL. Hence, it was further verified that all titration curves were fully reversible and independent of the dendrimer concentration within experimental error, indicating that dendrimer–dendrimer interactions are negligible.Potentiometric acid–base titrations allow us to determine the average number of primary and tertiary amine groups of PAMAM–TRIS dendrimers. The initial pH of the aqueous PAMAM–TRIS dendrimer solutions was in the pH range of 9.7–10.0. According to back titration procedure, the initial pH of the solutions was adjusted to pH ∼ 2.0 and back titrated with standardized NaOH. In potentiometric titrations, two distinctive end points were observed (Fig. 6). One of these end points was for back titration of excess acid added to initial dendrimer solution while the second one was for the total number of mmoles of the observed tertiary amine groups. Sample potentiometric titration curves of 10–12 can be seen in Fig. 6. Second derivatives of the potentiometric titration curves were overlapped to show the end points clearly. These points are accepted as the inflection points. Diallo et al.27 accept these inflection points to be the pKa values of TRIS ended dendrimers. By using the same approach, pKa values of the tertiary amine groups of PAMAM–TRIS dendrimers 7–12 are presented in Table 4. It can be seen that the pKa values increase as the basicity of PAMAM–TRIS dendrimers increase in aqueous solution with an increasing number of surface hydroxyls (Tables 1 and 4).
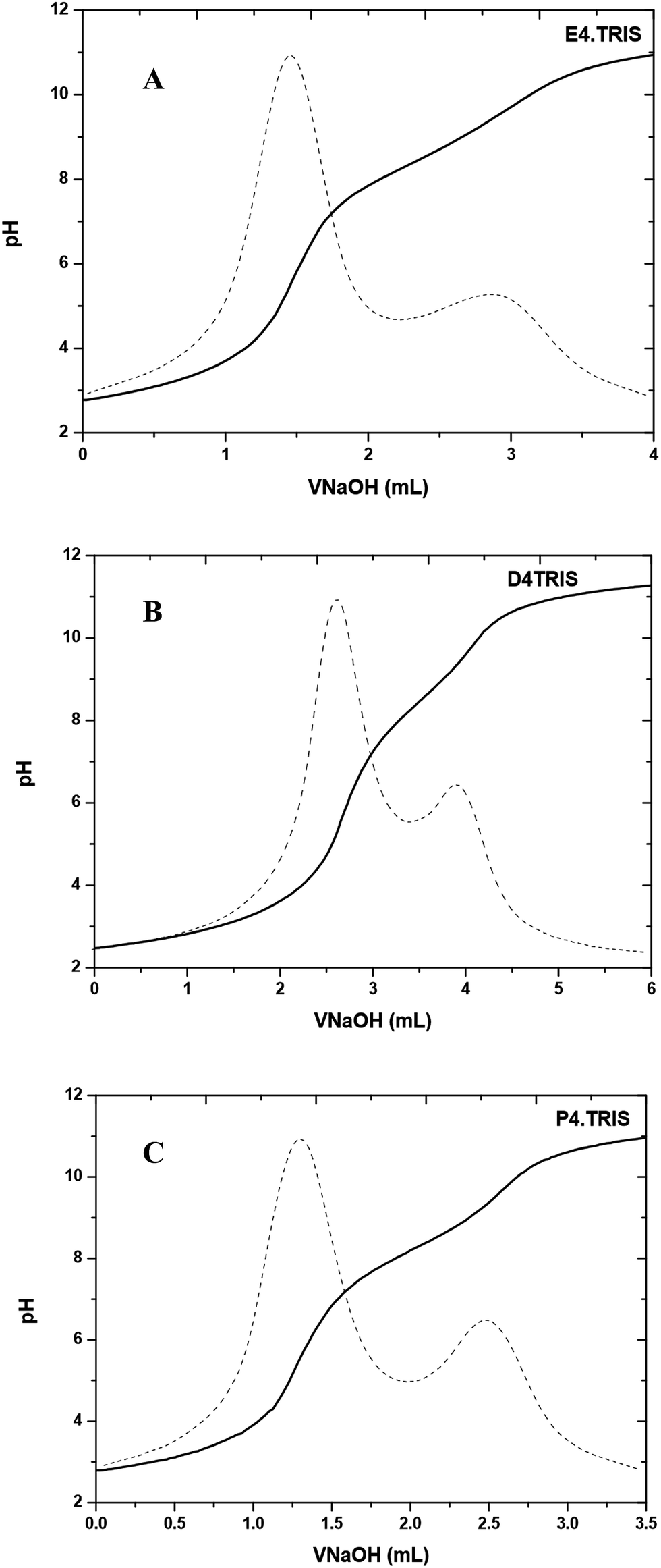 | ||
| Fig. 6 Potentiometric pH titration curve for (A) E4.TRIS (10), (B) D4.TRIS (11), and (C) P4.TRIS (12) at 100 mM ionic strength. | ||
PAMAM–TRIS dendrimers (7–12) have only tertiary amine groups (Table 1). For this reason, the number of experimentally observed tertiary amine groups were used as support to explain the potentiometric titration results for PAMAM–TRIS dendrimers. The difference between theoretical and experimental tertiary amine numbers gives us the information about the structural monodispersity. As can be seen from Fig. 6, only tertiary amine groups can be observed from potentiometric studies of PAMAM–TRIS dendrimers 10–12. Calculated numbers of tertiary amines (3N) present in PAMAM–TRIS dendrimers (7–12) are presented in Table 5. The results revealed that there exists a good correlation between the theoretical and practical numbers of tertiary amines in PAMAM–TRIS dendrimers (7–12). These results are also important to show that the synthesized dendrimers are almost pure and have ideal monodispersity and characteristics.
| PAMAM–TRIS dendrimer | 3N theoretical value | 3N practical value | % Correlation |
|---|---|---|---|
| a Results were calculated from potentiometric titrations for five repeated experiments. | |||
| 7 | 14 | 13.67 ± 0.32 | 97.64 |
| 8 | 18 | 17.80 ± 0.64 | 98.88 |
| 9 | 21 | 20.74 ± 0.30 | 98.76 |
| 10 | 30 | 30.82 ± 1.08 | 102.73 |
| 11 | 38 | 37.07 ± 0.67 | 97.55 |
| 12 | 45 | 42.99 ± 1.57 | 95.53 |
Spectroscopic titrations of PAMAM–TRIS dendrimers
Spectroscopic titrations of PAMAM–TRIS dendrimers (7–12) in the pH range of 2–12 revealed a prominent absorption band at 280–286 nm (Fig. 7). In Fig. 7, it can be observed that the intensity of the absorption band at λmax 280–286 nm decreases as the pH decreases, indicating the protonation of tertiary amine groups.28 We have observed this band for all the spectroscopic titrations of PAMAM–TRIS dendrimers. Titrations were carried out between the pH 2–12 at a constant ionic strength in order to prevent the shielding effect of the amino groups of the dendrimers. Between this pH range, PAMAM–TRIS dendrimers solutions displayed a characteristic λmax absorption band between 280–286 nm. It could be easily seen that this band does not disappear even at low pH < 3. All the PAMAM–TRIS dendrimers (7–12) have different numbers of absorbing sites which are tertiary amine groups (Table 1). That is, depending on the pH of the media, PAMAM–TRIS dendrimers can be found in different protonated conformations (absorbing species).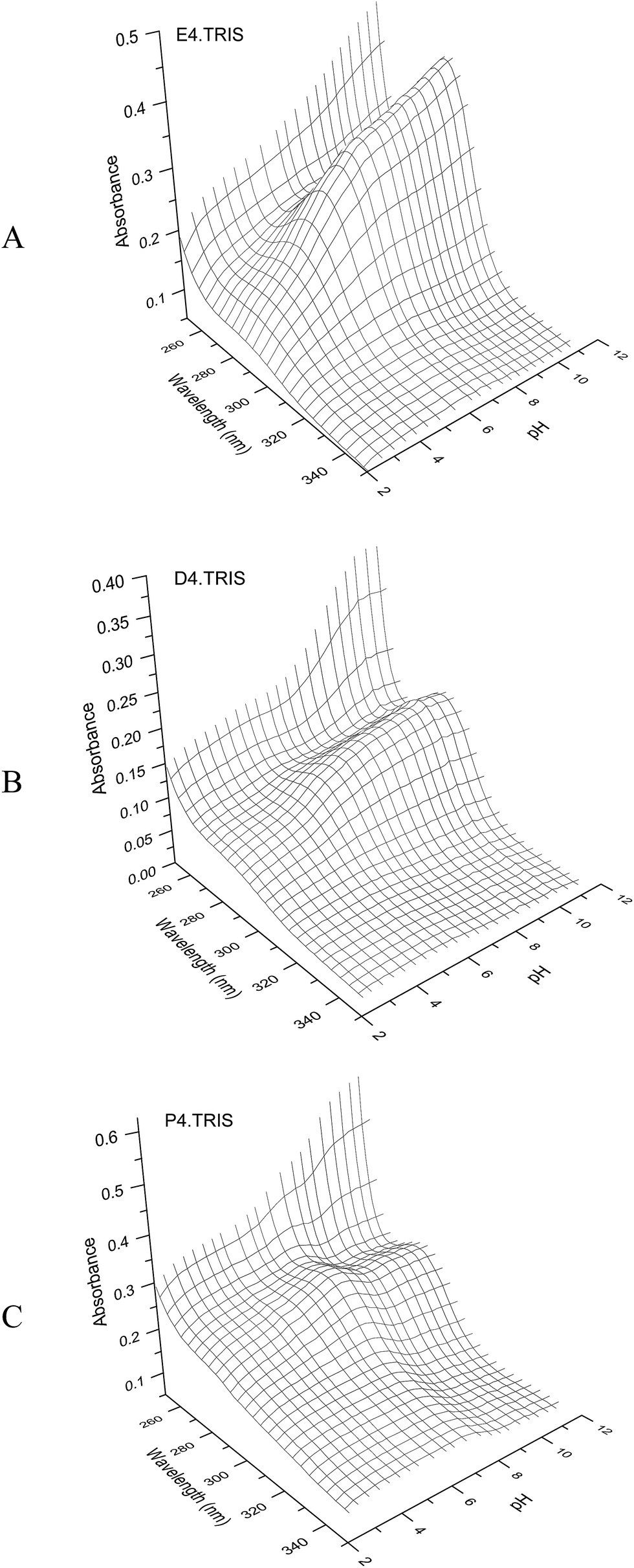 | ||
| Fig. 7 3D plots of UV spectroscopic titration of E4.TRIS (10) (7.08 × 10−4 M) (A), D4.TRIS (11) (3.70 × 10−4 M) (B), P4.TRIS (12) (6.06 × 10−4 M) (C) solution with 0.098 N HCl at 25 ± 0.1 °C, I = 100 mM NaCl. | ||
Synthesis and characterization of Cu(II)–PAMAM–TRIS intradendrimer complexes
:4 dendrimer to tertiary amine molar ratio.31
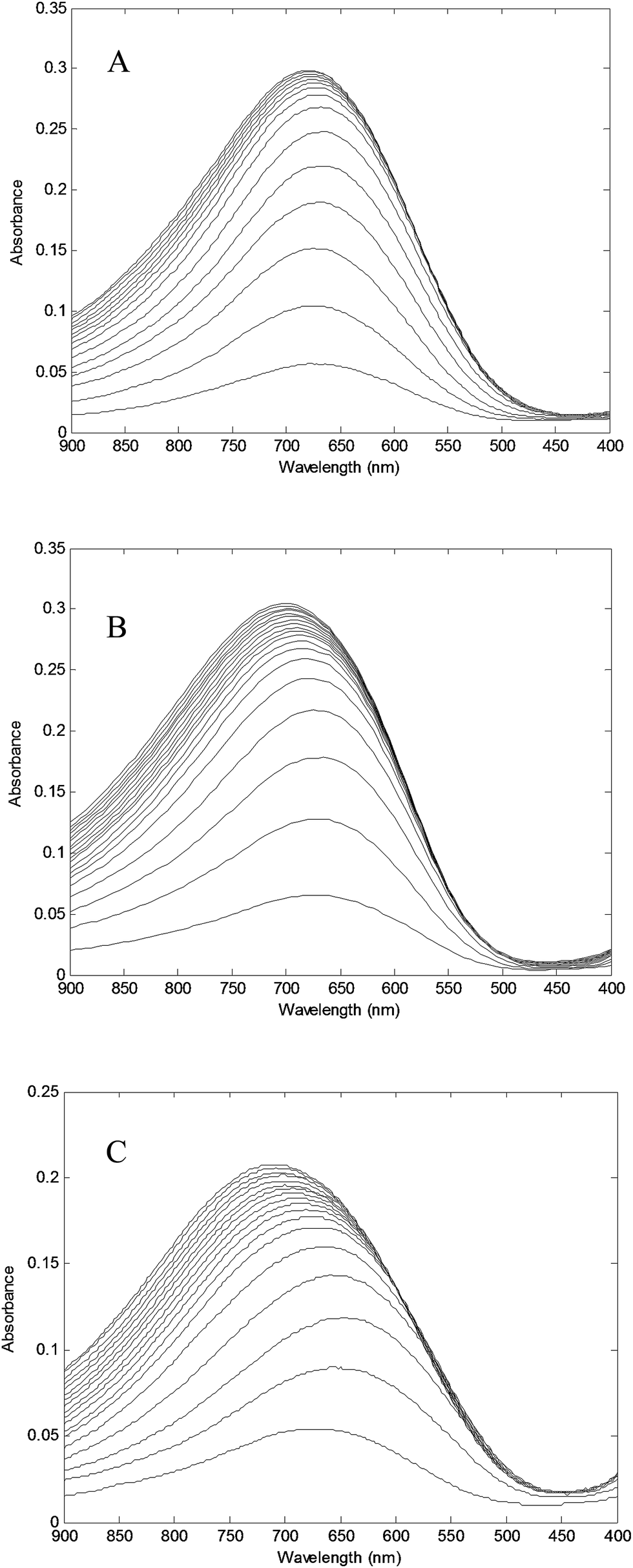 | ||
| Fig. 8 Absorption spectra of PAMAM–TRIS dendrimers at 680 nm (A) 10 (0.228 mM) solution titrated with Cu2+ (81.54 mM); (B) 11 (0.202 mM) solution titrated with Cu2+ (80.14 mM); (C) 12 (0.148 mM) solution titrated with Cu2+ (81.54 mM). | ||
Absorbance at λmax = 680 nm versus Cu2+/PAMAM–TRIS dendrimer molar ratio plots were used to determine experimental end points at where PAMAM–TRIS dendrimers (7–12) could bind the maximum number of Cu2+ ions. That is, the maximum molar excess of Cu2+ that can be loaded onto PAMAM–TRIS dendrimers was calculated from these plots (Fig. 9). The maximum molar excess of Cu2+ ions that PAMAM–TRIS dendrimers (10–12) can bind are shown on the spectroscopic titration curves (Fig. 9) and summarized in Table 6. The results revealed that the number of tertiary amine numbers observed from spectroscopic titration data were in good agreement with the calculated ones. It can be concluded that PAMAM–TRIS dendrimers (7–12) absorb the number of Cu2+ ions equivalent to the number of tertiary amines. This correlation also indicates that the structure of PAMAM–TRIS dendrimers is at the desired monodispersity and highly pure. Thus, it could be also concluded that each Cu2+ ion is coordinated by four tertiary amine groups.
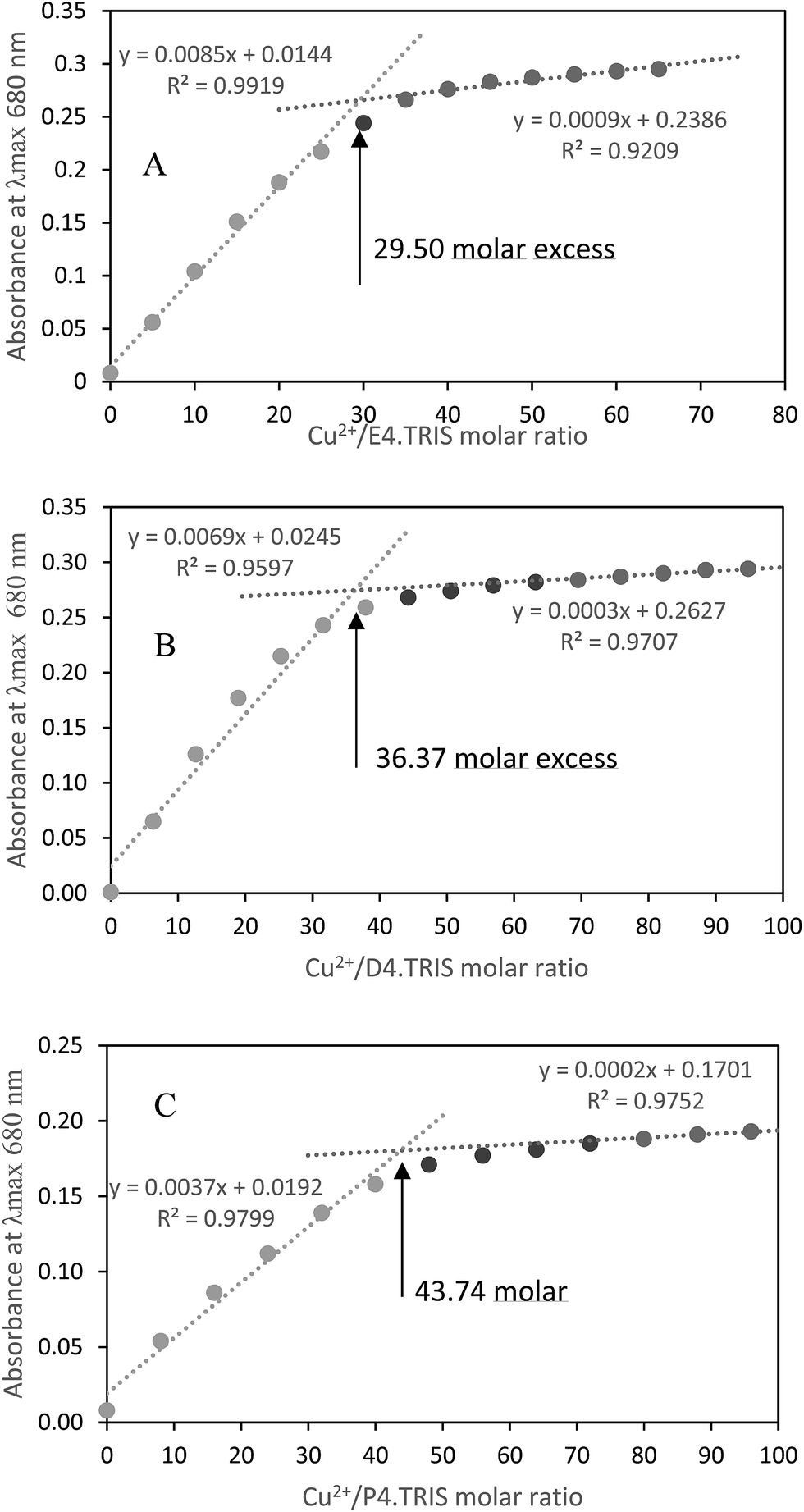 | ||
| Fig. 9 Spectroscopic titration curve of PAMAM–TRIS dendrimers (A) 10; (B) 11; (C) 12 with Cu2+ ions. | ||
Transition d–d complex bands resulting from the coordination of internal amine groups of ethanol amine terminated PAMAM dendrimers were reported at λmax at 605 nm.30 This band was reported in the range of 600–800 nm depending on surface modification with TRIS31 and could not be observed at low concentrations. Upon the addition of the appropriate calculated molar ratio of CuSO4 solutions to PAMAM–TRIS dendrimer solutions, a strong band at around 270–280 nm for Cu(II)–PAMAM–TRIS dendrimers (7–12) complexes occurred. This band is assigned to ligand to metal charge transfer (LMCT) bands.32–34 In addition, a d–d copper transition band at around 680 nm for all Cu(II)–PAMAM–TRIS complex solutions were observed. Sample spectra of PAMAM–TRIS dendrimers (10–12) evidencing the complexation by color and UV-Vis spectra change can be seen in Fig. 10.
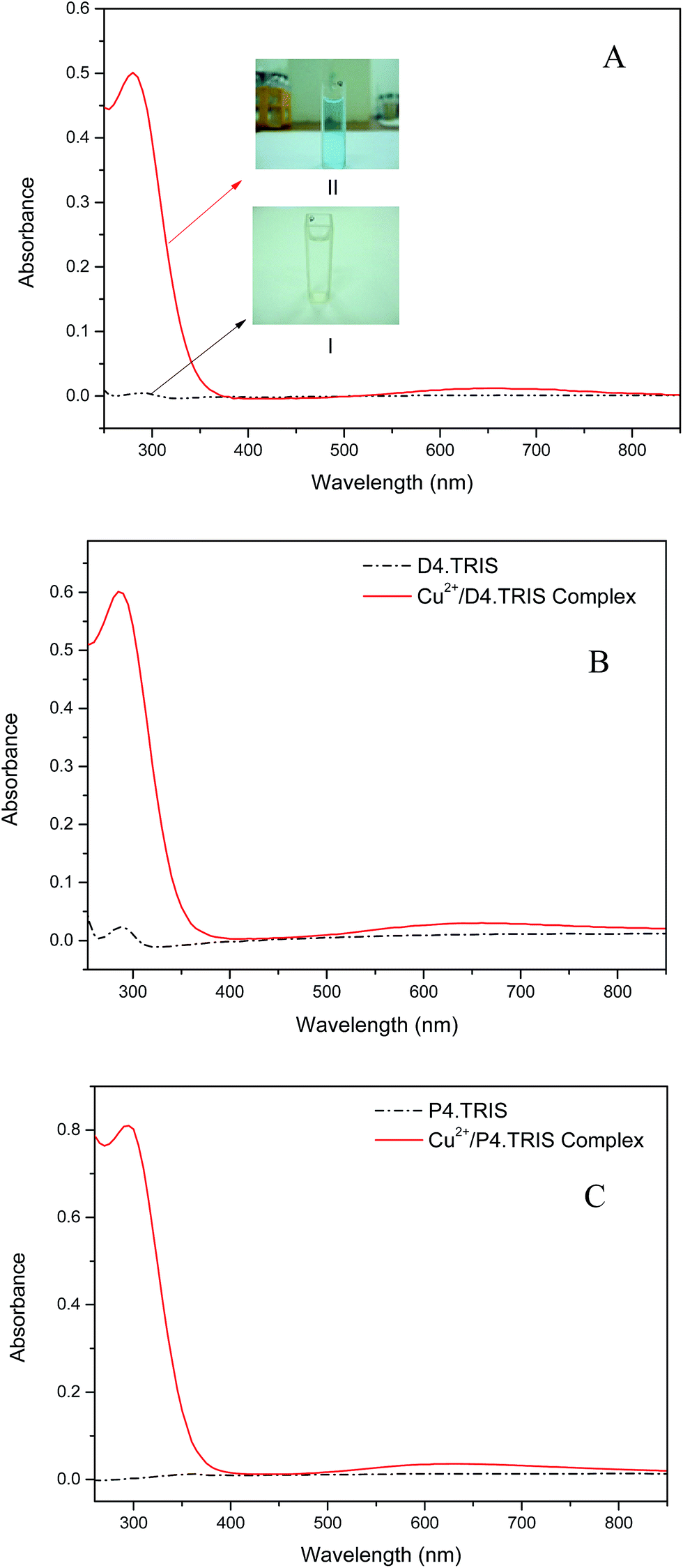 | ||
| Fig. 10 Change in color and UV-Vis absorption spectra during the complexation of Cu2+–E4.TRIS: (I) aqueous solution of E4.TRIS, (II) Cu2+ dendrimer complex solution (A); Cu2+–D4.TRIS (B); Cu2+–P4.TRIS (C). Color change was observed similarly in all complexations. | ||
Conclusion
This article has reported the surface modification of PAMAM–OCH3 dendrimers with TRIS. PAMAM–TRIS dendrimers were synthesized by both conventional and microwave methods and the best reaction conditions were optimized. The newly developed microwave method (Method B) allows the synthesis of PAMAM–TRIS dendrimers in 110–140 minutes rather than three to four days15,25 as an alternative to Method A.With the use of new developed MAS method, the fast, efficient, easy, and one-pot synthesis of six water-soluble PAMAM–TRIS dendrimers, five or which are novel, with different cores of ethylenediamine, diethylenetriamine and Jeffamine® T-403 were performed. The other PAMAM–OCH3 dendrimers (2, 5), which are precursors for PAMAM–TRIS dendrimers (8, 11) are novel molecules. Furthermore, the synthesized molecules are water-soluble except for PAMAM–OCH3 dendrimers (1–6). The synthesized water-soluble PAMAM–TRIS dendrimers (7–12) were purified by the LPR technique and characterized by 1H-NMR, 13C-NMR, ATR (IR), EA, UV-Vis spectroscopy, potentiometric and spectroscopic titrations. Spectroscopic titration studies of the PAMAM–TRIS dendrimers with Cu2+ ions revealed that all of the PAMAM–TRIS dendrimers are of good purity and in high yield. As a result, they could be used as possible potential drug carriers and templates for the synthesis of dendrimer-encapsulated metal nanoparticles as catalysts in future studies at the desired purity and monodispersity.
Experimental
Materials
Jeffamine® T-403 Mn 440 was purchased from Aldrich. Methyl acrylate, ethylenediamine, diethylenetriamine, methanol, n-butanol, tris(hydroxymethyl)aminomethane were purchased from Merck. NaOH, 37% HCl, NaH2PO4, NaCl, KHP, NaBH4, were supplied from Merck. All solutions were prepared using 18.2 MΩ Millipore Milli-Q deionized water. NaOH solutions were used as titrant after being standardized with primary grade KHP. Standardized HCl against KHP was used as excess acid in order to adjust initial pH of dendrimer solutions. pH 4.0, 7.0, 11.0 buffer solutions for the calibration were supplied from Merck. Dendrimer solutions were stored at 4 °C. Unless otherwise stated all chemicals were analytical grade and used without further purification. Liquid-phase polymer-based retention (LPR) ultrafiltration membranes, Amicon 8000 Stirred Cell and dialysis membranes having a molecular cut of size (MWCO) 1–3 kDa were supplied from Millipore.Instrumentation and software
A CEM Focused Microwave™ synthesis system, model, Discover (CEM Corporation, North Carolina, USA) with a continuous microwave power output from 0–300 watts (±30 watts) programmable in 1 watt increments, infrared temperature control system programmable from 25–250 °C, and 5–125 mL vessel capacity, was used as microwave reactor.The IR spectra (4000–400 cm−1, resolution 4 cm−1) were recorded with a Perkin-Elmer Spectrum One (Serial no.: C68739) in ATR. The NMR spectra were recorded on a Bruker Avance 500 MHz Spectrometer. A Thermo Scientific Flash EA 2000 Series (Organic Elemental Analyzer) CHN/S was used for the determination of the main organics. The UV-Vis absorbance spectra were obtained using a PG T 70 Spectrometer (PG Instruments, England) and a quartz cuvette having an optical path length of 1.00 cm.
Potentiometric titrations were carried out automatically by using a TitroLine® 7000 (SI Analytics GmbH, Hattenbergstraβe, Germany) autotitrator and thermostated titration vessel under nitrogen media. The temperature was kept at room temperature (25 ± 0.1 °C) using a Polyscience® digital temperature controller circulating bath (Polyscience, Illinois, USA). The titrator was controlled by a personal computer with Schott Instruments, Titrisoft® 2.73 software. pH data were collected with an IoLine ultra precise glass electrode with iodine/iodide reference system. The glass electrode was calibrated with Merck pH 4.0, 7.0, 11.0 buffer solutions.
Spectroscopic titrations were carried out automatically by using TitroLine® 7000 autotitrator equipped with thermostated titration vessel under nitrogen media and PG TG 70 UV-Vis spectrophotometer equipped with UVWin5 Software v5.0.5, together.
General procedure for the synthesis of PAMAM–OCH3 dendrimers (1–6)
PAMAM–OCH3 dendrimers (1–6) were synthesized by following the procedure reported in our recent study17 and briefly summarized hereafter. This method involves alkylation and amidation steps. In the alkylation step, excess methyl acrylate (2.5 M eq. per terminal amine) was added to a methanolic solution of dendrimer core (C) (ethylenediamine (E), diethylenetriamine (D) and Jeffamine® T-403 (P)) or full generation PAMAM dendrimers (Cn). The reaction mixture was stirred for 24 h at room temperature and excess reagents and solvents were removed under vacuum at 65 °C bath temperature and purified by means of LPR. The resulting PAMAM–OCH3 dendrimers (Cn.5) E0.5, D0.5 and P0.5 were colorless oils.In the amidations, excess E (10 M eq. of E per ester branched half generation) was added to the stirred methanolic solution of PAMAM–OCH3 dendrimers (Cn.5). The final mixture was irradiated with MW at 200 W for 60 min. Final traces of E were firstly removed under vacuum below a bath temperature of 65 °C by using 50 mL of n-butanol as hydrogen competitive reagent three times. The resulting product was purified by means of LPR. The final methanolic solution of retained product was removed under vacuum below a bath temperature of 65 °C. The final products were full generation amine terminated PAMAM dendrimers (PAMAM–NH2 dendrimers) (Cn) E1, D1, and P1. By repeating the above cycle, E2.5, E3.5, D2.5, D3.5, P2.5 and P3.5 were synthesized. Yields were in the range of 92–96% (Table 7).
| PAMAM–OCH3 dendrimers (Cn.5) | R-aminea (Cn) g (mmol) | MA g (mmol) | MeOH (mL) | Time (h) | Yield (%) |
|---|---|---|---|---|---|
| a R-amine refers to full generation precursor.17 | |||||
| E2.5 (1) | 15.64 (10.93) | 18.82 (218) | 40 | 24 | 95 |
| E3.5 (2) | 14.60 (4.48) | 15.43 (179) | 40 | 24 | 93 |
| D2.5 (3) | 6.76 (3.72) | 8.02 (93) | 40 | 24 | 96 |
| D3.5 (4) | 8.12 (1.98) | 8.53 (100) | 40 | 24 | 95 |
| P2.5 (5) | 13.48 (5.40) | 13.95 (162) | 40 | 24 | 96 |
| P3.5 (6) | 12.00 (2.29) | 11.84 (138) | 40 | 24 | 92 |
General procedure for the conventional synthesis of PAMAM–TRIS dendrimers (7–12) with Method A
A methanolic solution of PAMAM–OCH3 dendrimer (C2.5, C3.5) was added to a stirred suspension of TRIS (1.2 M equiv. per terminal ester) and anhydrous potassium carbonate (1.5 M equiv. per terminal ester) in 10–15 mL of MeOH. The resulting mixture was vigorously stirred at 90 °C in a preheated oil bath for 7–8 hours. The final reaction mixture was filtered to remove excess potassium carbonate and the filtrate collected. Then the product was purified with LPR by dialyzing against 50% aqueous methanol solution under 15 psi nitrogen (N2) gas pressure for 24 hours by using Millipore ultrafiltration disks having a molecular cut of size 1 kDa. The final methanolic solution of retained product (Cn.TRIS) was concentrated under vacuum below a bath temperature of 65 °C. Yields were in range of 90–95%.General procedure for the microwave-assisted synthesis of PAMAM–TRIS dendrimers (7–12) with Method B
A methanolic solution of PAMAM–OCH3 dendrimer (C2.5, C3.5) was added to a stirred suspension of TRIS (1.2 M equiv. per terminal ester) and anhydrous potassium carbonate (1.5 M equiv. per terminal ester) in 10–15 mL of MeOH. The resulting mixture was irradiated at 200 W for 110–140 min by refluxing at a bulk temperature of 70–90 °C. The final reaction mixture was filtered to remove excess solid reagents, and the filtrate collected. Then product was purified with the LPR method. In other words, it was continuously dialyzed with 50% aqueous methanol solution under 15 psi nitrogen (N2) gas pressure for 24 hours by using Millipore ultrafiltration disks having a molecular cut of size 1 kDa. The final methanolic solution of retained product (Cn.TRIS) was concentrated under vacuum below a bath temperature of 65 °C. Yields were in the range of 93–96%.LPR experiments
The appropriate Millipore ultrafiltration membrane disk was equipped with an Amicon 8000 stirred cell was used for the LPR method. A (1:1) MeOH:methanolic aqueous solution of crude product was transferred into the cell. Depending on the expected size of the product, membrane disks were selected in the range of MWCO 1–3 kDA. The solution was diluted to 200 mL inside the cell. Methanol–water mixture was used as the feeding solvent. Continuous dialysis was performed under 15 psi nitrogen pressure for 24 hours. Finally, methanol water mixture was evaporated under vacuum.
Potentiometric titrations of PAMAM–TRIS dendrimers
Acid base titrations of PAMAM–TRIS dendrimers were conducted according to literature.35,36 Analytical grade NaCl was added to a precisely weighed portion of PAMAM–TRIS dendrimer solution to prevent shielding of amine groups from each other because of interactions, and keep the ionic strength (I) constant at 100 mM. Titrations were carried out automatically by using an autotitrator and thermostated titration vessel under continuous N2 stream. The temperature was kept at room temperature (25 ± 0.1 °C). PAMAM–TRIS dendrimers (18–22 mg) were dissolved in 20 mL of 0.10 M NaCl solution to give a final concentration of 0.9–1.1 mg mL−1 the PAMAM–TRIS dendrimers solutions were titrated with standard HCl solution and back titrated with standard NaOH solution, respectively (Table 8).| PAMAM–TRIS dendrimer (Cn.TRIS) | Total weight (mg) | pH range | Initial volume (mL) | Excess acid (mL) | HCl (mol L−1) | NaOH (mol L−1) | Data points |
|---|---|---|---|---|---|---|---|
| a 25 μL increments, error in reading is 1 μL, I = 100 mm at 25 ± 0.1 °C, error in pH reading is 0.002. | |||||||
| E3.TRIS (7) | 19.53 | 2.78–11.00 | 20.0 | 1.325 | 0.098 | 0.050 | 148 |
| D3.TRIS (8) | 19.76 | 2.47–11.20 | 20.0 | 1.125 | 0.098 | 0.050 | 248 |
| P3.TRIS (9) | 18.46 | 2.81–11.00 | 20.0 | 1.425 | 0.099 | 0.048 | 152 |
| E4.TRIS (10) | 21.00 | 2.77–11.00 | 20.0 | 1.700 | 0.098 | 0.050 | 167 |
| D4.TRIS (11) | 19.76 | 2.47–11.30 | 20.0 | 1.950 | 0.098 | 0.050 | 246 |
| P4.TRIS (12) | 21.10 | 2.79–11.00 | 20.0 | 1.300 | 0.099 | 0.048 | 146 |
Spectroscopic titrations of PAMAM–TRIS dendrimers
All of the UV-Vis measurements were taken between the wavelength ranges of 250–350 nm with 10 mm quartz UV cells. Precision of titration was increased ± 0.01 pH unit by using a TitroLine® 7000 (SI Analytics GmbH, Hattenbergstraβe, Germany) autotitrator supported with Titrisoft® 2.73 software. After each pH adjustment, dendrimer solution was transferred into the cuvette and the absorption spectrum is recorded. The pH meter was calibrated with at least three buffer solutions at pH 4.0, 7.0 and 10.0 before each experiment. Ionic strength was maintained by adding an appropriate amount of 0.10 M NaCl.Spectroscopic titrations of PAMAM–TRIS dendrimers with Cu2+ ions and synthesis of Cu(II)–PAMAM–TRIS complexes
E, D and P cored PAMAM–TRIS dendrimers have many metal binding sites. In order to determine the maximum metal loading capacity of each dendrimer, the number of metal ions that can be coordinated with the tertiary amine groups of PAMAM–TRIS dendrimers was determined by spectroscopic Cu2+ titrations. Spectroscopic titrations of PAMAM–TRIS dendrimers with Cu2+ ions and the synthesis of Cu(II)–PAMAM–TRIS dendrimer complexes were adapted from the literature.29,30 Before each titration, the pH of the aqueous unbuffered PAMAM–TRIS dendrimers solution was adjusted to pH ∼ 8.0 and then unbuffered CuSO4 (pH ∼ 4.6) was used to titrate dendrimer solutions (Table 9).| PAMAM–TRIS dendrimer (Cn.TRIS) | Dendrimer conc. (mM) | Conc. of CuSO4 (mM) | CuSO4 (mM) increment (μL) |
|---|---|---|---|
| E3.TRIS (7) | 0.441 | 80.14 | 43.00 |
| D3.TRIS (8) | 0.351 | 93.12 | 20.00 |
| P3.TRIS (9) | 0.269 | 81.54 | 35.00 |
| E4.TRIS (10) | 0.228 | 81.54 | 37.00 |
| D4.TRIS (11) | 0.202 | 80.14 | 42.00 |
| P4.TRIS (12) | 0.148 | 81.54 | 38.00 |
In general, spectroscopic titrations were carried out by the addition of a 10.0 mL aqueous PAMAM–TRIS dendrimer solution to a thermostated vessel at 25 ± 0.1 °C. Then, identical aliquots of CuSO4 solution were added to the vessel each time while a stir bar vigorously stirred the solution. 15–20 seconds were usually allowed to provide sufficient time for Cu2+ to bind to the dendrimer before an absorbance measurement was acquired. The UV-Vis spectrum of the solution was recorded in the wavelength range of 400–900 nm with 5.00 nm intervals. λmax = 680 nm was the wavelength, which was associated with the complexation of Cu2+ ions with the internal tertiary amines of PAMAM–TRIS dendrimers. When the excess of Cu2+ ions was added after the equivalence point, the increase in the absorbance is levelled off indicating the maximum metal Cu2+ ions loading capacity of dendrimer has been reached. Finally, spectroscopic titration plots, on which the absorbance at the peak maximum of 680 nm as a function of the Cu2+ ions per PAMAM–TRIS dendrimers, were plotted. In order to determine the maximum metal loading capacity of each PAMAM–TRIS dendrimer, the titration end point was estimated as the extrapolated intersection of the linear regions of the curve before and after the equivalence point. The small absorbance beyond the equivalence point is due to a small absorbance contributed by the titrant.
Characterization
O), 1644 (HNCO), 1544 (HNCO). 1H NMR δH(400 MHz; D2O) 2.37 (32H, bm, NR2CH2CH2COOCH3), 2.45 (32H, t, NR2CH2CH2COOCH3), 2.64 (16H, bm, CONHCH2CH2NR2), 3.15 (16H, bm, CONHCH2CH2NR2), 3.61 (48H, s, COOCH3). 13C NMR δC(100 MHz; D2O) 31.96 (CH2CH2COOCH3), 47.67 (CH2CH2COOCH3), 48.18 (CH2CH2NR2), 49.54 (COOCH3), 170.56, 170.87 (NCH2CH2CONH), 171.56 (COOCH3).O). 1H NMR δH(300 MHz; DMSO-d6) 2.62 (40H, m, NR2CH2CH2COOCH3), 2.85 (40H, m, NR2CH2CH2COOCH3), 2.87 (40H, m, CONHCH2CH2NR2), 3.29 (40H, brm, CONHCH2CH2NR2), 3.78 (60H, s, COOCH3). 13C NMR δC(75 MHz; DMSO-d6) 32.66 (20C, NR2CH2CH2COOCH3), 37.28 (10C, CONHCH2CH2NR2), 50.48 (20C, NR2CH2CH2COOCH3), 51.83 (10C, CONHCH2CH2NR2), 51.87 (20C, COOCH3), 172.89, 173.13, 173.19 (10C, NR2CH2CH2CONH), 174.84 (20C, COOCH3).O), 1644 (HNCO), 1535 (HNCO). 1H-NMR δH(400 MHz; DMSO) 2.42 (48H, t, CH2CH2COOCH3), 2.74 (48H, t, CH2CH2COOCH3), 3.65 (72H, s, COOCH3). 13C NMR δC(100 MHz; DMSO) 172.86 (COOCH3), 75.41 (COOCH3), 51.48 (CH2CH2NR2), 49.15 (CH2CH2COOCH3), 32.63 (CH2CH2COOCH3).O), 1638 (HNCO), 1557 (HNCO). 1H NMR δH(400 MHz; D2O) 2.40 (64H, bm, NR2CH2CH2COOCH3), 2.52 (64H, t, NR2CH2CH2COOCH3), 2.67 (32H, bm, CONHCH2CH2NR2), 3.17 (32H, bm, CONHCH2CH2NR2), 3.62 (96H, s, COOCH3). 13C NMR δC(100 MHz; D2O) 31.32 (CH2CH2COOCH3), 47.41 (CH2CH2COOCH3), 48.47 (CH2CH2NR2) 50.35 (COOCH3), 171.02, 171.15, 171.43 (NCH2CH2CONH), 172.01 (COOCH3).O). 1H NMR δH(300 MHz; DMSO-d6) 2.60 (80H, m, NR2CH2CH2COOCH3), 2.86 (80H, brm, NR2CH2CH2COOCH3), 2.89 (80H, brm, CONHCH2CH2NR2), 3.27 (80H, brm, CONHCH2CH2NR2), 3.78 (120H, s, COOCH3). 13C NMR δC(75 MHz; DMSO-d6) 32.66 (40C, NR2CH2CH2COOCH3), 37.31 (20C, CONHCH2CH2NR2), 51.83 (40C, NR2CH2CH2COOCH3), 51.87 (40C, COOCH3), 52.92 (20C, CONHCH2CH2NR2), 171.71, 171.77, 171.85 (20C, NCH2CH2CONH), 173.13 (40C, COOCH3).O), 1642 (HNCO), 1535 (HNCO). 1H-NMR δH(400 MHz; DMSO) 2.42 (96H, t, CH2CH2COOCH3), 2.68 (96H, t, CH2CH2COOCH3), 3.59 (144H s, CH2CH2COOCH3). 13C NMR δC(100 MHz; DMSO) 172.43 (COOCH3), 51.15 (CH2CH2COOCH3), 48.87 (CH2CH2COOCH3), 32.00 (CH2CH2COOCH3).O), 1558 (HNCO), 1392 (O–H). 1H NMR δH(400 MHz; CD3OD) 2.56 (32H, bm, CH2CH2CONHCR3), 2.76 (32H, bm, CH2CH2CONHCR3), 3.57 (96H, bs, CH2OH). 13C NMR δC(100 MHz; CD3OD) 32.22 (CH2CH2CONHCR3), 48.70 (CH2CH2CONHCR3), 49.24 (CH2CH2NR2), 56.51 (CONHCR2CH2OH), 63.26 (CONHCR2CH2OH), 174.66, 174.82, 180.73, 181.01 (NCH2CH2CONH).O), 1554 (HNCO), 1392 (O–H). 1H NMR δH(300 MHz; CD3OD) 2.61 (40H, m, CH2CH2CONHR3), 2.78 (40H, bm, CH2CH2CONHR3), 2.77 (40H, bm, CONHCH2CH2NR2), 3.23 (40H, bm, CONHCH2CH2NR2), 3.65 (120H, s, CH2OH). 13C NMR δC(75 MHz; CD3OD) 33.67 (20C, NR2CH2CH2COOCH3), 38.23 (10C, CONHCH2CH2NR2), 50.33 (20C, NR2CH2CH2COOCH3), 51.22 (10C, CONHCH2CH2NR2), 56.47 (15C, CONHCCH2OH), 63.69 (48C, CONHCCH2OH), 174.83, 175.35, 180.48, 181.12 (20C, NCH2CH2CONH).O), 1563 (HNCO), 1393 (O–H). 1H NMR δH(400 MHz; CD3OD) 2.58 (48H, bm, CH2CH2CONHCR3), 2.77 (48H, t, CH2CH2CONHCR3), 3.54 (192H, bs, CH2OH). 13C NMR δC(100 MHz; CD3OD) 32.64 (CH2CH2CONHCR3), 48.91 (CH2CH2CONHCR3), 52.08 (CH2CH2NR2), 56.30 (CONHCR2CH2OH), 63.12 (CONHCR2CH2OH) 174.61, 175.53, 181.12 (NCH2CH2CONH).O), 1558 (HNCO), 1394 (O–H). 1H NMR δH(400 MHz; CD3OD) 2.52 (64H, bm, CH2CH2CONHCR3), 2.70 (64H, bm, CH2CH2CONHCR3), 3.65 (192H, bs, CH2OH). 13C NMR δC(100 MHz; CD3OD) 32.62 (CH2CH2CONHCR3), 48.88 (CH2CH2CONHCR3), 49.02 (CH2CH2NR2), 56.37 (CONHCR2CH2OH), 63.06 (CONHCR2CH2OH), 174.62, 174.92, 180.93, 181.21 (NCH2CH2CONH).O), 1554 (HNCO), 1388 (O–H). 1H NMR δH(300 MHz; CD3OD) 2.55 (80H, bm, CH2CH2CONHR3), 2.70 (80H, bm, CH2CH2CONHR3), 2.82 (80H, bm, CONHCH2CH2NR2), 3.29 (80H, bm, CONHCH2CH2NR2), 3.71 (240H, s, CH2OH). 13C NMR δC(75 MHz; CD3OD) 33.55 (40C, CH2CH2CONHR3), 38.60 (20C, CONHCH2CH2NR2), 50.03 (40C, CH2CH2CONHR3), 51.10 (20C, CONHCH2CH2NR2), 56.37 (30C, CONHCR2CH2OH), 63.6 (96C, CONHCR2CH2OH), 174.93, 175.44, 180.4, 181.04 (40C, NCH2CH2CONH).O), 1558 (HNCO), 1392 (O–H). 1H NMR δH(400 MHz; CD3OD) 2.5 (96H, bm, CH2CH2CONHC), 2.68 (96H, t, CH2CH2CONHC), 3.63 (288H, bs, CH2OH). 13C NMR δC(100 MHz; CD3OD) 32.59 (CH2CH2CONHC), 48.97 (CH2CH2CONHC), 51.18 (CH2CH2NR2), 56.38 (CONHCR2CH2OH), 63.02 (CONHCR2CH2OH) 174.65, 175.55, 181.09 (NCH2CH2CONH).Acknowledgements
This research has been supported by Yıldız Technical University Scientific Research Projects Coordination Department. Project Numbers (2011-01-02-KAP04, 2011-01-02-KAP05, 2011-01-02-KAP06 and 2012-01-02-DOP05).References
- R. Esfand and D. A. Tomalia, Drug Discovery Today, 2001, 6, 427–436 CrossRef CAS.
- R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS PubMed.
- L. Cheng, G. E. Pacey and J. A. Cox, Electrochim. Acta, 2001, 46, 4223–4228 CrossRef CAS.
- M. Tülü and A. S. Ertürk, in A Search for Antibacterial Agents, ed. V. Bobbarala, In Tech, 2012, ch. 6, pp. 89–106, DOI:10.5772/46051.
- K. Winnicka, K. Sosnowska, P. Wieczorek, P. T. Sacha and E. Tryniszewska, Biol. Pharm. Bull., 2011, 34, 1129–1133 CAS.
- K. Winnicka, M. Wroblewska, P. Wieczorek, P. T. Sacha and E. Tryniszewska, Molecules, 2012, 17, 4612–4624 CrossRef CAS PubMed.
- J. F. Kukowska-Latallo, A. U. Bielinska, J. Johnson, R. Spindler, D. A. Tomalia and J. R. Baker, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4897–4902 CrossRef CAS.
- J. D. Eichman, A. U. Bielinska, J. F. Kukowska-Latallo and J. R. Baker Jr, Pharm. Sci. Technol. Today, 2000, 3, 232–245 CrossRef CAS.
- D. Luo, K. Haverstick, N. Belcheva, E. Han and W. M. Saltzman, Macromolecules, 2002, 35, 3456–3462 CrossRef CAS.
- A. K. Patri, I. J. Majoros and J. R. Baker Jr, Curr. Opin. Chem. Biol., 2002, 6, 466–471 CrossRef CAS.
- U. Boas and P. M. H. Heegaard, Chem. Soc. Rev., 2004, 33, 43–63 RSC.
- A. E. Beezer, A. S. H. King, I. K. Martin, J. C. Mitchell, L. J. Twyman and C. F. Wain, Tetrahedron, 2003, 59, 3873–3880 CrossRef CAS.
- J. W. J. Knapen, d. M. A. W. van, W. J. C. de, L. P. W. N. M. van, P. Wijkens, D. M. Grove and K. G. van, Nature, 1994, 372, 659–663 CrossRef CAS PubMed.
- D. A. Tomalia and P. R. Dvornic, Nature, 1994, 372, 617–618 CrossRef CAS PubMed.
- G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendritic Molecules: Concepts, Syntheses, Perspectives, Wiley, 2008 Search PubMed.
- WO2007149501A2, 2007.
- A. S. Ertürk, M. Tülü, A. E. Bozdoğan and T. Parali, Eur. Polym. J., 2014, 52, 218–226 CrossRef PubMed.
- M. Godoi, G. V. Botteselle, J. Rafique, M. S. T. Rocha, J. M. Pena and A. L. Braga, Asian J. Org. Chem., 2013, 2, 746–749 CrossRef CAS PubMed.
- F. Zhang and G. Zhang, Green Chem., 2011, 13, 178–184 RSC.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117–132 CrossRef CAS.
- M. Xu, Q. R. Chen, D. Kumar, S. A. Stass and A. J. Mixson, Mol. Genet. Metab., 1998, 64, 193–197 CrossRef CAS PubMed.
- N. Malik, R. Wiwattanapatapee, R. Klopsch, K. Lorenz, H. Frey, J. W. Weener, E. W. Meijer, W. Paulus and R. Duncan, J. Controlled Release, 2000, 65, 133–148 CrossRef CAS.
- K. Öztürk, A. S. Ertürk, C. Sarısözen, M. Tulu and S. Çalış, Journal of Microencapsulation, 2014, 31, 127–136 CrossRef PubMed.
- I. J. Majoros, C. B. Mehta and J. R. Baker, J. Comput. Theor. Nanosci., 2004, 1, 193–198 CrossRef CAS PubMed.
- G. R. Newkome, G. R. Baker, S. Arai, M. J. Saunders, P. S. Russo, K. J. Theriot, C. N. Moorefield, L. E. Rogers and J. E. Miller, et al., J. Am. Chem. Soc., 1990, 112, 8458–8465 CrossRef CAS.
- R. Esfand and D. A. Tomalia, in Dendrimers and Other Dendritic Polymers, John Wiley & Sons, Ltd, 2002, pp. 587–604, DOI:10.1002/0470845821.ch25.
- M. S. Diallo, S. Christie, P. Swaminathan, L. Balogh, X. Shi, W. Um, C. Papelis, W. A. Goddard III and J. H. Johnson Jr., Langmuir, 2004, 20, 2640–2651 CrossRef CAS.
- S. Pande and R. M. Crooks, Langmuir, 2011, 27, 9609–9613 CrossRef CAS PubMed.
- Z. V. Feng, J. L. Lyon, J. S. Croley, R. M. Crooks, D. A. Vanden Bout and K. J. Stevenson, J. Chem. Educ., 2009, 86, 368 CrossRef CAS.
- M. Zhao, L. Sun and R. M. Crooks, J. Am. Chem. Soc., 1998, 120, 4877–4878 CrossRef CAS.
- P. Chen, Y. Yang, P. Bhattacharya, P. Wang and P. C. Ke, J. Phys. Chem. C, 2011, 115, 12789–12796 CAS.
- H. Yokoi and T. Isobe, Bull. Chem. Soc. Jpn., 1969, 42, 2187–2193 CrossRef CAS.
- B. P. Kennedy and A. B. P. Lever, J. Am. Chem. Soc., 1973, 95, 6907–6913 CrossRef CAS.
- A. R. Amundsen, J. Whelan and B. Bosnich, J. Am. Chem. Soc., 1977, 99, 6730–6739 CrossRef CAS.
- I. J. Majoros, B. Keszler, S. Woehler, T. Bull and J. R. Baker, Macromolecules, 2003, 36, 5526–5529 CrossRef CAS.
- I. J. Majoros, T. P. Thomas, C. B. Mehta and J. R. Baker, J. Med. Chem., 2005, 48, 5892–5899 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |